
Histamine H1 Receptor-Mediated JNK Phosphorylation Is Regulated by Gq Protein-Dependent but Arrestin-Independent Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Histamine Induces JNK Phosphorylation in CHO Cells Expressing S487TR but Not S487A
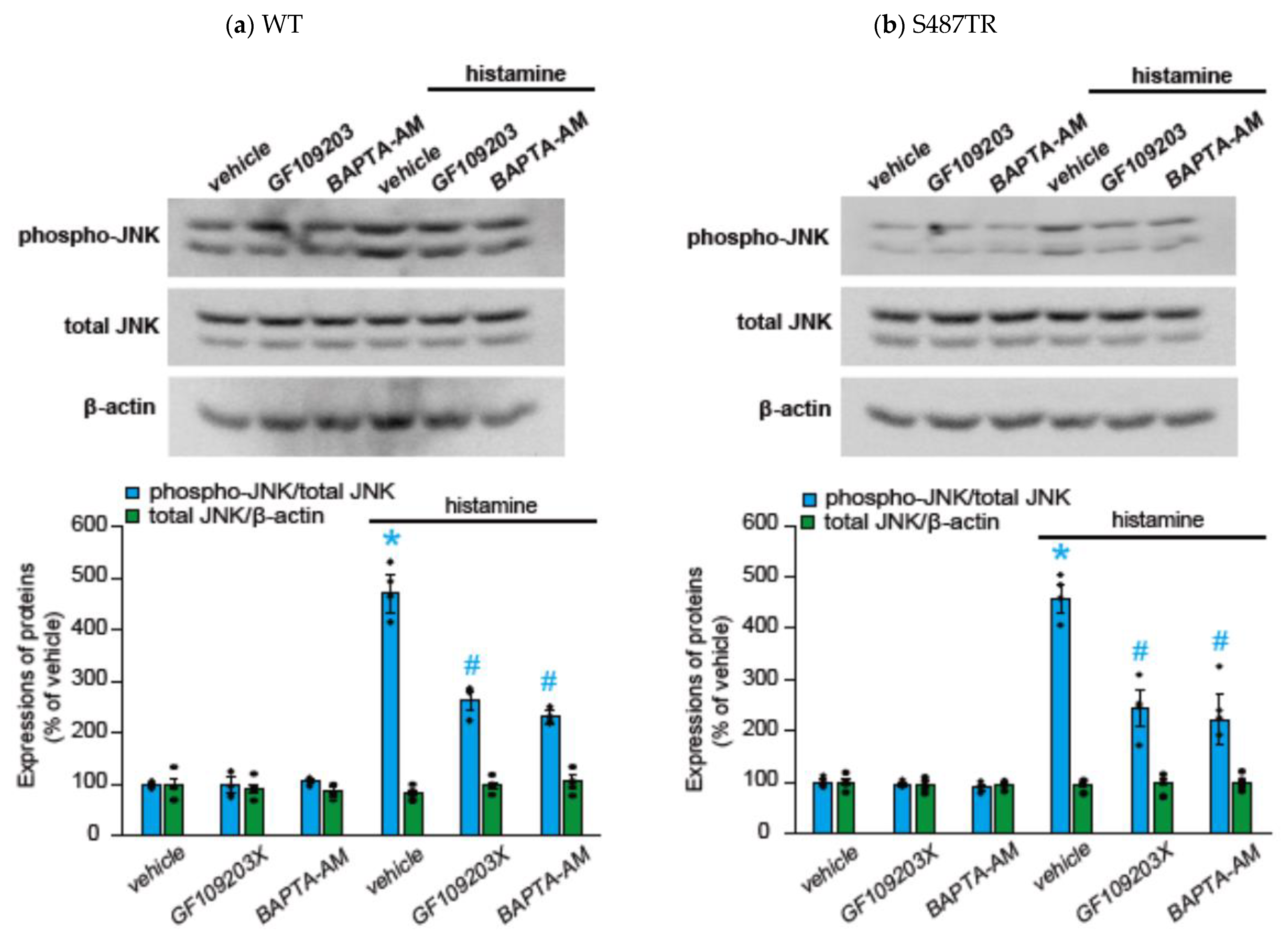
2.2. Histamine-Induced JNK Phosphorylation Is Mediated by Gq Protein/Ca2+/PKC-Dependent but GRK/Arrestin/Clathrin/Dynamin-Independent Pathway via Activation of H1 Receptors in CHO Cells Expressing WT and S487TR
3. Discussion
3.1. Mechanisms of H1 Receptor-Mediated Phosphorylation of JNK in CHO Cells
3.2. Physiological and Pathophysiological Roles of JNK Phosphorylation
4. Materials and Methods
4.1. Preparation of CHO Cells Expressing WT and Mutant Human Histamine H1 Receptors
4.2. Drug Treatments
4.3. Immunoblotting
4.4. β-Arrestin2 Knockdown
4.5. Immunocytochemistry
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CHO | Chinese hamster ovary |
| ERK | Extracellular signal-regulated kinase |
| GPCR | G protein-coupled receptor |
| GRK | G protein-coupled receptor kinase |
| JNK | c-Jun N-terminal kinase |
| MAP | Mitogen-activated protein |
| PBS | Phosphate-buffered saline |
References
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.C.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. Seven transmembrane receptors: Something old, something new. Acta Physiol. 2007, 190, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Lefkowitz, R.J. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2001, 2, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Shenoy, S.K.; Lefkowitz, R.J. Trafficking of G protein-coupled receptors. Circ. Res. 2006, 99, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Jean-Charles, P.Y.; Kaur, S.; Shenoy, S.K. G protein-coupled receptor signaling through beta-arrestin-dependent mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling regulation: The role of GRKs and arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Jiang, H.; Galtes, D.; Wang, J.; Rockman, H.A. G protein-coupled receptor signaling: Transducers and effectors. Am. J. Physiol. Cell Physiol. 2022, 323, C731–C748. [Google Scholar] [CrossRef]
- Wolfe, B.L.; Trejo, J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic 2007, 8, 462–470. [Google Scholar] [CrossRef]
- Perrais, D. Cellular and structural insight into dynamin function during endocytic vesicle formation: A tale of 50 years of investigation. Biosci. Rep. 2022, 42, BSR20211227. [Google Scholar] [CrossRef] [PubMed]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef]
- Liggett, S.B. Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci. Signal. 2011, 4, pe36. [Google Scholar] [CrossRef]
