
Roles of DNA Methylation in Color Alternation of Eastern Honey Bees (Apis cerana) Induced by the Royal Jelly of Western Honey Bees (Apis mellifera)


Abstract
1. Introduction
2. Results
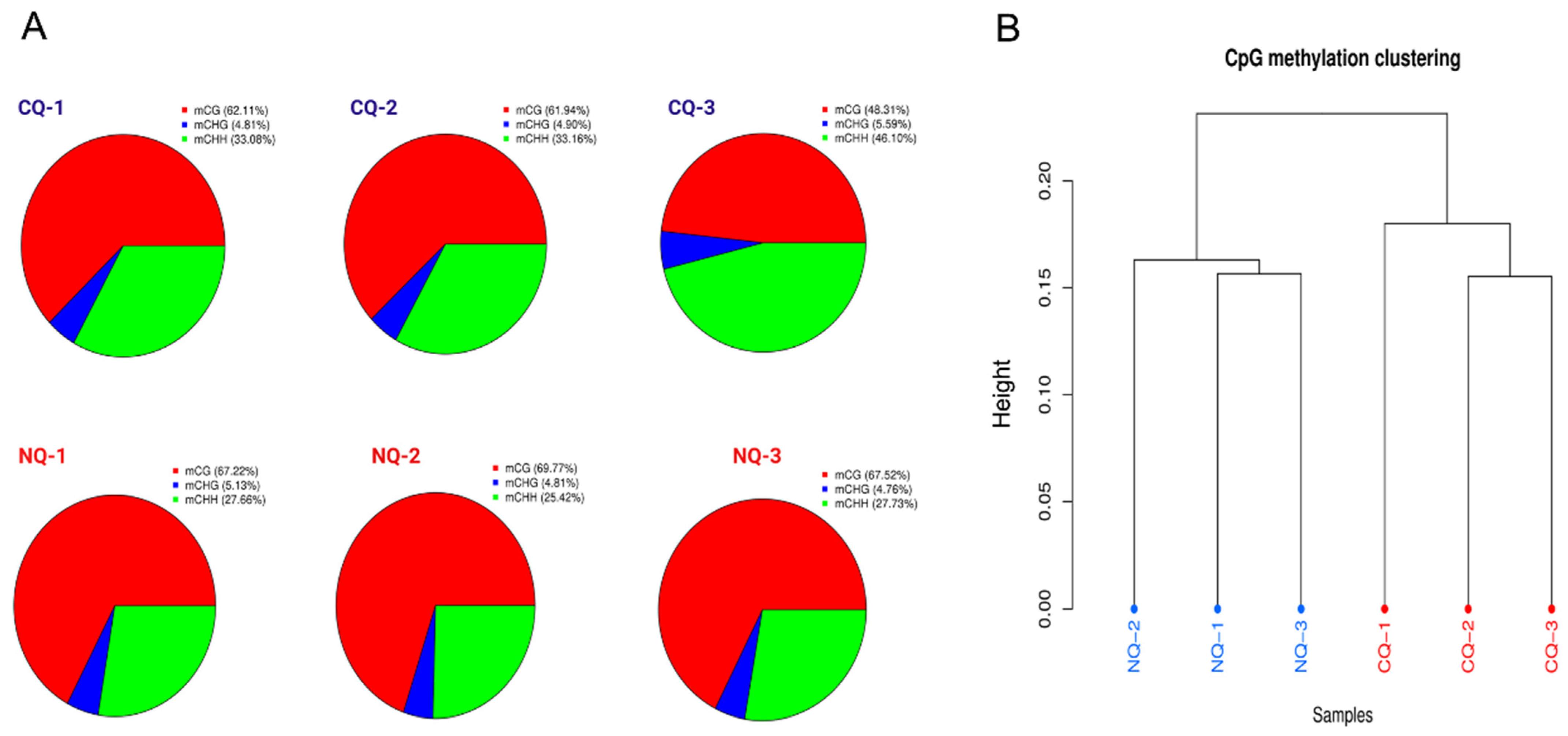
2.1. Overview of Whole-Genome Bisulfite Sequencing and DNA Methylation
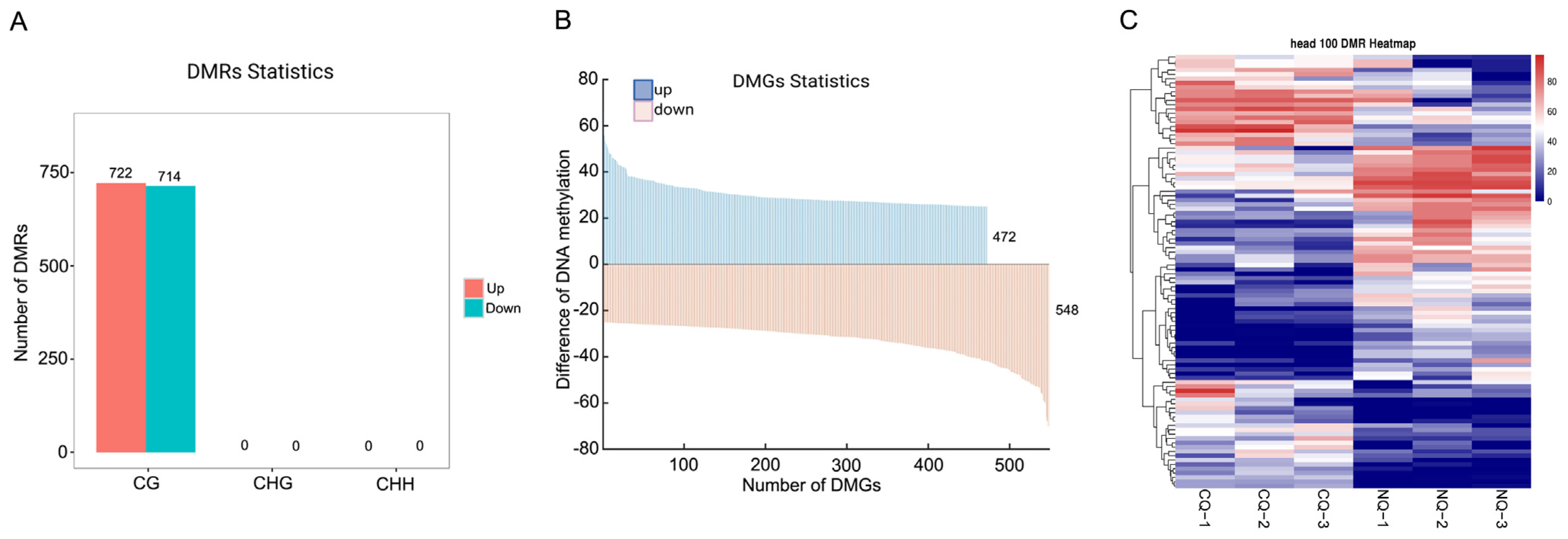
2.2. DMRs and DMGs between NQs and CQs
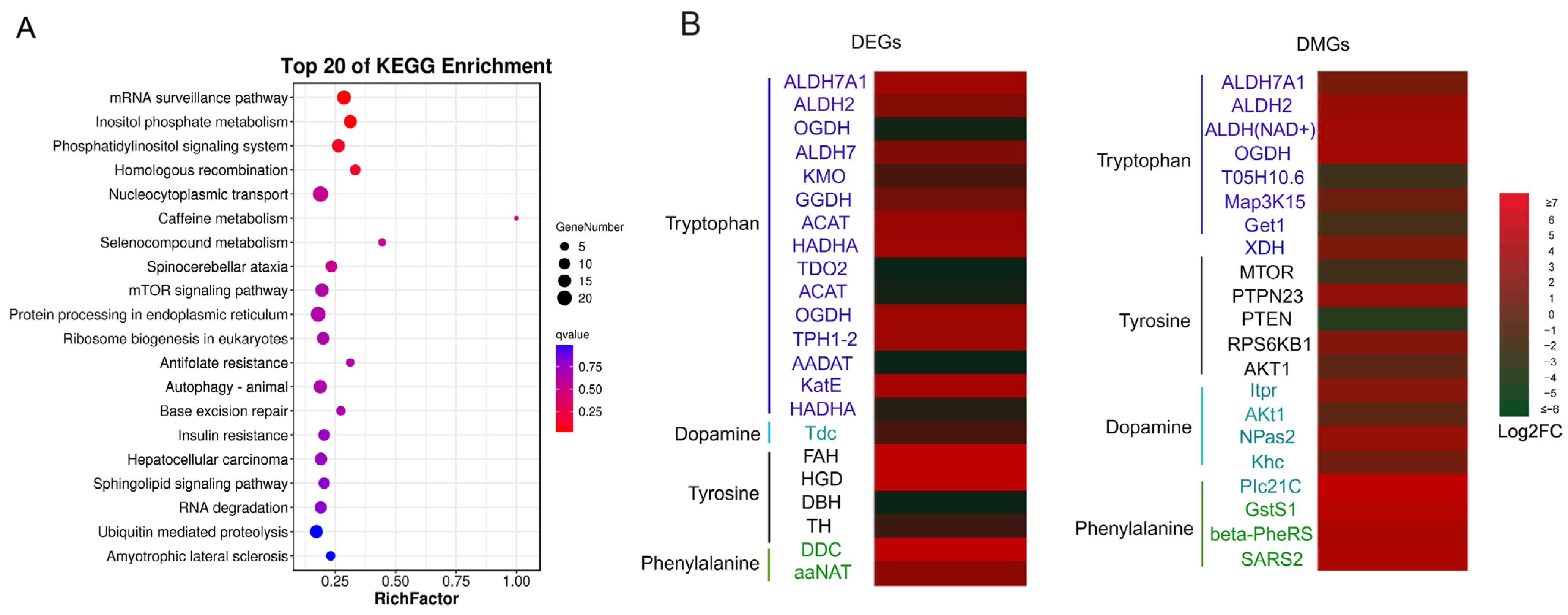
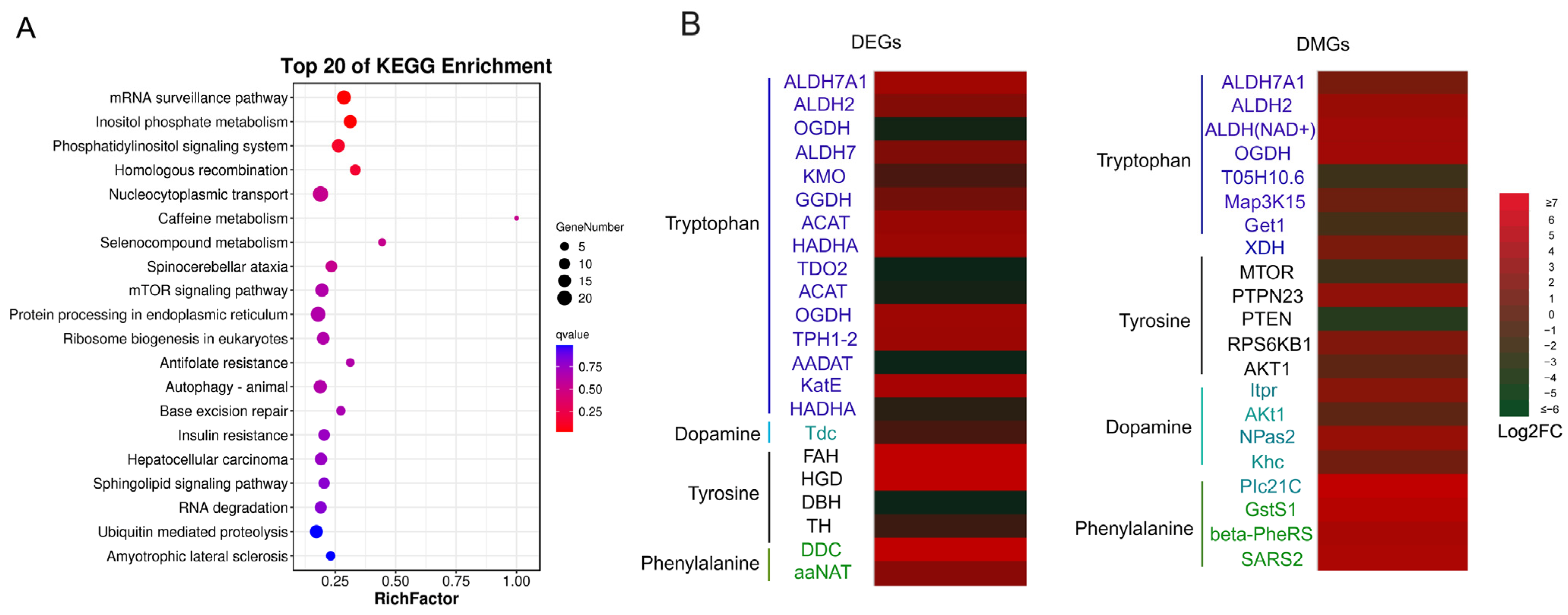
2.3. GO and KEGG Enrichment
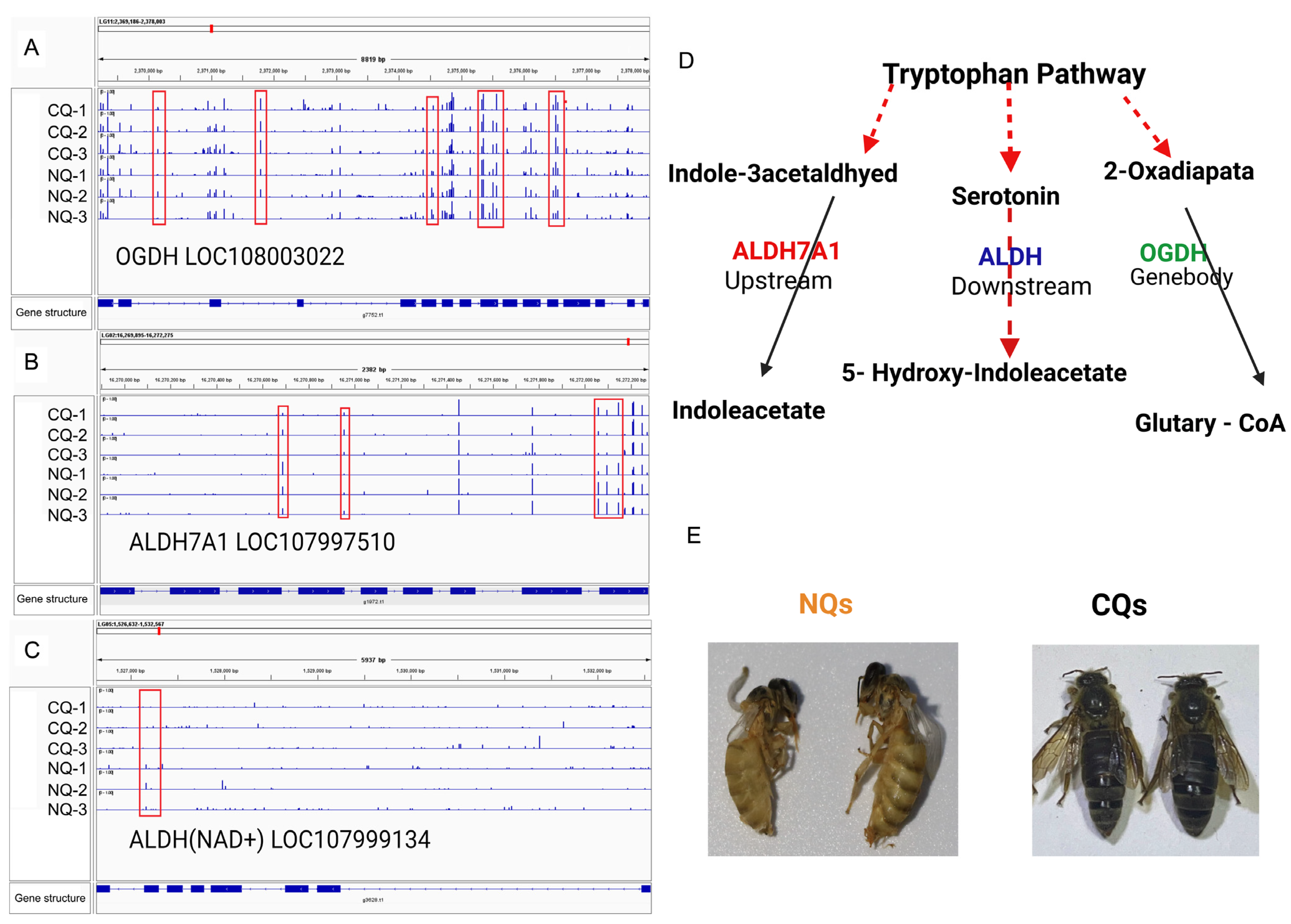
2.4. DMGs Related to Body Color Regulation
3. Discussion
4. Materials and Methods
4.1. Queen Rearing and Sampling
4.2. A. cerana Nutrient Crossbred Queen Rearing
4.3. Control Queen Rearing
4.4. Genome-Wide Methylation Analysis
4.5. Library Construction and Sequencing
4.6. Data Filtering
4.7. Mapping Reads to the Reference Genome
4.8. Bisulfite Treatment Efficiency Detection
4.9. Differential Methylation Analysis
4.10. Functional Enrichment Analysis of DMGs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharma, U.; Conine, C.C.; Shea, J.M.; Boskovic, A.; Derr, A.G.; Bing, X.Y.; Belleannee, C.; Kucukural, A.; Serra, R.W.; Sun, F. Biogenesis and Function of TRNA Fragments during Sperm Maturation and Fertilization in Mammals. Science 2016, 351, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Wedd, L.; Kucharski, R.; Maleszka, R. DNA Methylation in Honey Bees and the Unresolved Questions in Insect Methylomics BT—DNA Methyltransferases—Role and Function. In DNA Methyltransferases—Role and Function; Jeltsch, A., Jurkowska, R.Z., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 159–176. ISBN 978-3-031-11454-0. [Google Scholar]
- Richard, G.; Jaquiéry, J.; Le Trionnaire, G. Contribution of Epigenetic Mechanisms in the Regulation of Environmentally-Induced Polyphenism in Insects. Insects 2021, 12, 649. [Google Scholar] [CrossRef] [PubMed]
- Smaragdova, N. Study on the Brood Food of Worker of the Bees Apis mellifera L., Apis mellifera caucasica Gorb., and of Their Crossbreds. In Proceedings of the International Beekeeping Congress, Prague, Czech Republic, 12–17 August 1963; Volume 19, pp. 109–110. [Google Scholar]
