
Effective Targeting of Melanoma Cells by Combination of Mcl-1 and Bcl-2/Bcl-xL/Bcl-w Inhibitors
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
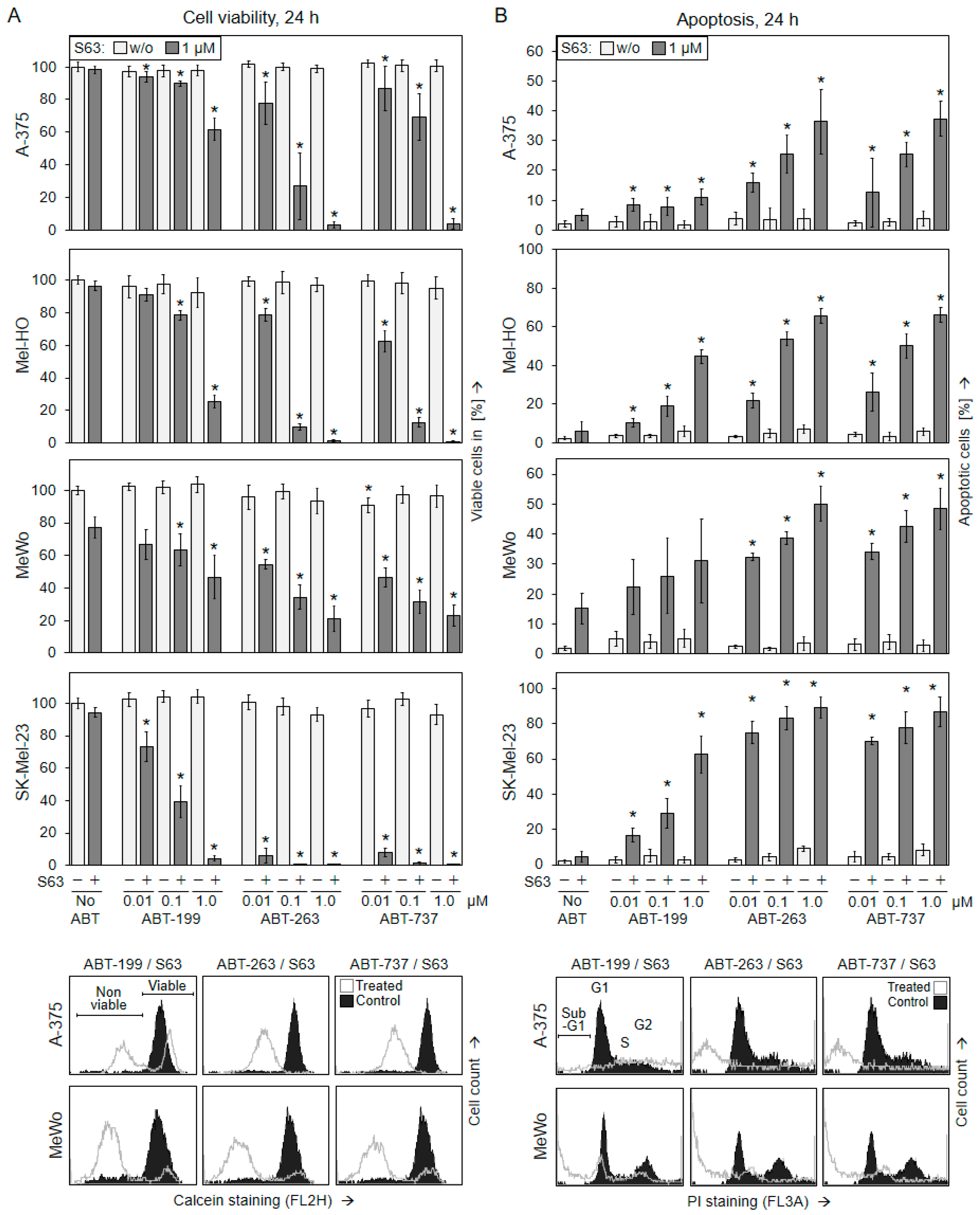
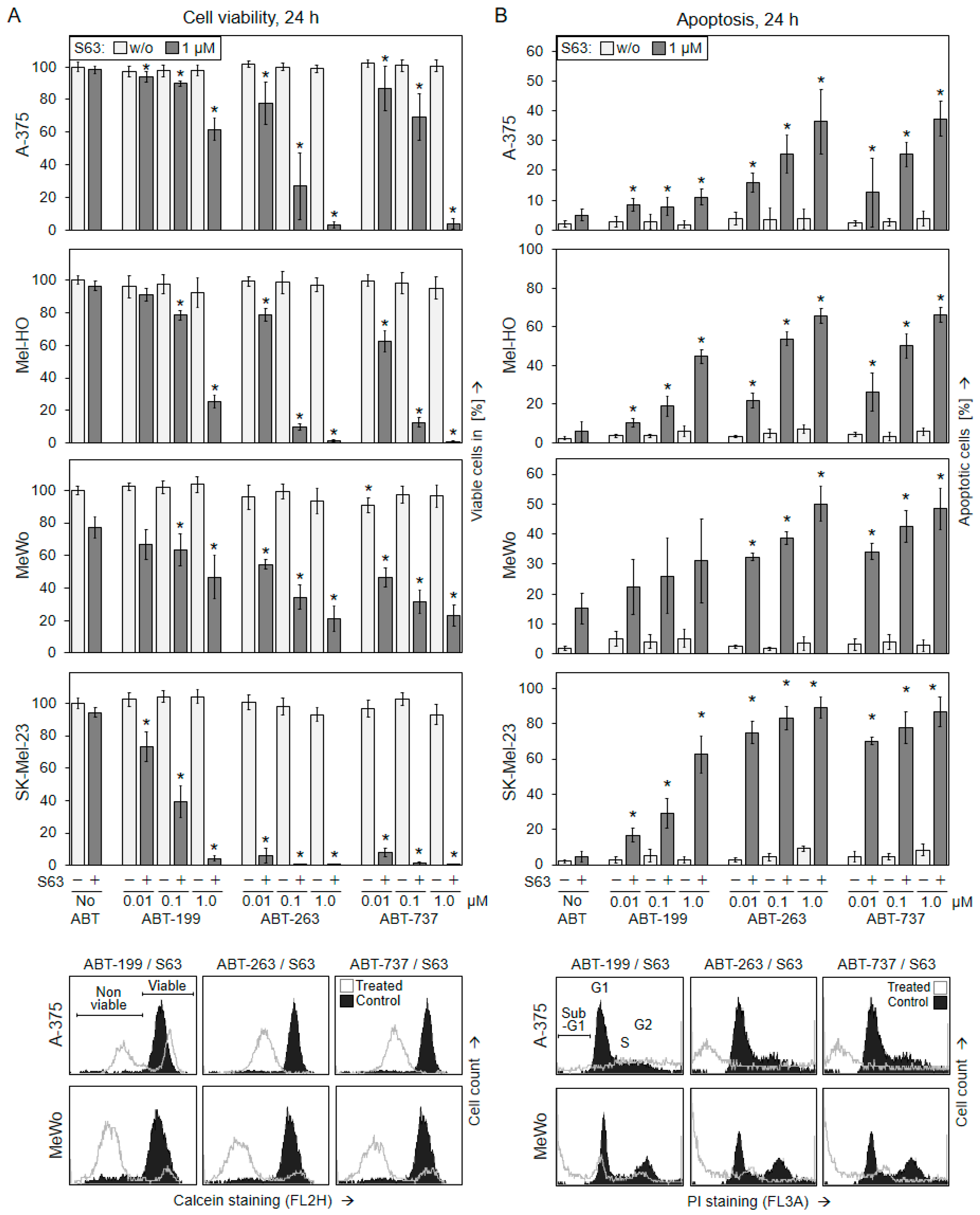
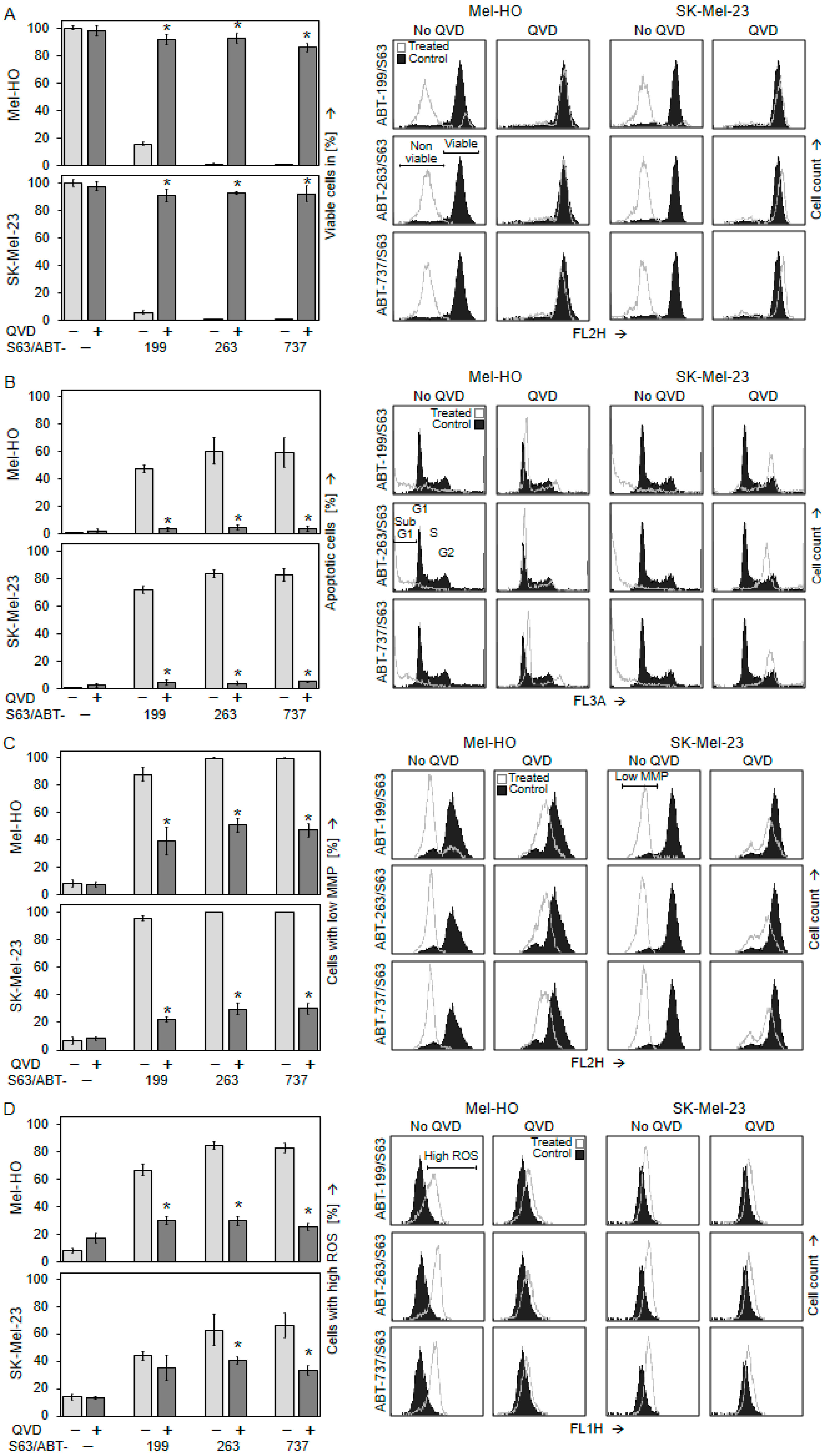
2.1. Significant Decrease in Cell Viability Only after Combination of BH3 Mimetics
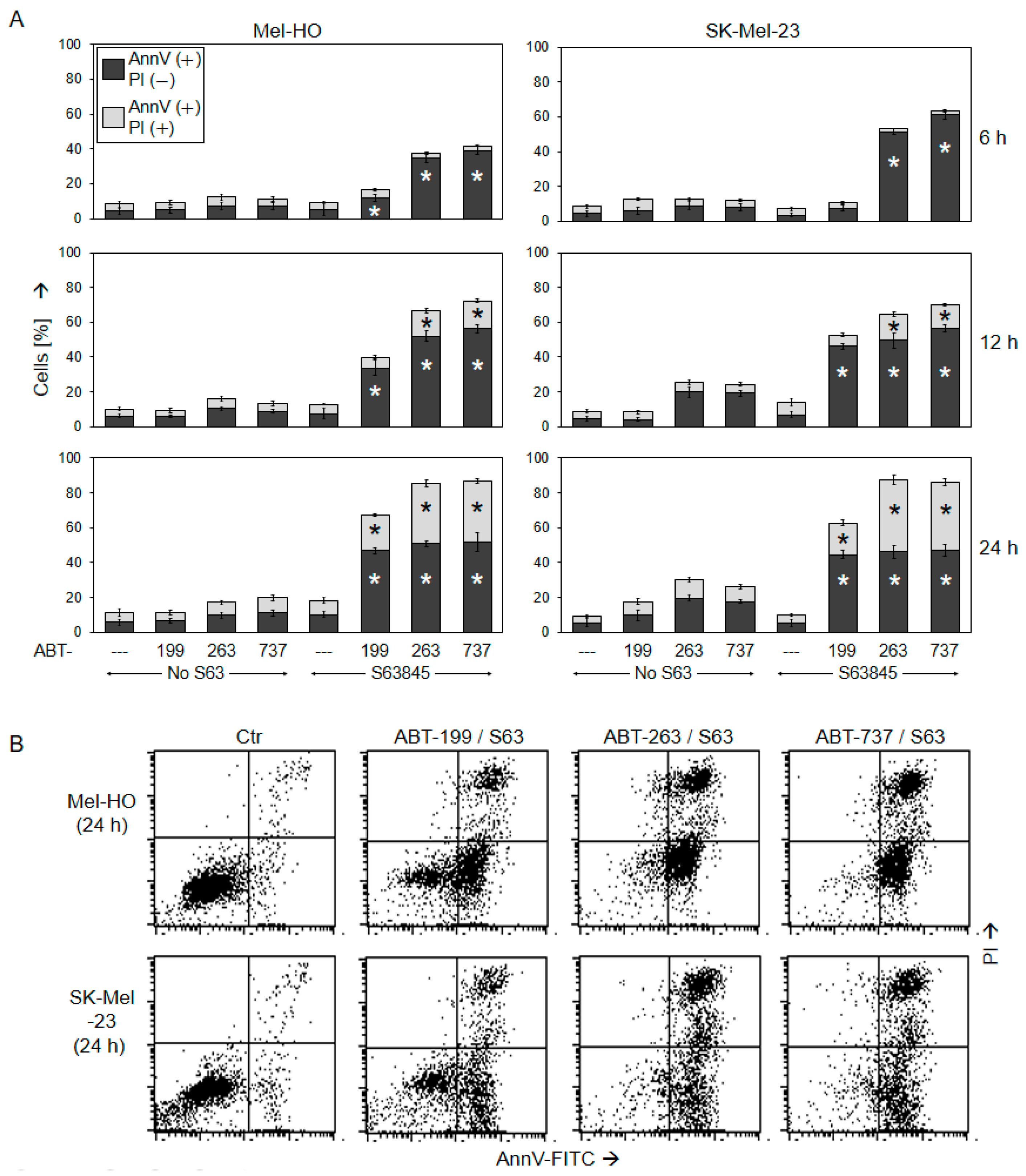
2.2. Decreased Cell Viability Correlates with Induction of Apoptosis
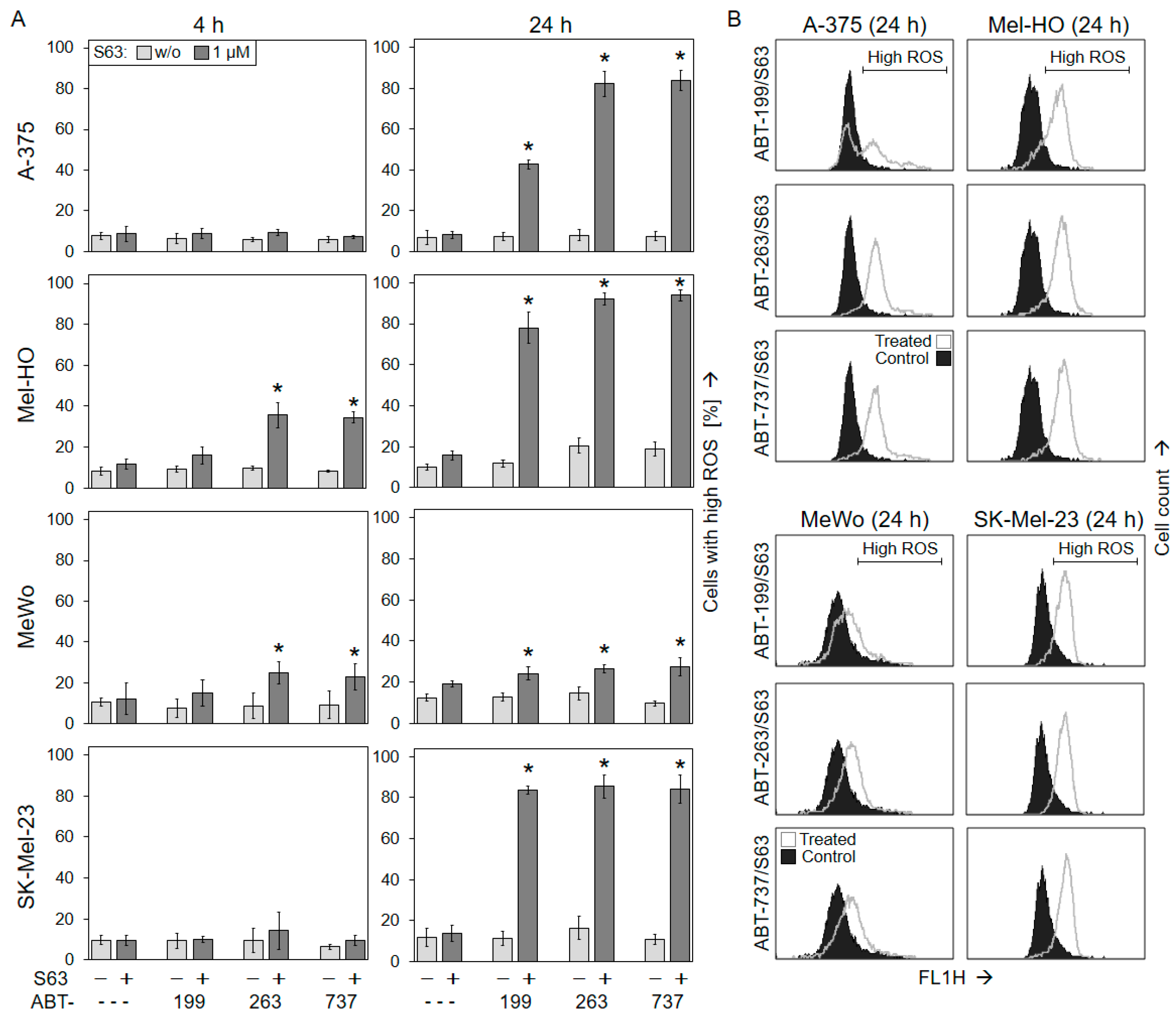
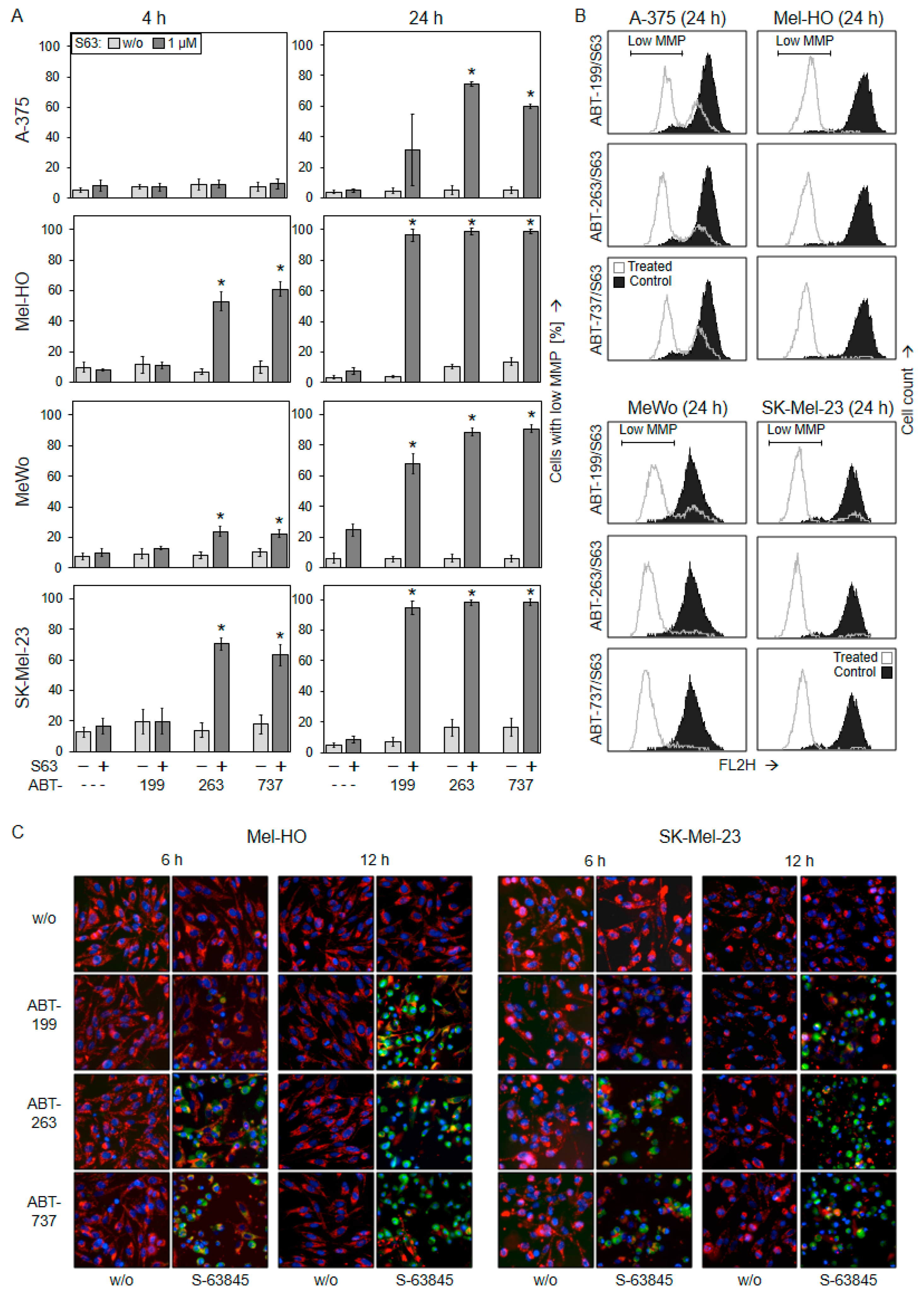
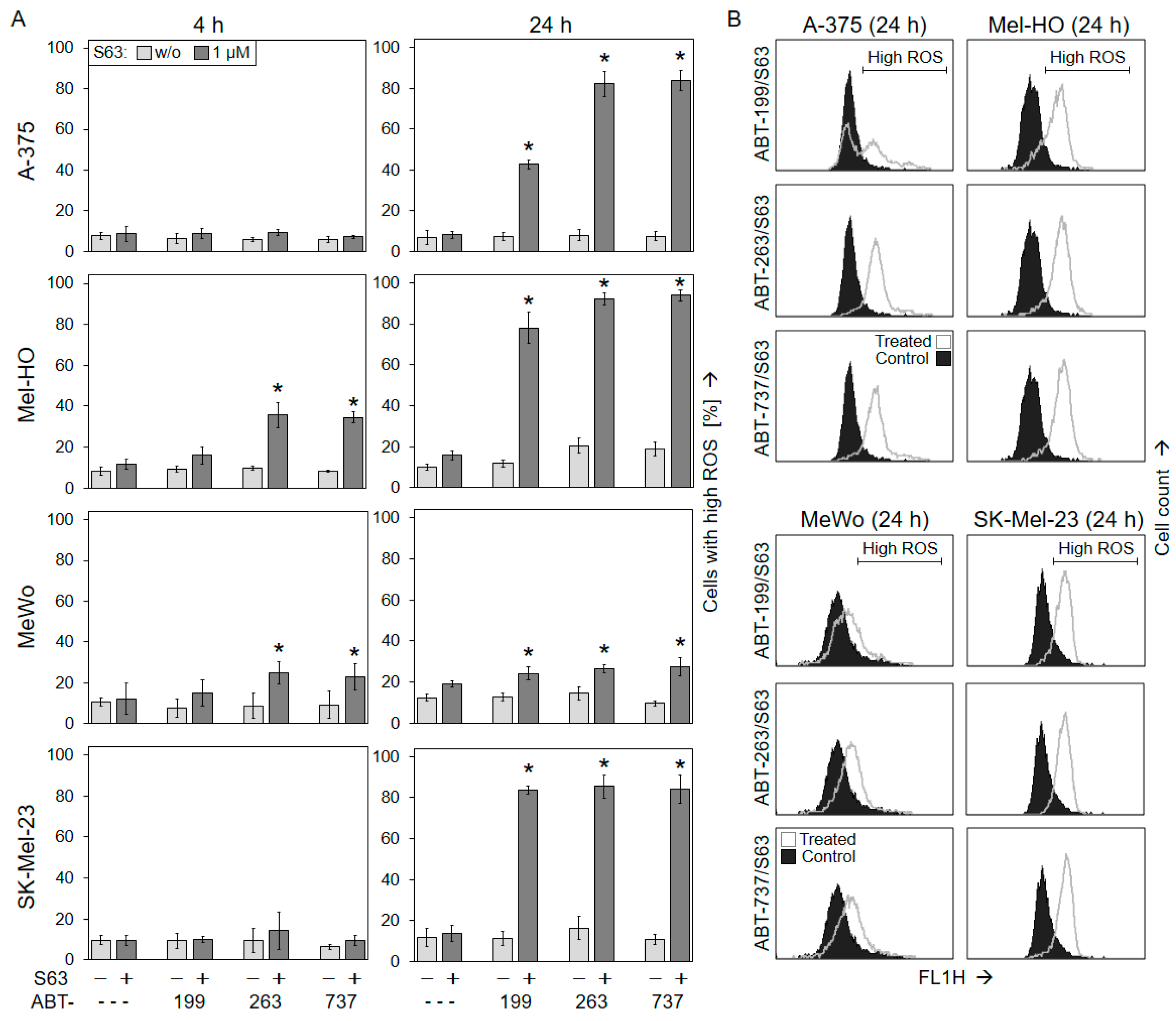
2.3. Activation of Mitochondrial Apoptosis Pathways
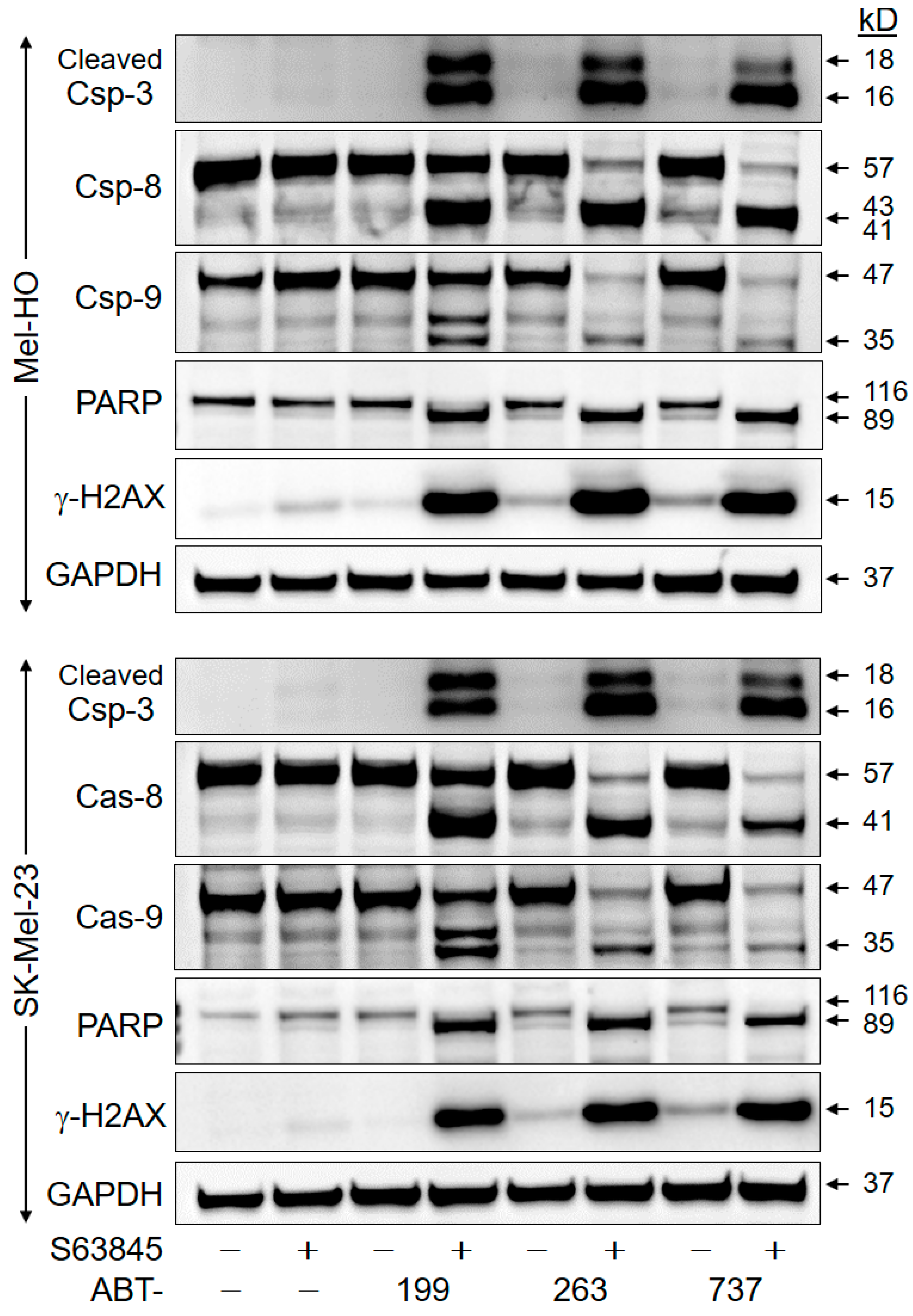
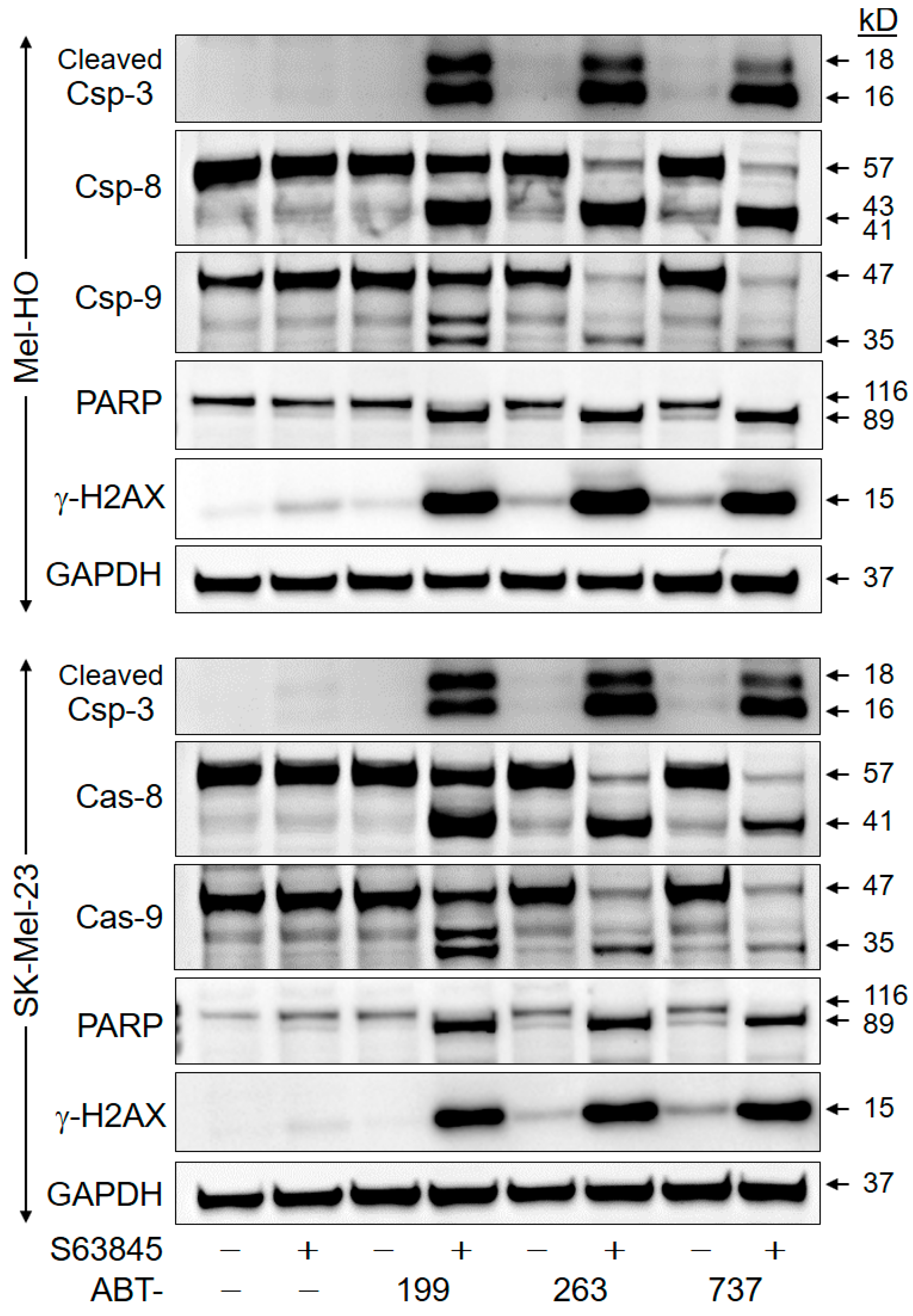
2.4. Crucial Role of Caspase-Mediated Pathways
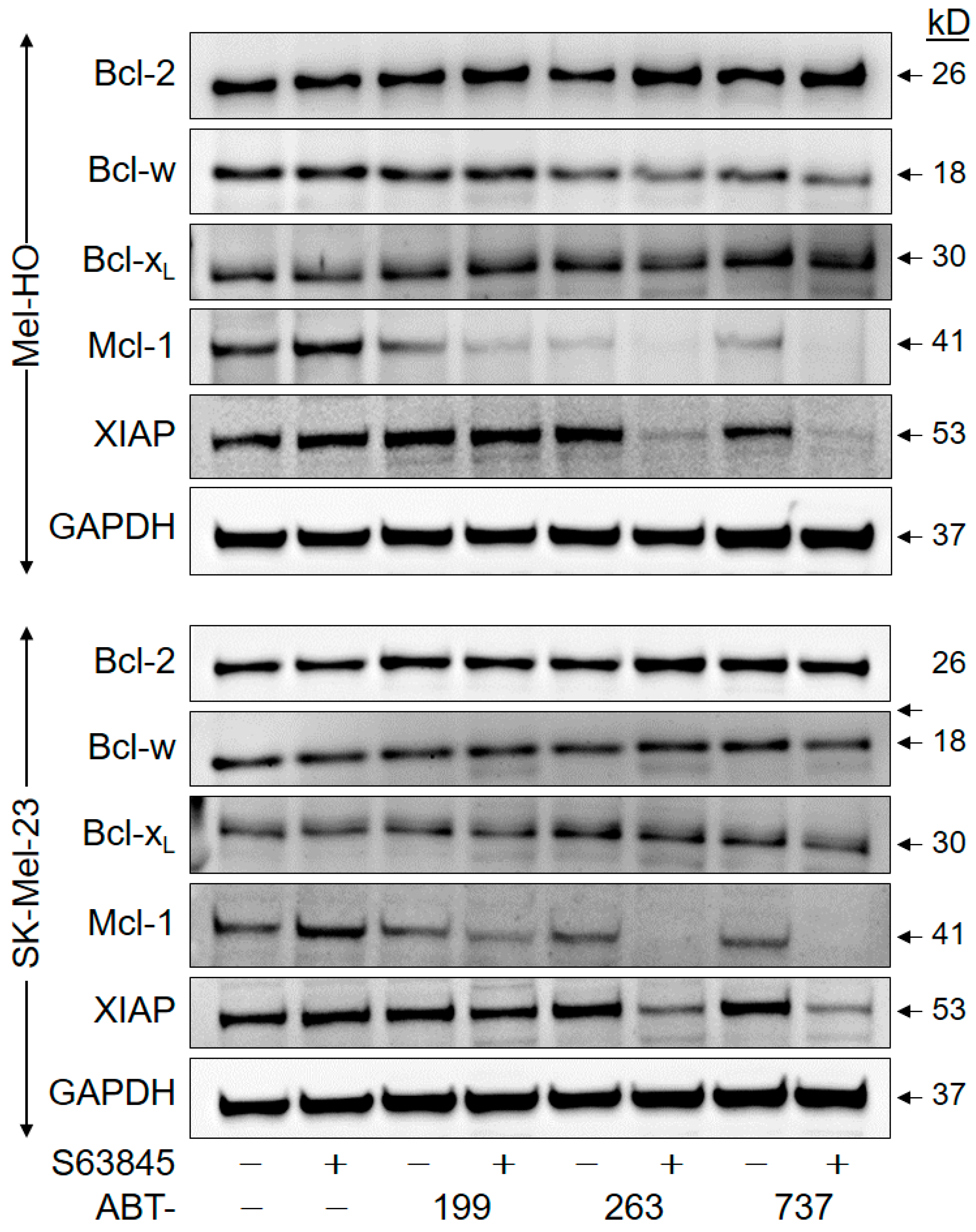
2.5. Downregulation of Anti-Apoptotic Factors
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Quantification of Cell Viability and Apoptosis
4.3. Assays for Mitochondrial Membrane Potential and Reactive Oxygen Species (ROS)
4.4. Western Blotting
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dreno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Long, G.V. Systemic treatment for BRAF-mutant melanoma: Where do we go next? Lancet Oncol. 2014, 15, e371–e381. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dreno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment—Update 2022. Eur. J. Cancer 2022, 170, 256–284. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes With Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients With Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.; Klairmont, M.; Sharfman, W.H.; Kaufman, H.L. Interleukin-2, Ipilimumab, and Anti-PD-1: Clinical management and the evolving role of immunotherapy for the treatment of patients with metastatic melanoma. Cancer Biol. Ther. 2021, 22, 513–526. [Google Scholar] [CrossRef]
- Johnpulle, R.A.; Johnson, D.B.; Sosman, J.A. Molecular Targeted Therapy Approaches for BRAF Wild-Type Melanoma. Curr. Oncol. Rep. 2016, 18, 6. [Google Scholar] [CrossRef]
- Dimitriou, F.; Long, G.V.; Menzies, A.M. Novel adjuvant options for cutaneous melanoma. Ann. Oncol. 2021, 32, 854–865. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Eberle, J.; Fecker, L.F.; Forschner, T.; Ulrich, C.; Rowert-Huber, J.; Stockfleth, E. Apoptosis pathways as promising targets for skin cancer therapy. Brit. J. Dermatol. 2007, 156, 18–24. [Google Scholar] [CrossRef]
