
Whole-Genome Survey Analyses of Five Goby Species Provide Insights into Their Genetic Evolution and Invasion-Related Genes

Abstract
1. Introduction
2. Results
2.1. Whole-Genome Sequencing
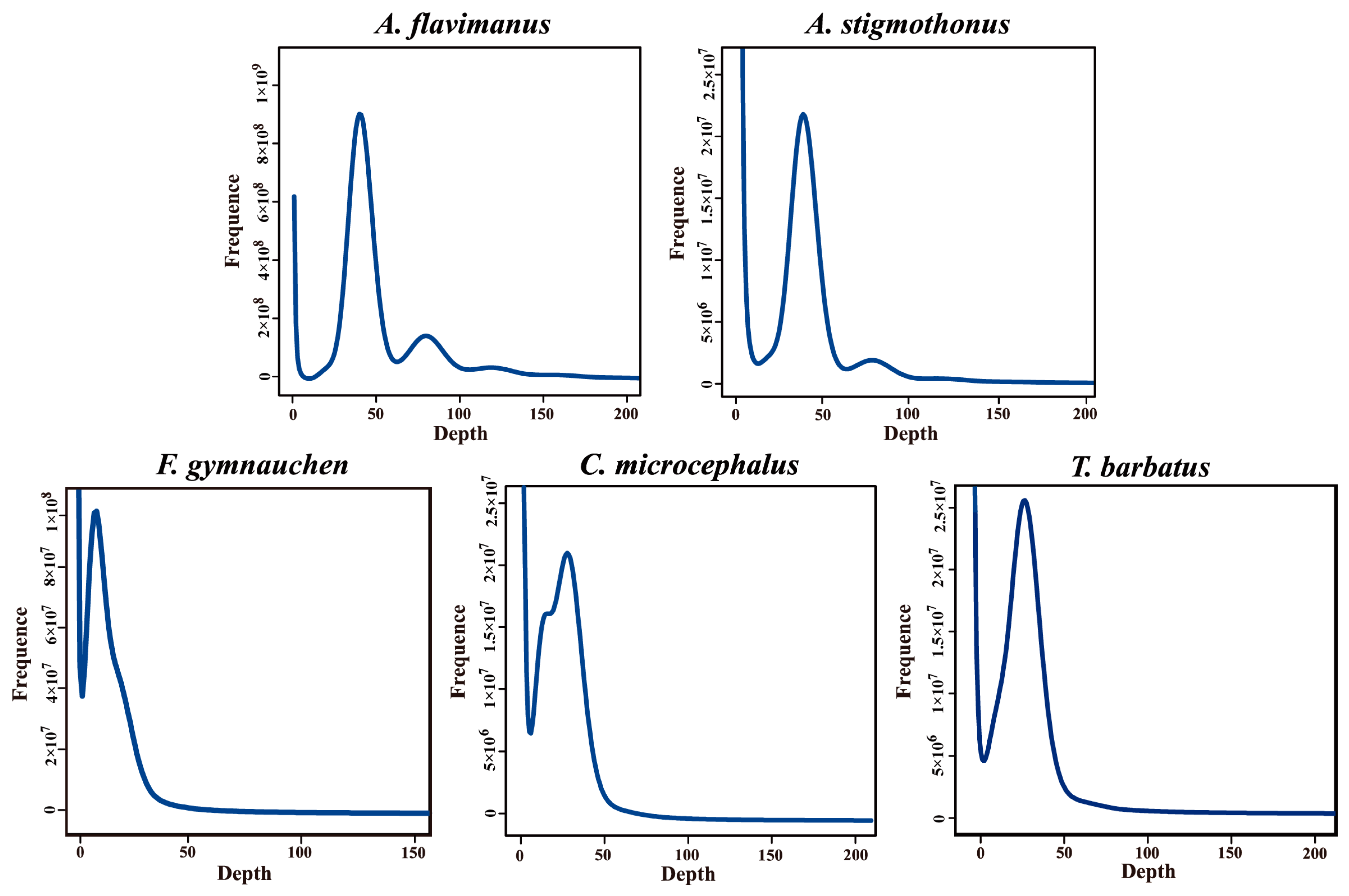