- Prihandoko, R.; Bradley, S.J.; Tobin, A.B.; Butcher, A.J. Determination of GPCR Phosphorylation Status: Establishing a Phosphorylation Barcode. Curr. Protoc. Pharmacol. 2015, 69, 2.13.1–2.13.26. [Google Scholar] [CrossRef]
- Bakker, R.A.; Timmerman, H.; Leurs, R. Histamine receptors: Specific ligands, receptor biochemistry, and signal transduction. Clin. Allergy Immunol. 2002, 17, 27–64. [Google Scholar]
- Neumann, D.; Schneider, E.H.; Seifert, R. Analysis of histamine receptor knockout mice in models of inflammation. J. Pharmacol. Exp. Ther. 2014, 348, 2–11. [Google Scholar] [CrossRef]
- Monczor, F.; Fernandez, N. Current knowledge and perspectives on histamine H1 and H2 receptor pharmacology: Functional selectivity, receptor crosstalk, and repositioning of classic histaminergic ligands. Mol. Pharmacol. 2016, 90, 640–648. [Google Scholar] [CrossRef]
- Ohtsu, H.; Seike, M. Histamine and histamine receptors in allergic dermatitis. Handb. Exp. Pharmacol. 2017, 241, 333–345. [Google Scholar]
- Tiligada, E.; Ennis, M. Histamine pharmacology: From Sir Henry Dale to the 21st century. Br. J. Pharmacol. 2020, 177, 469–489. [Google Scholar] [CrossRef]
- Panula, P. Histamine receptors, agonists, and antagonists in health and disease. Handb. Clin. Neurol. 2021, 180, 377–387. [Google Scholar]
- Yoshikawa, T.; Nakamura, T.; Yanai, K. Histaminergic neurons in the tuberomammillary nucleus as a control centre for wakefulness. Br. J. Pharmacol. 2021, 178, 750–769. [Google Scholar] [CrossRef]
- Ueda, Y.; Hirai, S.; Osada, S.; Suzuki, A.; Mizuno, K.; Ohno, S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J. Biol. Chem. 1996, 271, 23512–23519. [Google Scholar] [CrossRef]
- Robinson, A.J.; Dickenson, J.M. Activation of the p38 and p42/p44 mitogen-activated protein kinase families by the histamine H1 receptor inDDT1MF-2 cells. Br. J. Pharmacol. 2001, 133, 1378–1386. [Google Scholar] [CrossRef]
- Cammarota, M.; Bevilaqua, L.R.; Rostas, J.A.; Dunkley, P.R. Histamine activates tyrosine hydroxylase in bovine adrenal chromaffin cells through a pathway that involves ERK1/2 but not p38 or JNK. J. Neurochem. 2003, 84, 453–458. [Google Scholar] [CrossRef]
- Shimamura, K.; Takashiro, Y.; Akiyama, N.; Hirabayashi, T.; Murayama, T. Expression of adhesion molecules by sphingosine 1-phosphate and histamine in endothelial cells. Eur. J. Pharmacol. 2004, 486, 141–150. [Google Scholar] [CrossRef]
- Steffel, J.; Akhmedov, A.; Greutert, H.; Lüscher, T.F.; Tanner, F.C. Histamine induces tissue factor expression: Implications for acute coronary syndromes. Circulation 2005, 112, 341–349. [Google Scholar] [CrossRef]
- Matsubara, M.; Tamura, T.; Ohmori, K.; Hasegawa, K. Histamine H1 receptor antagonist blocks histamine-induced proinflammatory cytokine production through inhibition of Ca2+-dependent protein kinase C, Raf/MEK/ERK and IKK/IκB/NF-κB signal cascades. Biochem. Pharmacol. 2005, 69, 433–449. [Google Scholar] [CrossRef]