- Krammer, P.H.; Arnold, R.; Lavrik, I.N. Life and death in peripheral T cells. Nat. Rev. Immunol. 2007, 7, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Janicke, R.U.; Schulze-Osthoff, K. Many cuts to ruin: A comprehensive update of caspase substrates. Cell Death Differ. 2003, 10, 76–100. [Google Scholar] [CrossRef] [PubMed]
- Soldani, C.; Scovassi, A.I. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: An update. Apoptosis 2002, 7, 321–328. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhu, F.; Cho, Y.Y.; Tang, F.; Zykova, T.; Ma, W.Y.; Bode, A.M.; Dong, Z. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 2006, 23, 121–132. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Zhivkova, V.; Kiecker, F.; Langer, P.; Eberle, J. Crucial role of reactive oxygen species (ROS) for the proapoptotic effects of indirubin derivative DKP-073 in melanoma cells. Mol. Carcinog. 2019, 58, 258–269. [Google Scholar] [CrossRef]
- Quast, S.A.; Berger, A.; Eberle, J. ROS-dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL-induced apoptosis. Cell Death Dis. 2013, 4, e839. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef]
- Raisova, M.; Hossini, A.M.; Eberle, J.; Riebeling, C.; Wieder, T.; Sturm, I.; Daniel, P.T.; Orfanos, C.E.; Geilen, C.C. The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J. Investig. Dermatol. 2001, 117, 333–340. [Google Scholar] [CrossRef]
- Jansen, B.; Schlagbauer-Wadl, H.; Brown, B.D.; Bryan, R.N.; van Elsas, A.; Muller, M.; Wolff, K.; Eichler, H.G.; Pehamberger, H. bcl-2 antisense therapy chemosensitizes human melanoma in SCID mice. Nat. Med. 1998, 4, 232–234. [Google Scholar] [CrossRef]
- Eberle, J. Countering TRAIL Resistance in Melanoma. Cancers 2019, 11, 656. [Google Scholar] [CrossRef]
- Hanada, M.; Delia, D.; Aiello, A.; Stadtmauer, E.; Reed, J.C. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993, 82, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Krajewska, M.; Shabaik, A.; Wang, H.G.; Irie, S.; Fong, L.; Reed, J.C. Immunohistochemical analysis of in vivo patterns of Bcl-X expression. Cancer Res. 1994, 54, 5501–5507. [Google Scholar] [PubMed]
- Kozopas, K.M.; Yang, T.; Buchan, H.L.; Zhou, P.; Craig, R.W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 1993, 90, 3516–3520. [Google Scholar] [CrossRef]