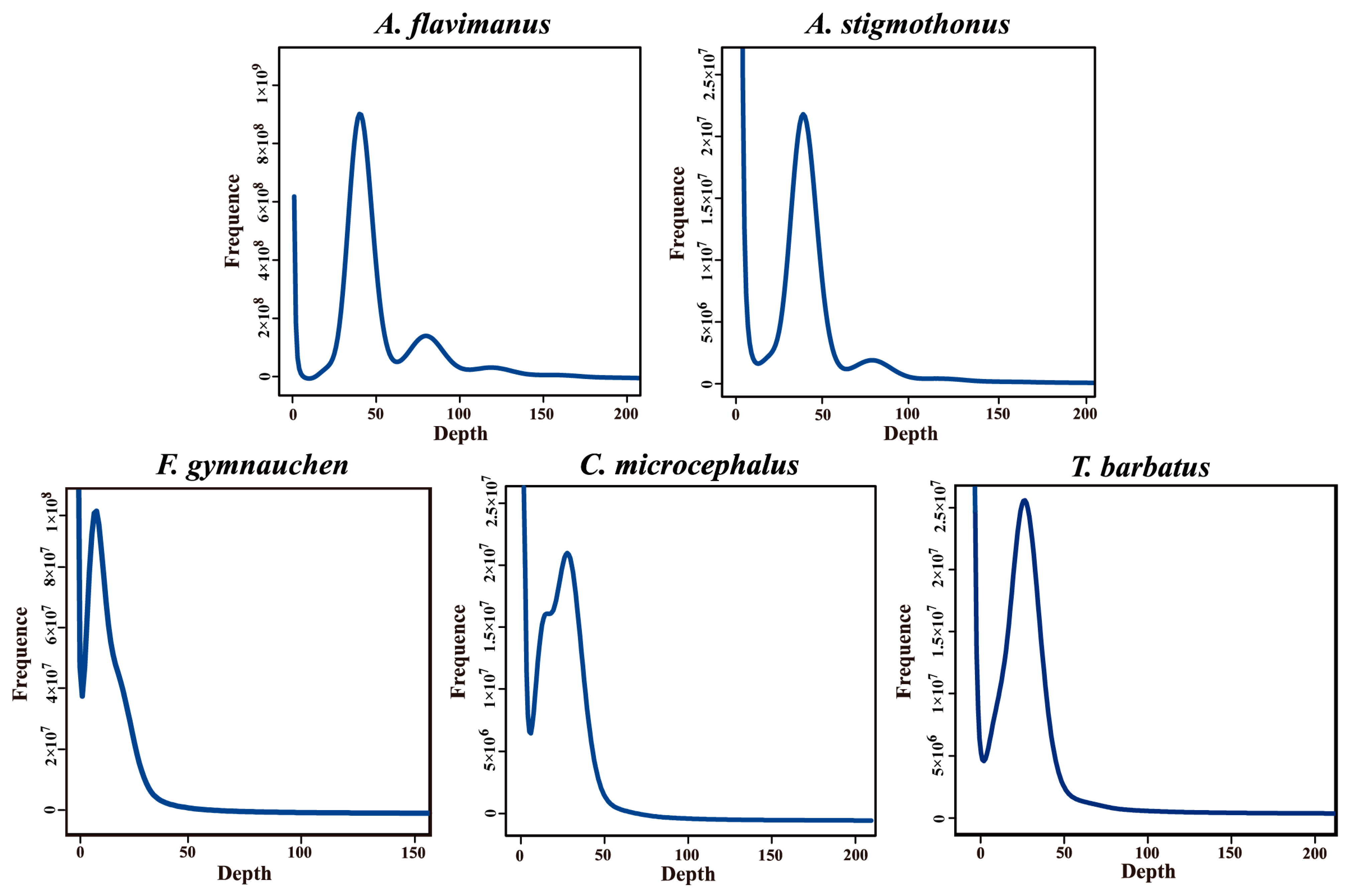
2.2. K-mer Analysis
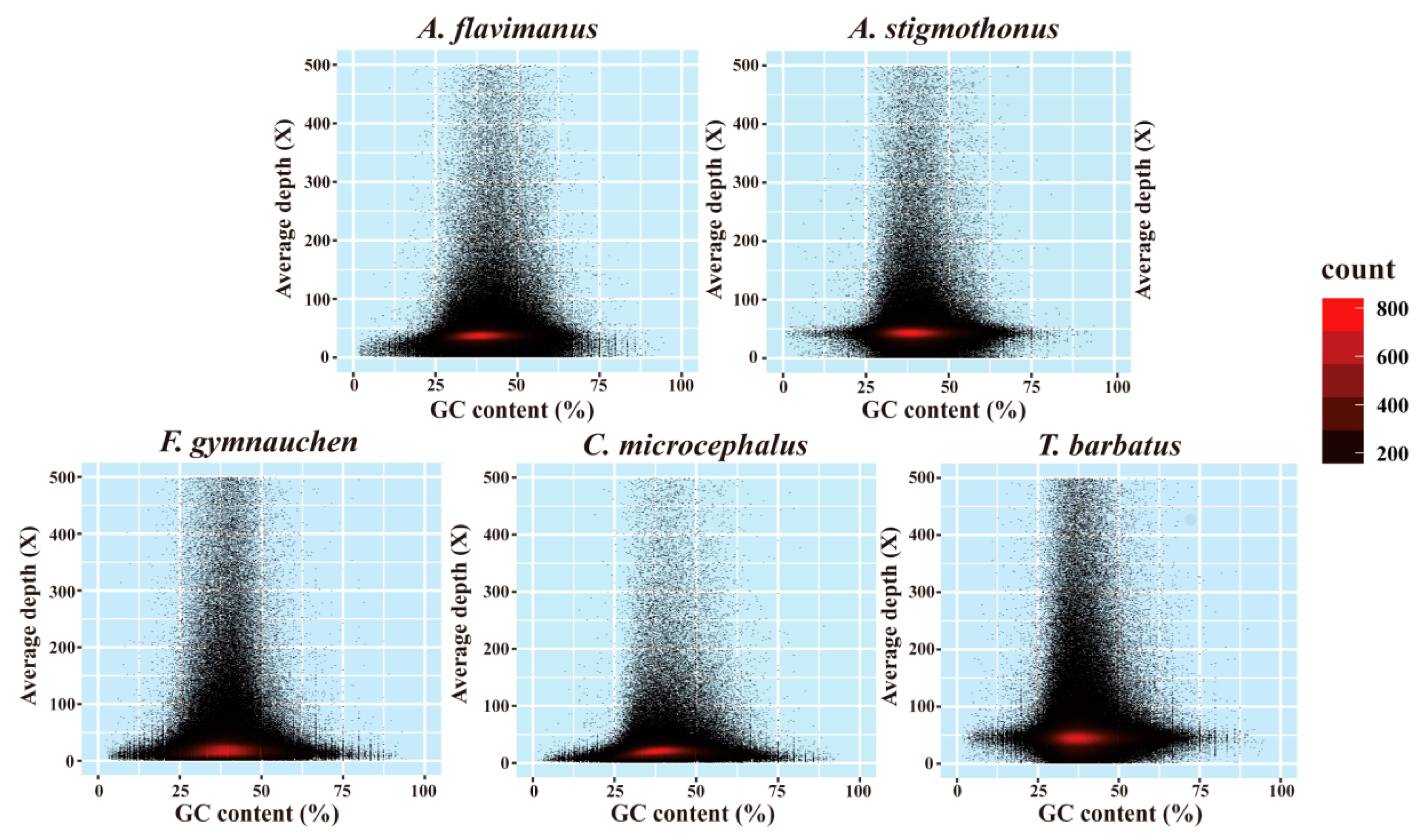
2.3. Genome Assembly and GC Content