- Rinderer, T.E.; Hellmich, R.L.; Danka, R.G.; Collins, A.M. Male Reproductive Parasitism: A Factor in the Africanization of European Honey-Bee Populations. Science 1985, 228, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Li, M.S. Discuss on Breeding of Cross-Feeding between Apis cerana cerana and Apis mellifera ligustica. Apic. Sci. Technol. 1982, 1, 42–44. [Google Scholar]
- Zhu, M. Study on cross-feeding between Apis cerana cerana and Apis mellifera ligustica. J. Bee 1985, 3, 14. [Google Scholar]
- Xie, X.B.; Peng, W.J.; Zeng, Z.J. Breeding the Mite-Resistant Honeybee by Nutritional Crossbreed Technology. Agric. Sci. China 2008, 7, 762–767. (In Chinese) [Google Scholar] [CrossRef]
- Chen, H.; Li, F.M.; Zheng, M.J.; Cao, D.D.; He, Z.M.; Yang, M.X. The effect of nutritional hybridization on the reproductive potential of the queen bee in China. Sichuan Anim. Husb. Vet. Med. 2015, 42, 24–26. (In Chinese) [Google Scholar]
- Wang, Z.; Li, Z.Y.; Zhu, H.W.; Li, J.L. Nutritional Crossbreed Technology of Honeybee and Its Application on Genetics and Breeding. J. Bee 2009, 7, 28–30. (In Chinese) [Google Scholar]
- Shi, Y.Y.; Huang, Z.Y.; Wu, X.B.; Wang, Z.L.; Yan, W.Y.; Zeng, Z.J. Changes in Alternative Splicing in Apis mellifera Bees Fed Apis cerana Royal Jelly. J. Apic. Sci. 2014, 58, 25–31. [Google Scholar] [CrossRef]
- Abdelmawla, A.; Yang, C.; Li, X.; Li, M.; Li, C.L.; Liu, Y.B.; He, X.J.; Zeng, Z.J. Feeding Asian Honeybee Queens with European Honeybee Royal Jelly Alters Body Color and Expression of Related Coding and Non-Coding RNAs. Front. Physiol. 2023, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Maleszka, R. Epigenetic Integration of Environmental and Genomic Signals in Honey Bees: The Critical Interplay of Nutritional, Brain and Reproductive Networks. Epigenetics 2008, 3, 188–192. [Google Scholar] [CrossRef]
- Elango, N.; Hunt, B.G.; Goodisman, M.A.; Yi, S.V. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc. Natl. Acad. Sci. USA 2009, 106, 11206–11211. [Google Scholar] [CrossRef]