- Diepstraten, S.T.; Anderson, M.A.; Czabotar, P.E.; Lessene, G.; Strasser, A.; Kelly, G.L. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat. Rev. Cancer 2022, 22, 45–64. [Google Scholar] [CrossRef]
- Leiter, U.; Schmid, R.M.; Kaskel, P.; Peter, R.U.; Krahn, G. Antiapoptotic bcl-2 and bcl-xL in advanced malignant melanoma. Arch. Dermatol. Res. 2000, 292, 225–232. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Desideri, M.; Ciuffreda, L.; Mottolese, M.; Ribatti, D.; Vacca, A.; Del Rosso, M.; Marcocci, L.; Zupi, G.; Del Bufalo, D. Bcl-2 overexpression in melanoma cells increases tumor progression-associated properties and in vivo tumor growth. J. Cell. Physiol. 2005, 205, 414–421. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Lee, E.F.; Harris, T.J.; Tran, S.; Evangelista, M.; Arulananda, S.; John, T.; Ramnac, C.; Hobbs, C.; Zhu, H.; Gunasingh, G.; et al. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death Dis. 2019, 10, 342. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. BCL-w: Apoptotic and non-apoptotic role in health and disease. Cell Death Dis. 2020, 11, 260. [Google Scholar] [CrossRef]
- Kim, Y.J.; Tsang, T.; Anderson, G.R.; Posimo, J.M.; Brady, D.C. Inhibition of BCL2 Family Members Increases the Efficacy of Copper Chelation in BRAFV600E-Driven Melanoma. Cancer Res. 2020, 80, 1387–1400. [Google Scholar] [CrossRef]
- Sarif, Z.; Tolksdorf, B.; Fechner, H.; Eberle, J. Mcl-1 targeting strategies unlock the proapoptotic potential of TRAIL in melanoma cells. Mol. Carcinog. 2020, 59, 1256–1268. [Google Scholar] [CrossRef]
- Placzek, W.J.; Wei, J.; Kitada, S.; Zhai, D.; Reed, J.C.; Pellecchia, M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010, 1, e40. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef]
- Peng, Z.; Gillissen, B.; Richter, A.; Sinnberg, T.; Schlaak, M.S.; Eberle, J. Enhanced Apoptosis and Loss of Cell Viability in Melanoma Cells by Combined Inhibition of ERK and Mcl-1 Is Related to Loss of Mitochondrial Membrane Potential, Caspase Activation and Upregulation of Proapoptotic Bcl-2 Proteins. Int. J. Mol. Sci. 2023, 24, 4961. [Google Scholar] [CrossRef]
- Torrens-Mas, M.; Cordani, M.; Mullappilly, N.; Pacchiana, R.; Riganti, C.; Palmieri, M.; Pons, D.G.; Roca, P.; Oliver, J.; Donadelli, M. Mutant p53 induces SIRT3/MnSOD axis to moderate ROS production in melanoma cells. Arch. Biochem. Biophys. 2020, 679, 108219. [Google Scholar] [CrossRef]