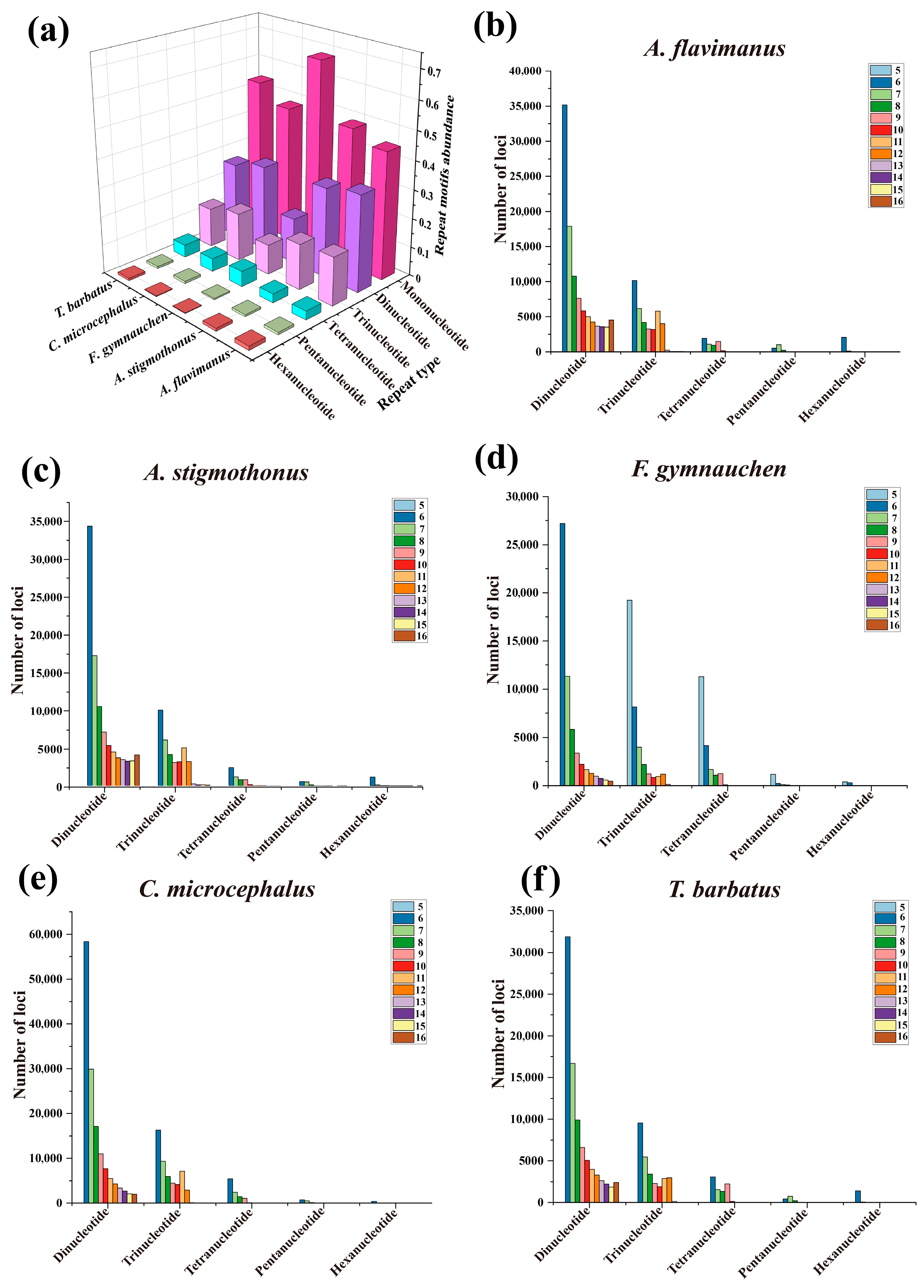
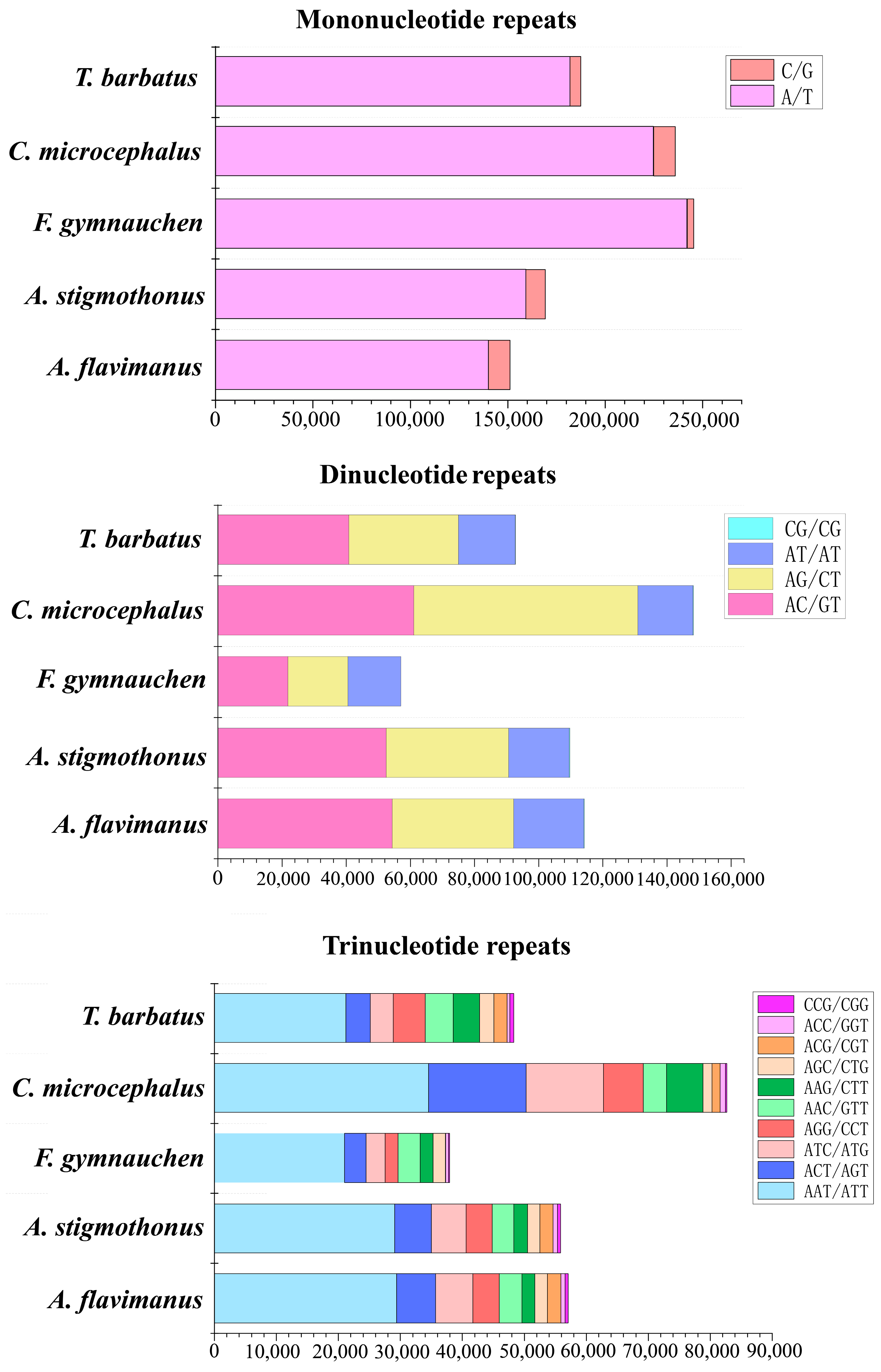
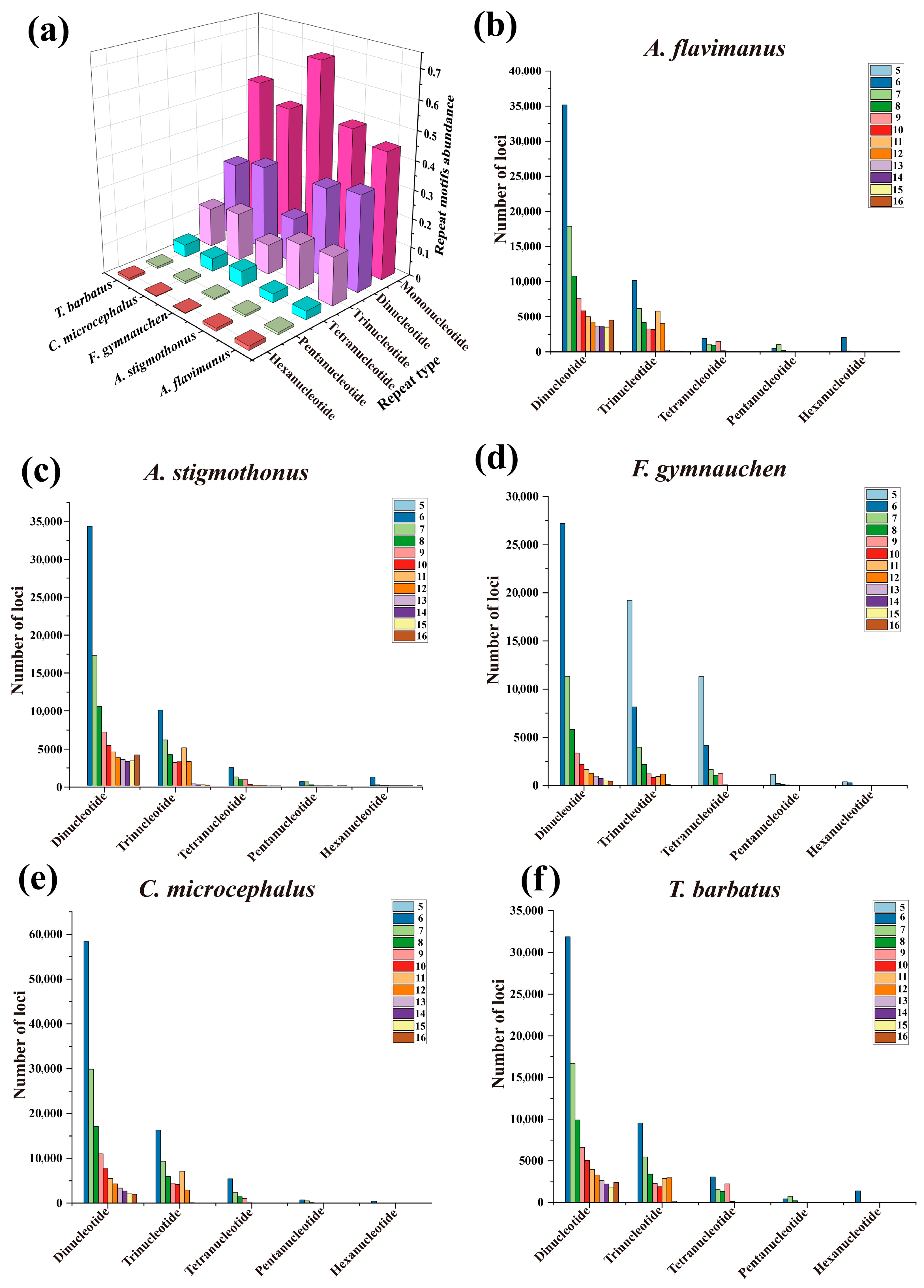