- Kucharski, R.; Maleszka, J.; Foret, S.; Maleszka, R. Nutritional Control of Reproductive Status in Honeybees via DNA Methylation. Science 2008, 319, 1827–1830. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, Z.; Wang, Y.; Ma, L.; Zhang, W.; Xu, B. Genome-Wide Differential DNA Methylation in Reproductive, Morphological, and Visual System Differences between Queen Bee and Worker Bee (Apis mellifera). Front. Genet. 2020, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; He, X.J.; Barron, A.B.; Liu, Y.B.; Wang, Z.L.; Yan, W.Y.; Zeng, Z.J. Transgenerational Accumulation of Methylome Changes Discovered in Commercially Reared Honey Bee (Apis mellifera) Queens. Insect Biochem. Mol. Biol. 2020, 127, 103476. [Google Scholar] [CrossRef] [PubMed]
- He, X.J.; Wei, H.; Jiang, W.J.; Liu, Y.B.; Wu, X.B.; Zeng, Z.J. Honeybee (Apis mellifera) Maternal Effect Causes Alternation of DNA Methylation Regulating Queen Development. Sociobiology 2021, 68, e5935. [Google Scholar] [CrossRef]
- Yi, Y.; Liu, Y.B.; Barron, A.B.; Zeng, Z.J. Effects of Commercial Queen Rearing Methods on Queen Fecundity and Genome Methylation. Apidologie 2021, 52, 282–291. [Google Scholar] [CrossRef]
- Minvielle, F.; Cecchi, T.; Passamonti, P.; Gourichon, D.; Renieri, C. Plumage colour mutations and melanins in the feathers of the Japanese quail: A first comparison. Anim. Genet. 2009, 40, 971–974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Fu, W.; Xu, W.; Zhang, H.; Chen, S.; Liu, W.; Peng, L.; Xiao, Y. Comparative Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying Skin Color Variations in Crucian Carp (Carassius carassius L.). BMC Genet. 2017, 18, 95. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal Genistein Alters Coat Color and Protects Avy Mouse Offspring from Obesity by Modifying the Fetal Epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, S.; Liu, M.; Zhao, B.; Yang, N.; Li, J.; Chen, Q.; Zhou, J.; Bao, G.; Wu, X. Analysis of Genome DNA Methylation at Inherited Coat Color Dilutions of Rex Rabbits. Front. Genet. 2021, 11, 603528. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.J.; Cunningham, C.B.; Dearden, P.K. Phenotypic Plasticity: What Has DNA Methylation Got to Do with It? Insects 2022, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. The Progress of Nutritional hybridization between Apis cerana cerana and Apis mellifera ligustica. Apic. China 2012, 63, 24–25. (In Chinese) [Google Scholar]
- Provataris, P. Evolution of DNA Methylation in Insects Panagiotis Provataris. Ph.D. Thesis, Universitäts-und Landesbibliothek Bonn, Bonn, Germany, 2021. [Google Scholar]
- Foret, S.; Kucharski, R.; Pellegrini, M.; Feng, S.; Jacobsen, S.E.; Robinson, G.E.; Maleszka, R. DNA Methylation Dynamics, Metabolic Fluxes, Gene Splicing, and Alternative Phenotypes in Honey Bees. Proc. Natl. Acad. Sci. USA 2012, 109, 4968–4973. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, Z.G.; Lin, Z.G.; Yin, L.; Gao, F.C.; Chen, G.H.; Ji, T. Epigenetic modifications may regulate the activation of the hypopharyngeal gland of honeybees (Apis mellifera) during winter. Front. Genet. 2020, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Simola, D.F.; Bonasio, R.; Liebig, J.; Berger, S.L.; Reinberg, D. Eusocial insects as emerging models for behavioural epigenetics. Nat. Rev. Genet. 2014, 15, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ma, C.; Chen, C.; Lu, Q.; Shi, W.; Liu, Z.; Wang, H.; Guo, H. Integration of LncRNA-MiRNA-MRNA Reveals Novel Insights into Oviposition Regulation in Honey Bees. PeerJ 2017, 5, e3881. [Google Scholar] [CrossRef]
- Lambrus, B.G.; Cochet-Escartin, O.; Gao, J.; Newmark, P.A.; Collins, E.-M.S.; Collins, J.J., 3rd. Tryptophan Hydroxylase Is Required for Eye Melanogenesis in the Planarian Schmidtea Mediterranea. PLoS ONE 2015, 10, e0127074. [Google Scholar] [CrossRef]
- Ahmad, S.; Mohammed, M.; Prasuna, M.; Chintalapati, S.; Chintalapati, V.R. Tryptophan, a non-canonical melanin precursor: New L-tryptophan based melanin production by Rubrivivax benzoatilyticus JA2. Sci. Rep. 2020, 10, 8925. [Google Scholar] [CrossRef]