- Tantawy, S.I.; Sarkar, A.; Hubner, S.; Tan, Z.; Wierda, W.G.; Eldeib, A.; Zhang, S.; Kornblau, S.; Gandhi, V. Mechanisms of MCL-1 Protein Stability Induced by MCL-1 Antagonists in B-Cell Malignancies. Clin. Cancer Res. 2023, 29, 446–457. [Google Scholar] [CrossRef]
- Herrant, M.; Jacquel, A.; Marchetti, S.; Belhacène, N.; Colosetti, P.; Luciano, F.; Auberger, P. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene 2004, 23, 7863–7873. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Mason, K.D.; Carpinelli, M.R.; Fletcher, J.I.; Collinge, J.E.; Hilton, A.A.; Ellis, S.; Kelly, P.N.; Ekert, P.G.; Metcalf, D.; Roberts, A.W.; et al. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef]
- Lok, S.W.; Whittle, J.R.; Vaillant, F.; Teh, C.E.; Lo, L.L.; Policheni, A.N.; Bergin, A.R.T.; Desai, J.; Ftouni, S.; Gandolfo, L.C.; et al. A Phase Ib Dose-Escalation and Expansion Study of the BCL2 Inhibitor Venetoclax Combined with Tamoxifen in ER and BCL2-Positive Metastatic Breast Cancer. Cancer Discov. 2019, 9, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, T.L.; Floros, K.V.; Naseri, M.; Powell, K.M.; Cook, W.; March, R.J.; Stein, G.T.; Greninger, P.; Maves, Y.K.; Saunders, L.R.; et al. Venetoclax Is Effective in Small-Cell Lung Cancers with High BCL-2 Expression. Clin. Cancer Res. 2018, 24, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Place, A.E.; Goldsmith, K.; Bourquin, J.P.; Loh, M.L.; Gore, L.; Morgenstern, D.A.; Sanzgiri, Y.; Hoffman, D.; Zhou, Y.; Ross, J.A.; et al. Accelerating drug development in pediatric cancer: A novel Phase I study design of venetoclax in relapsed/refractory malignancies. Future Oncol. 2018, 14, 2115–2129. [Google Scholar] [CrossRef]
- Kehr, S.; Vogler, M. It’s time to die: BH3 mimetics in solid tumors. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118987. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Skees, J.; Todd, K.J.; West, D.A.; Lambert, K.A.; Robinson, W.A.; Amato, C.M.; Couts, K.L.; Van Gulick, R.; MacBeth, M.; et al. MCL1 inhibitors S63845/MIK665 plus Navitoclax synergistically kill difficult-to-treat melanoma cells. Cell Death Dis. 2020, 11, 443. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.A.; Goldstein, N.B.; Johannes, W.U.; Walton, C.H.; Fujita, M.; Norris, D.A.; Shellman, Y.G. BH3 Mimetic ABT-737 and a Proteasome Inhibitor Synergistically Kill Melanomas through Noxa-Dependent Apoptosis. J. Investig. Dermatol. 