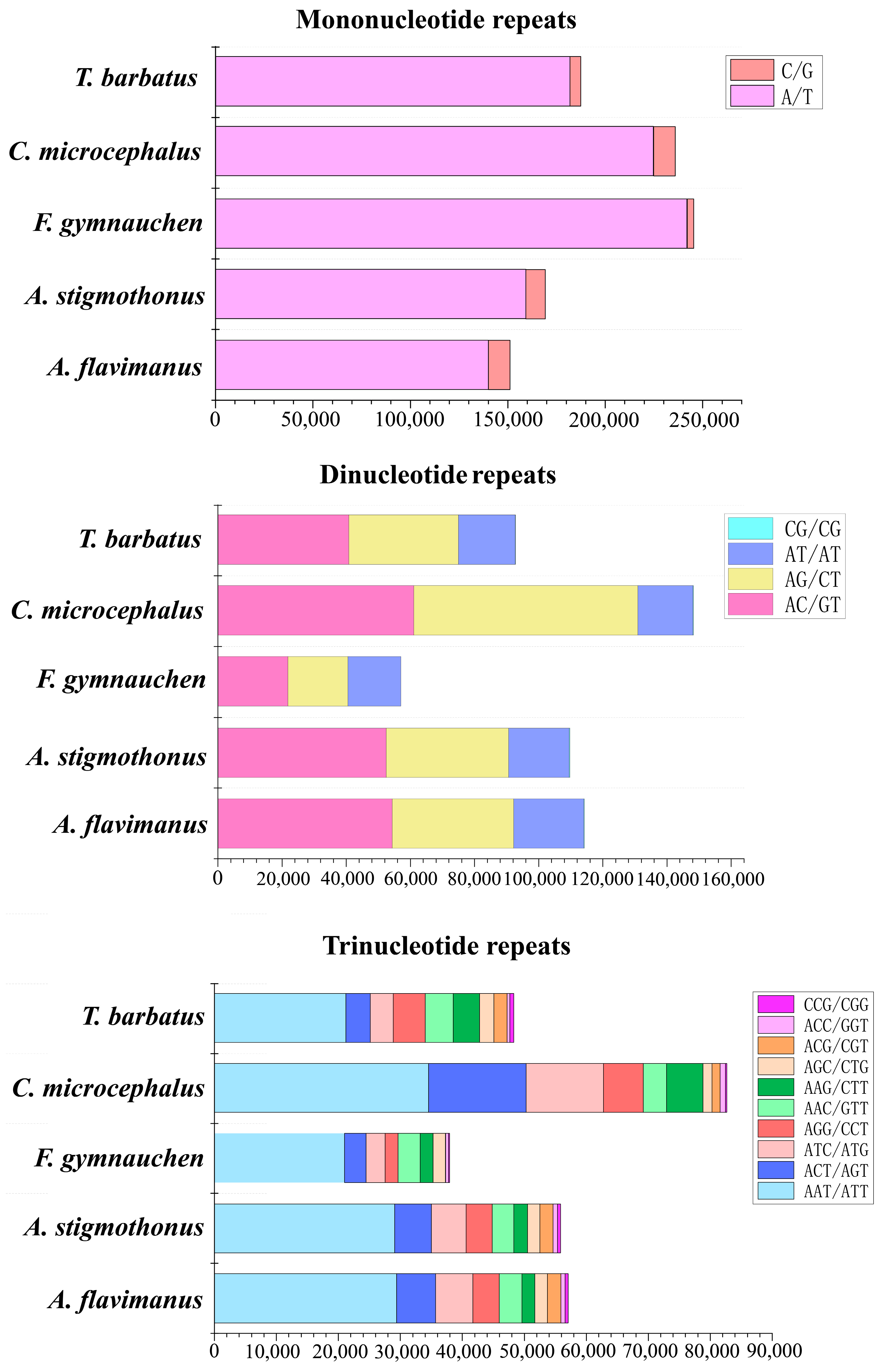
2.4. Microsatellite Profile of Five Gobiidae Fish Genomes
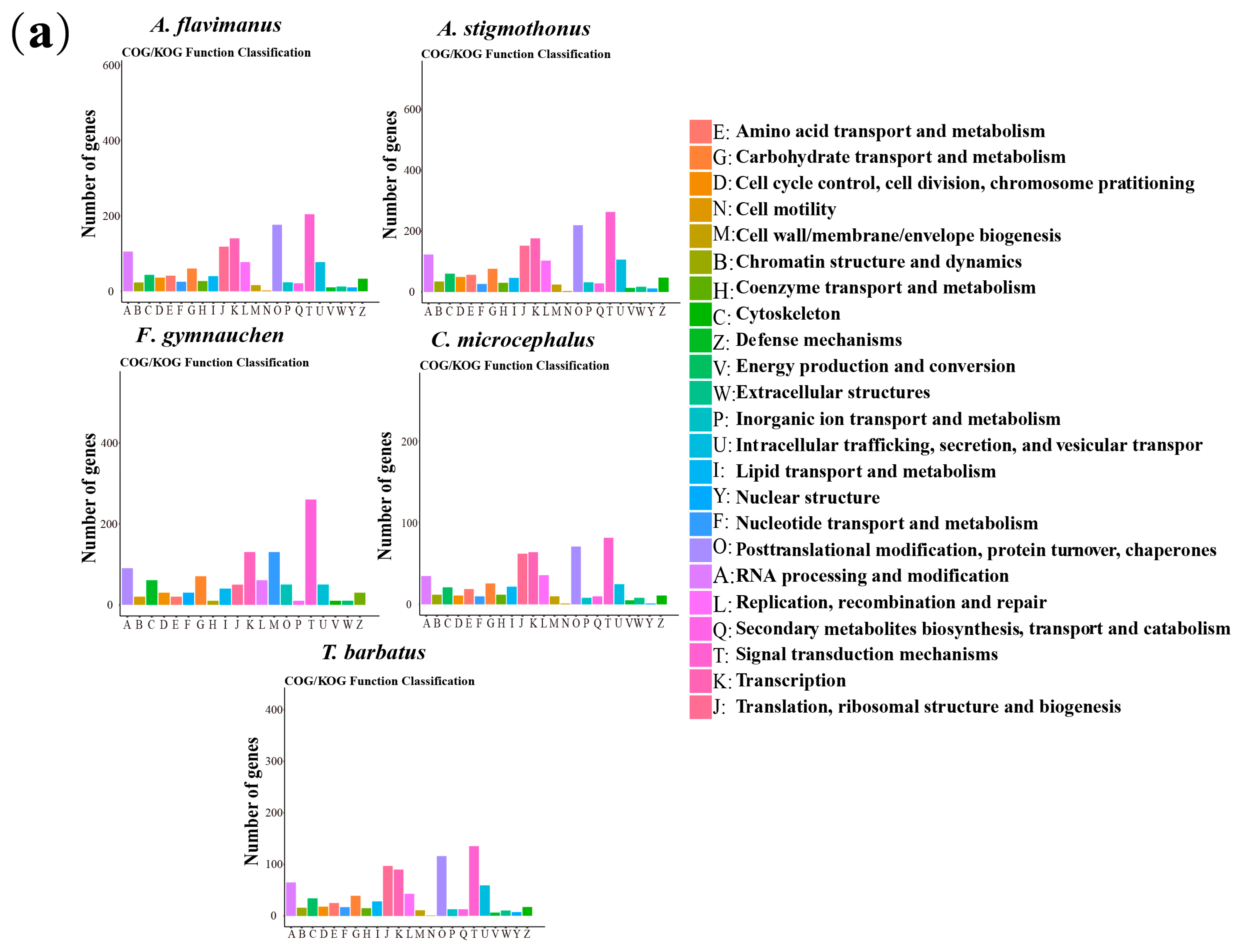