- Sasaki, K.; Yokoi, K.; Toga, K. Bumble bee queens activate dopamine production and gene expression in nutritional signaling pathways in the brain. Sci. Rep. 2021, 11, 5526. [Google Scholar] [CrossRef] [PubMed]
- Rabatel, A.; Febvay, G.; Gaget, K.; Duport, G.; Baa-Puyoulet, P.; Sapountzis, P.; Bendridi, N.; Rey, M.; Rahbé, Y.; Charles, H.; et al. Tyrosine pathway regulation is host-mediated in the pea aphid symbiosis during late embryonic and early larval development. BMC Genom. 2013, 14, 235. [Google Scholar] [CrossRef]
- Zhu, K.; Liu, M.; Fu, Z.; Zhou, Z.; Kong, Y.; Liang, H.; Lin, Z.; Luo, J.; Zheng, H.; Wan, P.; et al. Plant MicroRNAs in Larval Food Regulate Honeybee Caste Development. PLoS Genet. 2017, 13, e1006946. [Google Scholar] [CrossRef]
- Futahashi, R.; Koshikawa, S.; Okude, G.; Osanai-Futahashi, M. Diversity of melanin synthesis genes in insects. Adv. Insect Physiol. 2022, 62, 339–376. [Google Scholar]
- Futahashi, R.; Osanai-Futahashi, M. Pigments in Insects. In Pigments, Pigment Cells and Pigment Patterns; Hashimoto, H., Goda, M., Futahashi, R., Kelsh, R., Akiyama, T., Eds.; Springer: Singapore, 2021. [Google Scholar] [CrossRef]
- Miyazawa, S.; Watanabe, M.; Kondo, S. Theoretical Studies of Pigment Pattern; Springer: Singapore, 2021; ISBN 9789811614897. [Google Scholar]
- Kan, H.; Kim, C.H.; Kwon, H.M.; Park, J.W.; Roh, K.B.; Lee, H.; Park, B.J.; Zhang, R.; Zhang, J.; Söderhäll, K.; et al. Molecular control of phenoloxidase-induced melanin synthesis in an insect. J. Biol. Chem. 2008, 283, 25316–25323. [Google Scholar] [CrossRef] [PubMed]
- Mun, S.; Noh, M.Y.; Kramer, K.J.; Muthukrishnan, S.; Arakane, Y. Gene functions in adult cuticle pigmentation of the yellow mealworm, Tenebrio molitor. Insect Biochem. Mol. Biol. 2020, 117, 103291. [Google Scholar] [CrossRef]
- Arakane, Y.; Noh, M.Y.; Asano, T.; Karmer, K.J. Tyrosine Metabolism for Insect Cuticle Pigmentation and Sclerotization. In Extracellular Composite Matrices in Arthropods; Cohen, E., Moussian, B., Eds.; Spinger: Berlin/Heidelberg, Germany, 2016; pp. 156–220. (In Switzerland) [Google Scholar] [CrossRef]