2009, 129, 964–971. [Google Scholar] [CrossRef]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Brennan, M.S.; Chang, C.; Tai, L.; Lessene, G.; Strasser, A.; Dewson, G.; Kelly, G.L.; Herold, M.J. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood 2018, 132, 1573–1583. [Google Scholar] [CrossRef]
- Dastur, A.; Choi, A.; Costa, C.; Yin, X.Q.; Williams, A.; McClanaghan, J.; Greenberg, M.; Roderick, J.; Patel, N.U.; Boisvert, J.; et al. NOTCH1 Represses MCL-1 Levels in GSI-resistant T-ALL, Making them Susceptible to ABT-263. Clin. Cancer Res. 2019, 25, 312–324. [Google Scholar] [CrossRef]
- Guieze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernandez-Sanchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.E13. [Google Scholar] [CrossRef]
- Faber, A.C.; Farago, A.F.; Costa, C.; Dastur, A.; Gomez-Caraballo, M.; Robbins, R.; Wagner, B.L.; Rideout, W.M., 3rd; Jakubik, C.T.; Ham, J.; et al. Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E1288–E1296. [Google Scholar] [CrossRef]
- Williams, M.M.; Lee, L.; Hicks, D.J.; Joly, M.M.; Elion, D.; Rahman, B.; McKernan, C.; Sanchez, V.; Balko, J.M.; Stricker, T.; et al. Key Survival Factor, Mcl-1, Correlates with Sensitivity to Combined Bcl-2/Bcl-xL Blockade. Mol. Cancer Res. 2017, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Szlavik, Z.; Ondi, L.; Csekei, M.; Paczal, A.; Szabo, Z.B.; Radics, G.; Murray, J.; Davidson, J.; Chen, I.; Davis, B.; et al. Structure-Guided Discovery of a Selective Mcl-1 Inhibitor with Cellular Activity. J. Med. Chem. 2019, 62, 6913–6924. [Google Scholar] [CrossRef] [PubMed]
- Senichkin, V.V.; Streletskaia, A.Y.; Gorbunova, A.S.; Zhivotovsky, B.; Kopeina, G.S. Saga of Mcl-1: Regulation from transcription to degradation. Cell Death Differ. 2020, 27, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.Y.; Dreyer, J.; Emran, A.A.; Gunatilake, D.; Pirozyan, M.; Cullinane, C.; Dutton-Regester, K.; Rizos, H.; Hayward, N.K.; McArthur, G.; et al. Co-targeting bromodomain and extra-terminal proteins and MCL1 induces synergistic cell death in melanoma. Int. J. Cancer 2020, 147, 2176–2189. [Google Scholar] [CrossRef] [PubMed]
- Weeden, C.E.; Ah-Cann, C.; Holik, A.Z.; Pasquet, J.; Garnier, J.M.; Merino, D.; Lessene, G.; Asselin-Labat, M.L. Dual inhibition of BCL-XL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene 2018, 37, 4475–4488. [Google Scholar] [CrossRef] [PubMed]