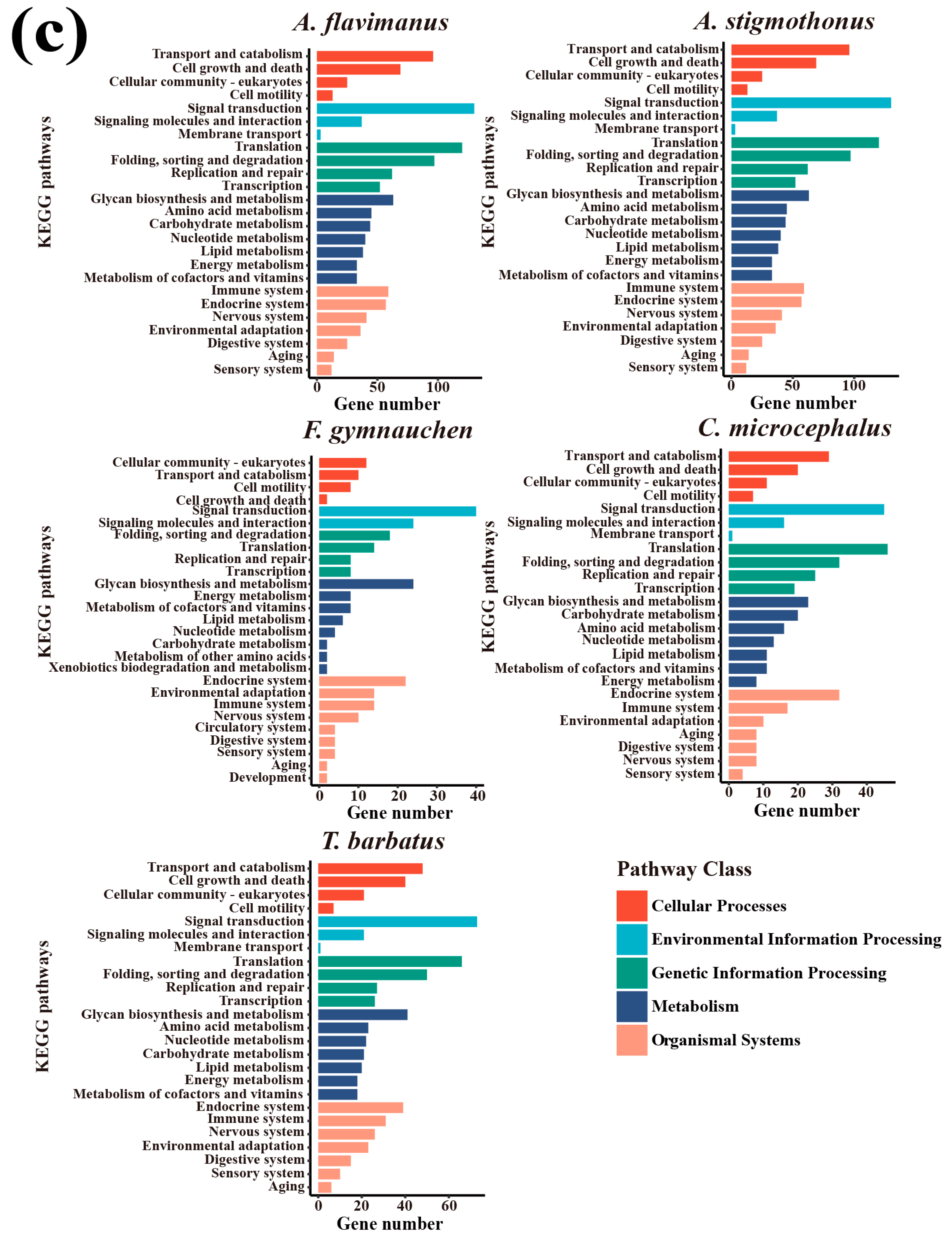
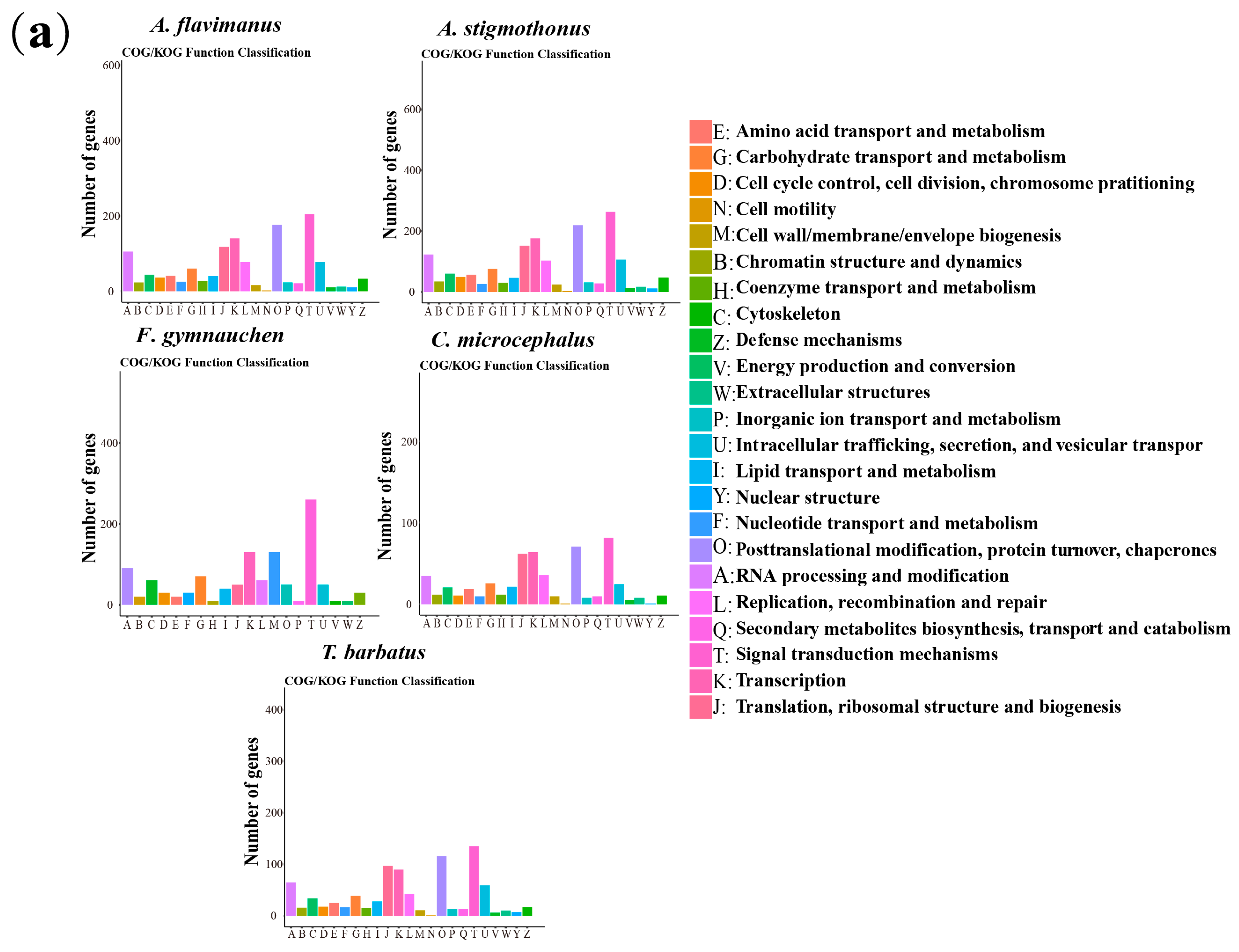
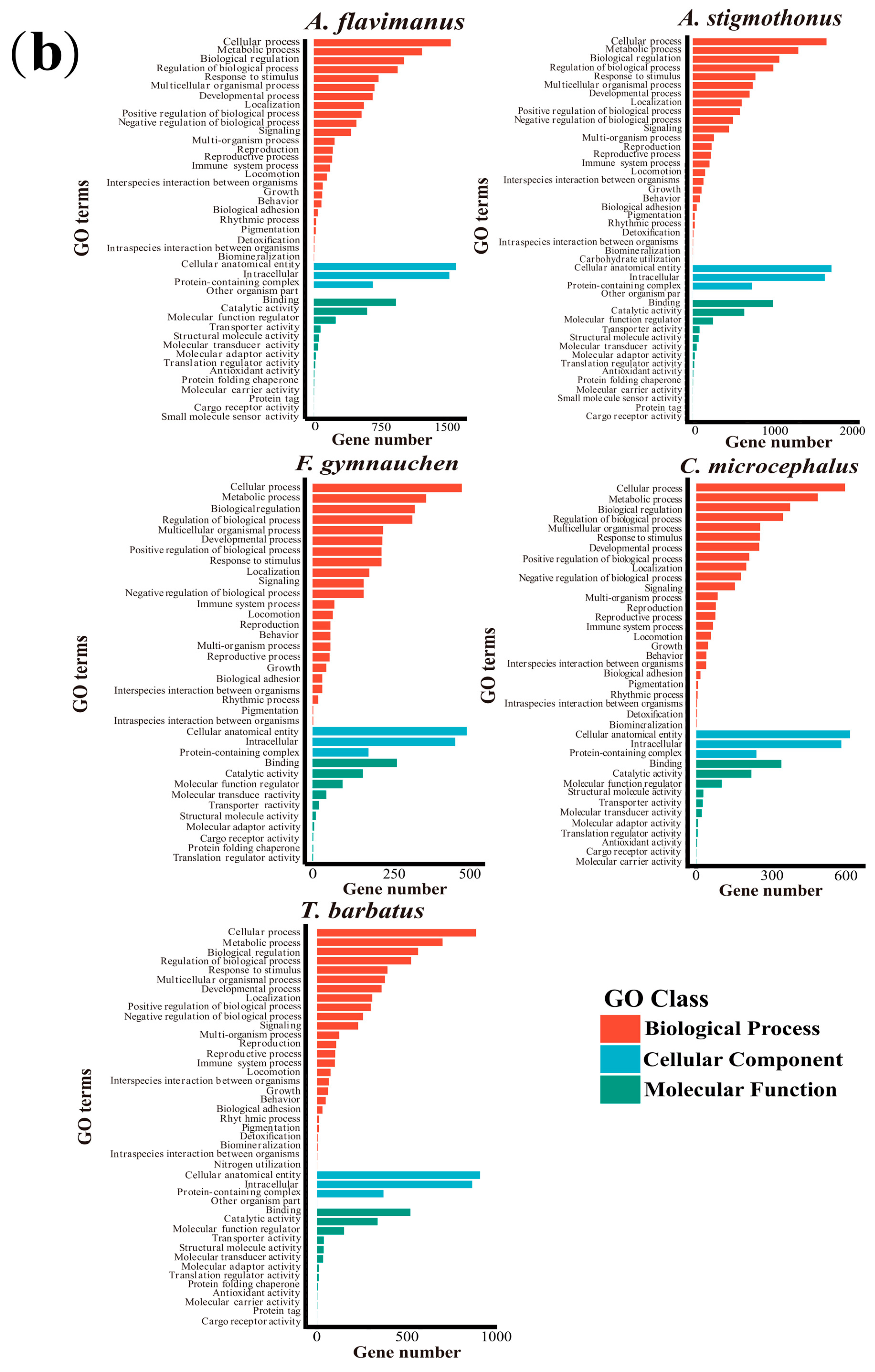