- Pereboom, J.J.M.; Biesmeijer, J.C. Thermal constraints for stingless bee foragers: The importance of body size and coloration. Oecologia 2003, 137, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Higgs, J.S.; Wattanachaiyingcharoen, W.; Oldroyd, B.P. A scientific note on a genetically-determined color morph of the dwarf honey bee, Apis andreniformis. Apidologie 2009, 40, 513–514. [Google Scholar] [CrossRef]
- Wittkopp, P.J.; True, J.R.; Carroll, S.B. Reciprocal functions of the Drosophila Yellow and Ebony proteins in the development and evolution of pigment patterns. Development 2002, 129, 1849–1858. [Google Scholar] [CrossRef]
- Nie, H.Y.; Liang, L.Q.; Li, Q.F.; Li, Z.H.Q.; Zhu, Y.N.; Guo, Y.K.; Zheng, Q.L.; Lin, Y.; Yang, D.L.; Li, Z.G.; et al. CRISPR/Cas9 mediated knockout of Amyellow-y gene results in melanization defect of the cuticle in adult Apis mellifera. J. Insect Physiol. 2021, 132, 104264. [Google Scholar] [CrossRef]
- Patterson Rosa, L.; Eimanifar, A.; Kimes, A.G.; Brooks, S.A.; Ellis, J.D. Attack of the dark clones the genetics of reproductive and color traits of South African honey bees (Apis mellifera spp.). PLoS ONE 2021, 16, e0260833. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Guo, X.; Su, S.; Geir, S.; Li, W.; Li, Z.; Zhang, S.; Chen, S.; Chen, R. Differential expression of miRNAs related to caste differentiation in the honey bee, Apis mellifera. Apidologie 2016, 47, 495–508. [Google Scholar] [CrossRef]
- Wojciechowski, M.; Lowe, R.; Maleszka, J.; Conn, D.; Maleszka, R.; Hurd, P.J. Phenotypically distinct female castes in honey bees are defined by alternative chromatin states during larval development. Genome Res. 2018, 28, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xiao, Y.; Li, Y.; Wang, X.; Qi, S.; Wang, Y.; Zhao, L.; Wang, K.; Peng, W.; Luo, G.Z. RNA m6A Modification Functions in Larval Development and Caste Differentiation in Honeybee (Apis mellifera). Cell Rep. 2021, 34, 108580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, X.J.; Barron, A.B.; Li, Z.; Jin, M.J.; Wang, Z.L.; Huang, Q.; Zhang, L.Z.; Wu, X.B.; Yan, W.Y.; et al. The diverging epigenomic landscapes of honeybee queens and workers revealed by multiomic sequencing. Insect Biochem. Mol. Biol. 2023, 155, 103929. [Google Scholar] [CrossRef] [PubMed]