- Kehr, S.; Haydn, T.; Bierbrauer, A.; Irmer, B.; Vogler, M.; Fulda, S. Targeting BCL-2 proteins in pediatric cancer: Dual inhibition of BCL-X(L) and MCL-1 leads to rapid induction of intrinsic apoptosis. Cancer Lett. 2020, 482, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.J.; Palmieri, M.; Riffkin, C.D.; Sakthianandeswaren, A.; Djajawi, T.M.; Hirokawa, Y.; Shuttleworth, V.; Segal, D.H.; White, C.A.; Nhu, D.; et al. Defining the susceptibility of colorectal cancers to BH3-mimetic compounds. Cell Death Dis. 2020, 11, 735. [Google Scholar] [CrossRef]
- Mukherjee, N.; Amato, C.M.; Skees, J.; Todd, K.J.; Lambert, K.A.; Robinson, W.A.; Van Gulick, R.; Weight, R.M.; Dart, C.R.; Tobin, R.P.; et al. Simultaneously Inhibiting BCL2 and MCL1 Is a Therapeutic Option for Patients with Advanced Melanoma. Cancers 2020, 12, 2182. [Google Scholar] [CrossRef]
- Griffioen, M.S.; de Leeuw, D.C.; Janssen, J.; Smit, L. Targeting Acute Myeloid Leukemia with Venetoclax; Biomarkers for Sensitivity and Rationale for Venetoclax-Based Combination Therapies. Cancers 2022, 14, 3456. [Google Scholar] [CrossRef] [PubMed]
- Bierbrauer, A.; Jacob, M.; Vogler, M.; Fulda, S. A direct comparison of selective BH3-mimetics reveals BCL-X(L), BCL-2 and MCL-1 as promising therapeutic targets in neuroblastoma. Br. J. Cancer 2020, 122, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Lu, Y.; Yu, R.; Xie, J.; Zhou, S. Synergistic Effects of TW-37 and ABT-263 on Renal Cell Carcinoma Cells. Cancer Manag. Res. 2021, 13, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Strosnider, A.; Vagher, B.; Lambert, K.A.; Slaven, S.; Robinson, W.A.; Amato, C.M.; Couts, K.L.; Bemis, J.G.T.; Turner, J.A.; et al. BH3 mimetics induce apoptosis independent of DRP-1 in melanoma. Cell Death Dis. 2018, 9, 907. [Google Scholar] [CrossRef]
- Hartman, M.L.; Gajos-Michniewicz, A.; Talaj, J.A.; Mielczarek-Lewandowska, A.; Czyz, M. BH3 mimetics potentiate pro-apoptotic activity of encorafenib in BRAF(V600E) melanoma cells. Cancer Lett. 2021, 499, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Fofaria, N.M.; Frederick, D.T.; Sullivan, R.J.; Flaherty, K.T.; Srivastava, S.K. Overexpression of Mcl-1 confers resistance to BRAFV600E inhibitors alone and in combination with MEK1/2 inhibitors in melanoma. Oncotarget 2015, 6, 40535–40556. [Google Scholar] [CrossRef]