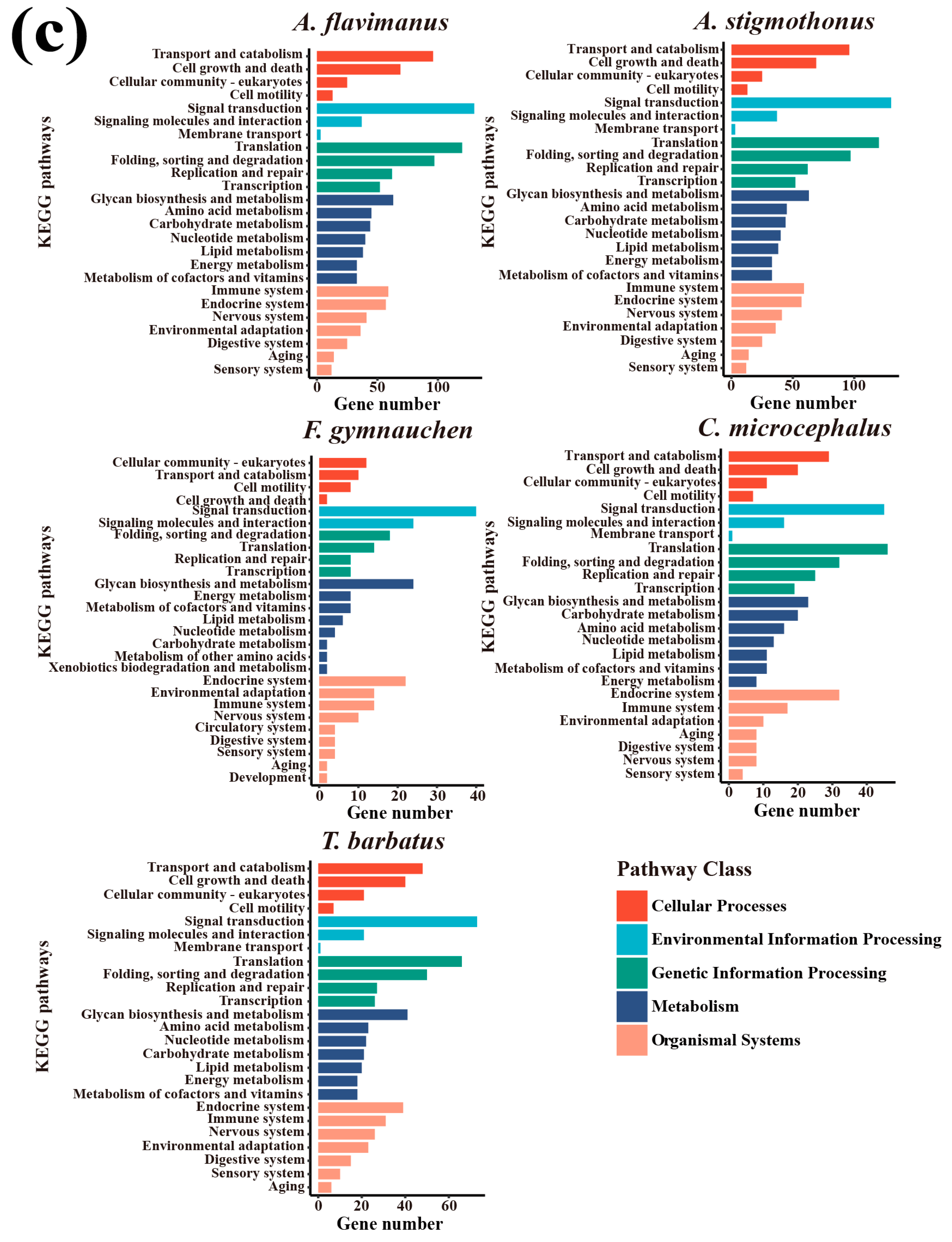
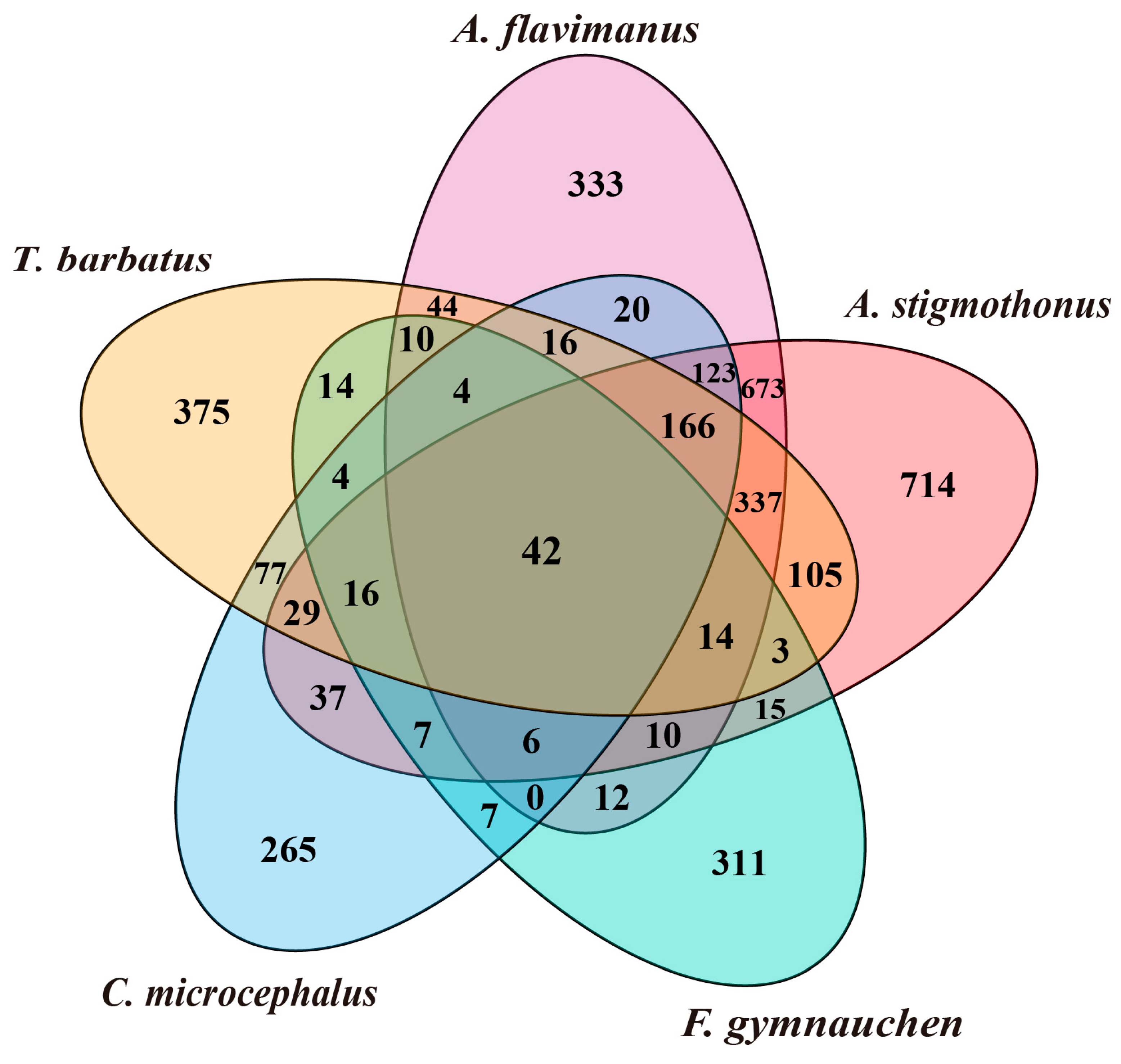
2.5. Genome Annotation