- Steffel, J.; Arnet, C.; Akhmedov, A.; Iseli, S.M.; Lüscher, T.F.; Tanner, F.C. Histamine differentially interacts with tumor necrosis factor-alpha and thrombin in endothelial tissue factor induction: The role of c-Jun NH2-terminal kinase. J. Thromb. Haemost. 2006, 4, 2452–2460. [Google Scholar] [CrossRef]
- Matsubara, M.; Ohmori, K.; Hasegawa, K. Histamine H1 receptor-stimulated interleukin 8 and granulocyte macrophage colony-stimulating factor production by bronchial epithelial cells requires extracellular signal-regulated kinase signaling via protein kinase C. Int. Arch. Allergy Immunol. 2006, 139, 279–293. [Google Scholar] [CrossRef]
- Lipnik-Stangelj, M. Multiple role of histamine H1-receptor-PKC-MAPK signalling pathway in histamine-stimulated nerve growth factor synthesis and secretion. Biochem. Pharmacol. 2006, 72, 1375–1381. [Google Scholar] [CrossRef]
- Hao, F.; Tan, M.; Xu, X.; Cui, M.Z. Histamine induces Egr-1 expression in human aortic endothelial cells via the H1 receptor-mediated protein kinase Cδ-dependent ERK activation pathway. J. Biol. Chem. 2008, 283, 26928–29636. [Google Scholar] [CrossRef]
- Notcovich, C.; Diez, F.; Tubio, M.R.; Baldi, A.; Kazanietz, M.G.; Davio, C.; Shayo, C. Histamine acting on H1 receptor promotes inhibition of proliferation via PLC, RAC, and JNK-dependent pathways. Exp. Cell Res. 2010, 316, 401–411. [Google Scholar] [CrossRef]
- Mizuguchi, H.; Terao, T.; Kitai, M.; Ikeda, M.; Yoshimura, Y.; Das, A.K.; Kitamura, Y.; Takeda, N.; Fukui, H. Involvement of PKCδ/Extracellular signal-regulated kinase/poly(ADP-ribose) polymerase-1 (PARP-1) signaling pathway in histamine- induced up-regulation of histamine H1 receptor gene expression in HeLa cells. J. Biol. Chem. 2011, 286, 30542–30551. [Google Scholar] [CrossRef]
- Brighton, P.J.; Rana, S.; Challiss, R.J.; Konje, J.C.; Willets, J.M. Arrestins differentially regulate histamine- and oxytocin-evoked phospholipase C and mitogen-activated protein kinase signalling in myometrial cells. Br. J. Pharmacol. 2011, 162, 1603–1617. [Google Scholar] [CrossRef]
- Beermann, S.; Vauth, M.; Hein, R.; Seifert, R.; Neumann, D. Distinct signalling pathways of murine histamine H1- and H4-receptors expressed at comparable levels in HEK293 cells. PLoS ONE 2014, 9, e107481. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, W.; Zeng, X.; Hu, G.; Zhang, H.; He, S.; Zhang, S. Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol. Neurobiol. 2014, 49, 1487–1500. [Google Scholar] [CrossRef]
- Beermann, S.; Bernhardt, G.; Seifert, R.; Buschauer, A.; Neumann, D. Histamine H1- and H4-receptor signaling cooperatively regulate MAPK activation. Biochem. Pharmacol. 2015, 98, 432–439. [Google Scholar] [CrossRef]