- Schmehl, D.R.; Tomé, H.V.V.; Mortensen, A.N.; Martins, G.F.; Ellis, J.D. Protocolo Para La Cría in Vitro de Obreras de Apis mellifera. J. Apic. Res. 2016, 55, 113–129. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y.; et al. Single base–resolutionmethylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Vaisvila, R.; Ponnaluri, V.C.; Sun, Z.; Bradley, W.; Langhorst, L.S.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. EM-Seq: Detection of DNA Methylation at Single Base Resolution from Picograms of DNA. bioRxiv 2019. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, 1–9. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A Web Tool for Plotting GO Annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, F.; Li, B.; Zhang, Y.; Chen, M.; Fan, T.; Wang, T. Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications16 in response to drought stress. Sci. Rep. 2020, 10, 8013. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A Web Server for Annotation and Identification of Enriched Pathways and Diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Li, B.; Hu, P.; Zhu, L.B.; You, L.L.; Cao, H.H.; Wang, J.; Zhang, S.Z.; Liu, M.H.; Toufeeq, S.; Huang, S.J.; et al. DNA methylation is correlated with gene expression during diapause termination of early embryonic development in the silkworm (Bombyx mori). Int. J. Mol. Sci. 2020, 21, 671. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Before Filter | After Filter | |||||||
|---|---|---|---|---|---|---|---|---|
| Samples | Clean Data (bp) | Q20% | N% | GC% | Clean Data (bp) | Q20% | N% | GC% |
| CQ-1 | 6,677,533,800 | 6,004,094,898 (89.91%) | 68,826 (0.0%) | 994,113,398 (14.88%) | 6,579,118,528 (98.53%) | 5,934,878,425 (90.21%) | 67,815 (0.0%) | 939,863,645 (14.28%) |
| CQ-2 | 7,590,253,800 | 6,973,265,799 (91.87%) | 97,377 (0.0%) | 1,088,876,017 (14.35%) | 7,533,897,309 (99.26%) | 6,935,328,057 (92.05%) | 96,666 (0.0%) | 1,068,174,594 (14.18%) |