- Haasler, L.; Kondadi, A.K.; Tsigaras, T.; von Montfort, C.; Graf, P.; Stahl, W.; Brenneisen, P. The BH3 mimetic (±) gossypol induces ROS-independent apoptosis and mitochondrial dysfunction in human A375 melanoma cells in vitro. Arch. Toxicol. 2021, 95, 1349–1365. [Google Scholar] [CrossRef]
- Jiang, C.C.; Wroblewski, D.; Yang, F.; Hersey, P.; Zhang, X.D. Human melanoma cells under endoplasmic reticulum stress are more susceptible to apoptosis induced by the BH3 mimetic obatoclax. Neoplasia 2009, 11, 945–955. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Franke, J.C.; Plotz, M.; Prokop, A.; Geilen, C.C.; Schmalz, H.G.; Eberle, J. New caspase-independent but ROS-dependent apoptosis pathways are targeted in melanoma cells by an iron-containing cytosine analogue. Biochem. Pharmacol. 2010, 79, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, M.; Bauer, J.A.; Martin de la Vega, C.; Wang, G.; Wolter, K.G.; Brenner, J.C.; Nikolovska-Coleska, Z.; Bengtson, A.; Nair, R.; Elder, J.T.; et al. A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 2006, 66, 11348–11359. [Google Scholar] [CrossRef]
- Howard, A.N.; Bridges, K.A.; Meyn, R.E.; Chandra, J. ABT-737, a BH3 mimetic, induces glutathione depletion and oxidative stress. Cancer Chemother. Pharmacol. 2009, 65, 41–54. [Google Scholar] [CrossRef]
- Winkler, M.; Friedrich, J.; Boedicker, C.; Dolgikh, N. Co-targeting MCL-1 and ERK1/2 kinase induces mitochondrial apoptosis in rhabdomyosarcoma cells. Transl. Oncol. 2022, 16, 101313. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; He, S.; Look, A.T. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia 2019, 33, 262–266. [Google Scholar] [CrossRef]
- Alcon, C.; Gomez Tejeda Zanudo, J.; Albert, R.; Wagle, N.; Scaltriti, M.; Letai, A.; Samitier, J.; Montero, J. ER+ Breast Cancer Strongly Depends on MCL-1 and BCL-xL Anti-Apoptotic Proteins. Cells 2021, 10, 1659. [Google Scholar] [CrossRef]
- Keuling, A.M.; Felton, K.E.; Parker, A.A.; Akbari, M.; Andrew, S.E.; Tron, V.A. RNA silencing of Mcl-1 enhances ABT-737-mediated apoptosis in melanoma: Role for a caspase-8-dependent pathway. PLoS ONE 2009, 4, e6651. [Google Scholar] [CrossRef]
- Bauer, D.; Werth, F.; Nguyen, H.A.; Kiecker, F.; Eberle, J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017, 8, e2594. [Google Scholar] [CrossRef]