- Mizuguchi, H.; Kitamura, Y.; Takeda, N.; Fukui, H. Molecular signaling and transcriptional regulation of histamine H1 receptor gene. Curr. Top. Behav. Neurosci. 2022, 59, 91–110. [Google Scholar]
- Hishinuma, S.; Young, J.M. Characteristics of the binding of [3H]-mepyramine to intact human U373 MG astrocytoma cells: Evidence for histamine-induced H1-receptor internalisation. Br. J. Pharmacol. 1995, 116, 2715–2723. [Google Scholar] [CrossRef]
- Hishinuma, S.; Naiki, A.; Tsuga, H.; Young, J.M. Ca2+/calmodulin-mediated regulation of agonist-induced sequestration of Gq protein-coupled histamine H1 receptors in human U373 MG astrocytoma cells. J. Neurochem. 1998, 71, 2626–2633. [Google Scholar] [CrossRef]
- Hishinuma, S.; Komazaki, H.; Fukui, H.; Shoji, M. Ubiquitin/proteasome-dependent down-regulation following clathrin-mediated internalization of histamine H1-receptors in Chinese hamster ovary cells. J. Neurochem. 2010, 113, 990–1001. [Google Scholar] [CrossRef]
- Hishinuma, S.; Sato, Y.; Akatsu, C.; Shoji, M. The affinity of histamine for Gq protein-coupled histamine H1-receptors is predominantly regulated by their internalization in human astrocytoma cells. J. Pharmacol. Sci. 2012, 119, 233–242. [Google Scholar] [CrossRef]
- Hishinuma, S.; Nozawa, H.; Akatsu, C.; Shoji, M. C-terminal of human histamine H1 receptors regulates their agonist-induced clathrin-mediated internalization and G-protein signaling. J. Neurochem. 2016, 139, 552–565. [Google Scholar] [CrossRef]
- Michinaga, S.; Nagata, A.; Ogami, R.; Ogawa, Y.; Hishinuma, S. Differential regulation of histamine H1 receptor-mediated ERK phosphorylation by Gq proteins and arrestins. Biochem. Pharmacol. 2023, 213, 115595. [Google Scholar] [CrossRef]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK signaling pathway in inflammatory skin disorders and cancer. Cells 2007, 9, 857. [Google Scholar] [CrossRef]
- Eynott, P.R.; Xu, L.; Bennett, B.L.; Noble, A.; Leung, S.Y.; Nath, P.; Groneberg, D.A.; Adcock, I.M.; Chung, K.F. Effect of an inhibitor of Jun N-terminal protein kinase, SP600125, in single allergen challenge in sensitized rats. Immunology 2004, 112, 446–453. [Google Scholar] [CrossRef]
- Zhang, J.; Lin, W.; Tang, M.; Zhao, Y.; Zhang, K.; Wang, X.; Li, Y. Inhibition of JNK ameliorates depressive-like behaviors and reduces the activation of pro-inflammatory cytokines and the phosphorylation of glucocorticoid receptors at serine 246 induced by neuroinflammation. Psychoneuroendocrinology 2020, 113, 104580. [Google Scholar] [CrossRef]
- Ahn, S.; Shenoy, S.K.; Wei, H.; Lefkowitz, R.J. Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 2004, 279, 35518–35525. [Google Scholar] [CrossRef]