| CQ-3 | 7,431,298,800 | 6,773,456,838 (91.15%) | 96,805 (0.0%) | 1,154,870,784 (15.54%) | 7,360,162,773 (99.04%) | 6,726,348,781 (91.39%) | 95,856 (0.0%) | 1,128,746,356 (15.34%) |
| NQ-1 | 8,955,201,900 | 8,178,295,710 (91.32%) | 97,416 (0.0%) | 1,375,753,231 (15.36%) | 8,862,330,769 (98.96%) | 8,114,049,174 (91.56%) | 92,520 (0.0%) | 1,332,182,306 (15.03%) |
| NQ-2 | 6,638,377,200 | 6,037,648,753 (90.95%) | 73,693 (0.0%) | 1,025,061,496 (15.44%) | 6,573,340,409 (99.02%) | 5,992,949,234 (91.17%) | 70,632 (0.0%) | 995,135,431 (15.14%) |
| NQ-3 | 7,353,264,300 | 6,695,053,336 (91.05%) | 81,142 (0.0%) | 1,040,993,533 (14.16%) | 7,298,867,089 (99.26%) | 6,658,844,959 (91.23%) | 78,310 (0.0%) | 1,020,328,299 (13.98%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelmawla, A.; Li, X.; Shi, W.; Zheng, Y.; Zeng, Z.; He, X. Roles of DNA Methylation in Color Alternation of Eastern Honey Bees (Apis cerana) Induced by the Royal Jelly of Western Honey Bees (Apis mellifera). Int. J. Mol. Sci. 2024, 25, 3368. https://doi.org/10.3390/ijms25063368
Abdelmawla A, Li X, Shi W, Zheng Y, Zeng Z, He X. Roles of DNA Methylation in Color Alternation of Eastern Honey Bees (Apis cerana) Induced by the Royal Jelly of Western Honey Bees (Apis mellifera). International Journal of Molecular Sciences. 2024; 25(6):3368. https://doi.org/10.3390/ijms25063368
Chicago/Turabian StyleAbdelmawla, Amal, Xin Li, Wenkai Shi, Yunlin Zheng, Zhijiang Zeng, and Xujiang He. 2024. "Roles of DNA Methylation in Color Alternation of Eastern Honey Bees (Apis cerana) Induced by the Royal Jelly of Western Honey Bees (Apis mellifera)" International Journal of Molecular Sciences 25, no. 6: 3368. https://doi.org/10.3390/ijms25063368
APA StyleAbdelmawla, A., Li, X., Shi, W., Zheng, Y., Zeng, Z., & He, X. (2024). Roles of DNA Methylation in Color Alternation of Eastern Honey Bees (Apis cerana) Induced by the Royal Jelly of Western Honey Bees (Apis mellifera). International Journal of Molecular Sciences, 25(6), 3368. https://doi.org/10.3390/ijms25063368