- Berger, A.; Quast, S.A.; Plotz, M.; Kammermeier, A.; Eberle, J. Sensitization of melanoma cells for TRAIL-induced apoptosis by BMS-345541 correlates with altered phosphorylation and activation of Bax. Cell Death Dis. 2013, 4, e477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate: A potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem. Pharmacol. 2003, 66, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef]
- Holzmann, B.; Lehmann, J.M.; Ziegler-Heitbrock, H.W.; Funke, I.; Riethmuller, G.; Johnson, J.P. Glycoprotein P3.58, associated with tumor progression in malignant melanoma, is a novel leukocyte activation antigen. Int. J. Cancer 1988, 41, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Bean, M.A.; Bloom, B.R.; Herberman, R.B.; Old, L.J.; Oettgen, H.F.; Klein, G.; Terry, W.D. Cell-mediated cytotoxicity for bladder carcinoma: Evaluation of a workshop. Cancer Res. 1975, 35, 2902–2913. [Google Scholar] [PubMed]
- Carey, T.E.; Takahashi, T.; Resnick, L.A.; Oettgen, H.F.; Old, L.J. Cell surface antigens of human malignant melanoma: Mixed hemadsorption assays for humoral immunity to cultured autologous melanoma cells. Proc. Natl. Acad. Sci. USA 1976, 73, 3278–3282. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Z.; Gillissen, B.; Richter, A.; Sinnberg, T.; Schlaak, M.S.; Eberle, J. Effective Targeting of Melanoma Cells by Combination of Mcl-1 and Bcl-2/Bcl-xL/Bcl-w Inhibitors. Int. J. Mol. Sci. 2024, 25, 3453. https://doi.org/10.3390/ijms25063453
Peng Z, Gillissen B, Richter A, Sinnberg T, Schlaak MS, Eberle J. Effective Targeting of Melanoma Cells by Combination of Mcl-1 and Bcl-2/Bcl-xL/Bcl-w Inhibitors. International Journal of Molecular Sciences. 2024; 25(6):3453. https://doi.org/10.3390/ijms25063453
Chicago/Turabian StylePeng, Zhe, Bernhard Gillissen, Antje Richter, Tobias Sinnberg, Max S. Schlaak, and Jürgen Eberle. 2024. "Effective Targeting of Melanoma Cells by Combination of Mcl-1 and Bcl-2/Bcl-xL/Bcl-w Inhibitors" International Journal of Molecular Sciences 25, no. 6: 3453. https://doi.org/10.3390/ijms25063453
APA StylePeng, Z., Gillissen, B., Richter, A., Sinnberg, T., Schlaak, M. S., & Eberle, J. (2024). Effective Targeting of Melanoma Cells by Combination of Mcl-1 and Bcl-2/Bcl-xL/Bcl-w Inhibitors. International Journal of Molecular Sciences, 25(6), 3453. https://doi.org/10.3390/ijms25063453