- Xiang, G.; Acosta-Ruiz, A.; Radoux-Mergault, A.; Kristt, M.; Kim, J.; Moon, J.D.; Broichhagen, J.; Inoue, A.; Lee, F.S.; Stoeber, M.; et al. Control of Gαq signaling dynamics and GPCR cross-talk by GRKs. Sci. Adv. 2022, 8, eabq3363. [Google Scholar] [CrossRef]
- Furuta, A.; Tanaka, M.; Omata, W.; Nagasawa, M.; Kojima, I.; Shibata, H. Microtubule disruption with BAPTA and dimethyl BAPTA by a calcium chelation-independent mechanism in 3T3-L1 adipocytes. Endocr. J. 2009, 56, 235–243. [Google Scholar] [CrossRef]
- Kim, J.; Lee, C.K.; Park, H.J.; Kim, H.J.; So, H.H.; Lee, K.S.; Lee, H.M.; Roh, H.Y.; Choi, W.S.; Park, T.K.; et al. Epidermal growth factor induces vasoconstriction through the phosphatidylinositol 3-kinase-mediated mitogen-activated protein kinase pathway in hypertensive rats. J. Pharmacol. Sci. 2006, 101, 135–143. [Google Scholar] [CrossRef]
- Lowe, J.D.; Sanderson, H.S.; Cooke, A.E.; Ostovar, M.; Tsisanova, E.; Withey, S.L.; Chavkin, C.; Husbands, S.M.; Kelly, E.; Henderson, G.; et al. Role of G protein-coupled receptor kinases 2 and 3 in μ-opioid receptor desensitization and internalization. Mol. Pharmacol. 2015, 88, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Barrias, E.S.; Reignault, L.C.; De Souza, W.; Carvalho, T.M. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS ONE 2010, 5, e7764. [Google Scholar] [CrossRef] [PubMed]
- Heuser, J.E.; Anderson, R.G. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J. Cell Biol. 1989, 108, 389–400. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michinaga, S.; Nagata, A.; Ogami, R.; Ogawa, Y.; Hishinuma, S. Histamine H1 Receptor-Mediated JNK Phosphorylation Is Regulated by Gq Protein-Dependent but Arrestin-Independent Pathways. Int. J. Mol. Sci. 2024, 25, 3395. https://doi.org/10.3390/ijms25063395
Michinaga S, Nagata A, Ogami R, Ogawa Y, Hishinuma S. Histamine H1 Receptor-Mediated JNK Phosphorylation Is Regulated by Gq Protein-Dependent but Arrestin-Independent Pathways. International Journal of Molecular Sciences. 2024; 25(6):3395. https://doi.org/10.3390/ijms25063395
Chicago/Turabian StyleMichinaga, Shotaro, Ayaka Nagata, Ryosuke Ogami, Yasuhiro Ogawa, and Shigeru Hishinuma. 2024. "Histamine H1 Receptor-Mediated JNK Phosphorylation Is Regulated by Gq Protein-Dependent but Arrestin-Independent Pathways" International Journal of Molecular Sciences 25, no. 6: 3395. https://doi.org/10.3390/ijms25063395
APA StyleMichinaga, S., Nagata, A., Ogami, R., Ogawa, Y., & Hishinuma, S. (2024). Histamine H1 Receptor-Mediated JNK Phosphorylation Is Regulated by Gq Protein-Dependent but Arrestin-Independent Pathways. International Journal of Molecular Sciences, 25(6), 3395. https://doi.org/10.3390/ijms25063395