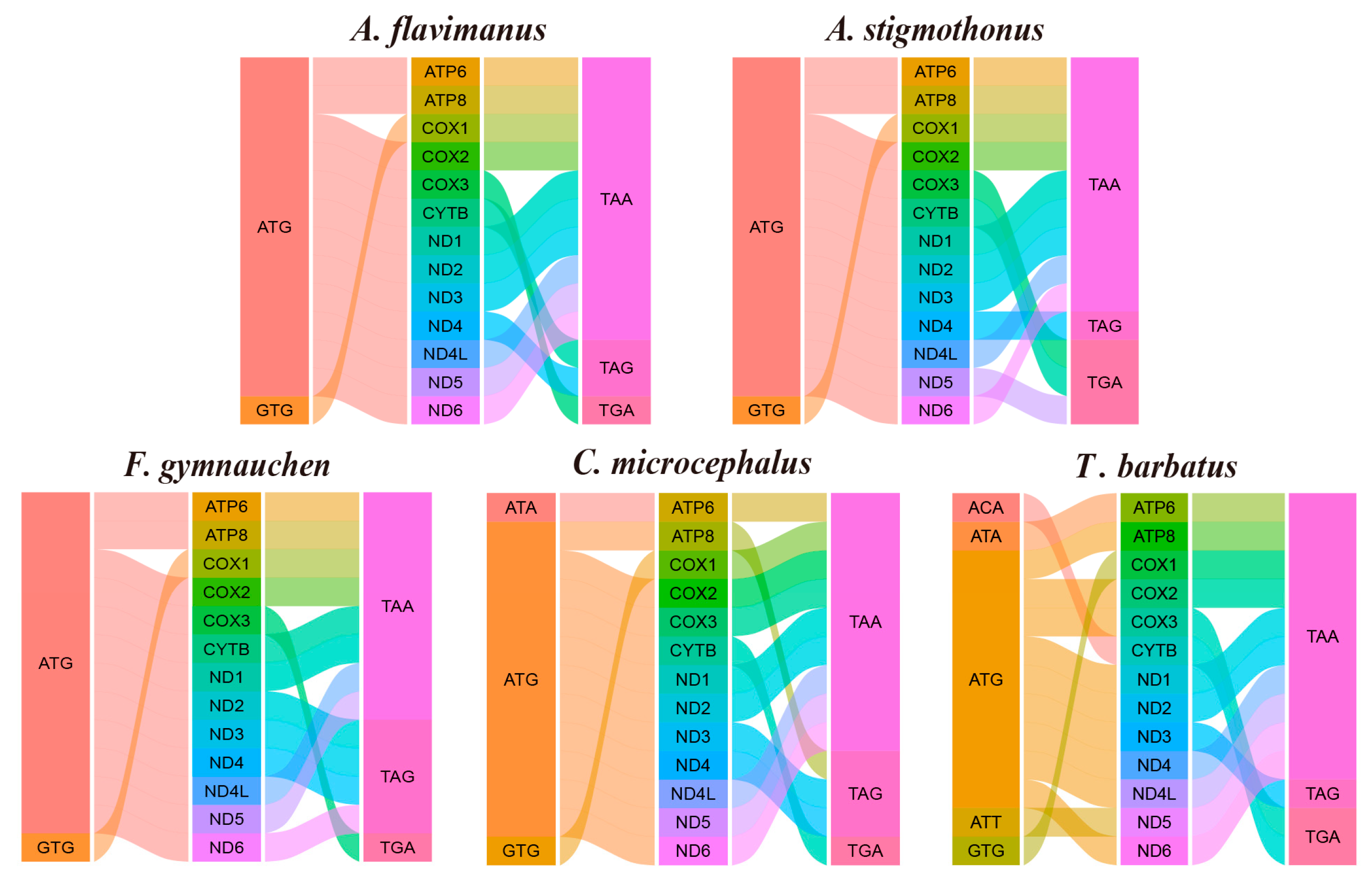
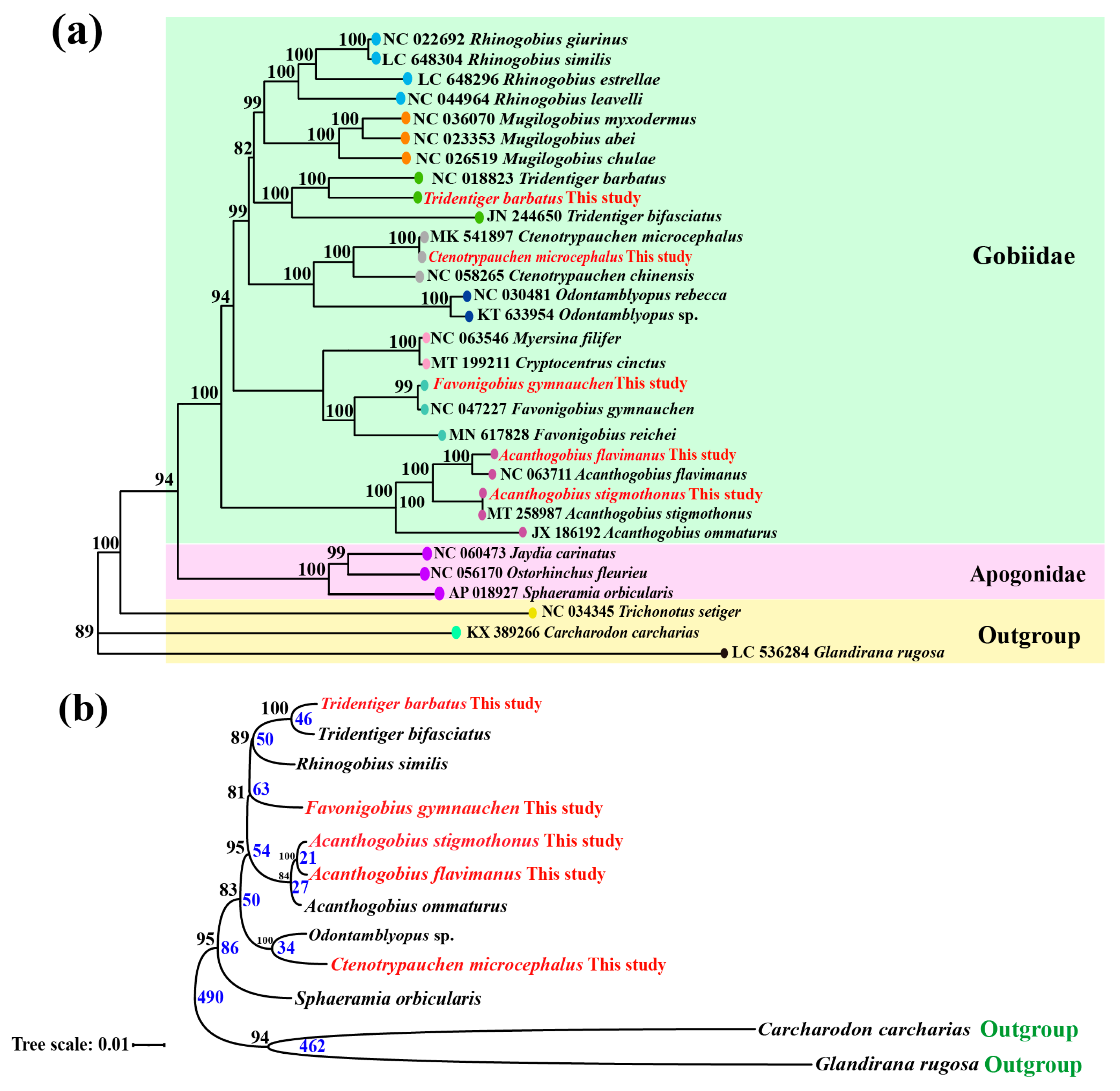
2.6. Mitochondrial Genome Assembly and Phylogenetic Analysis
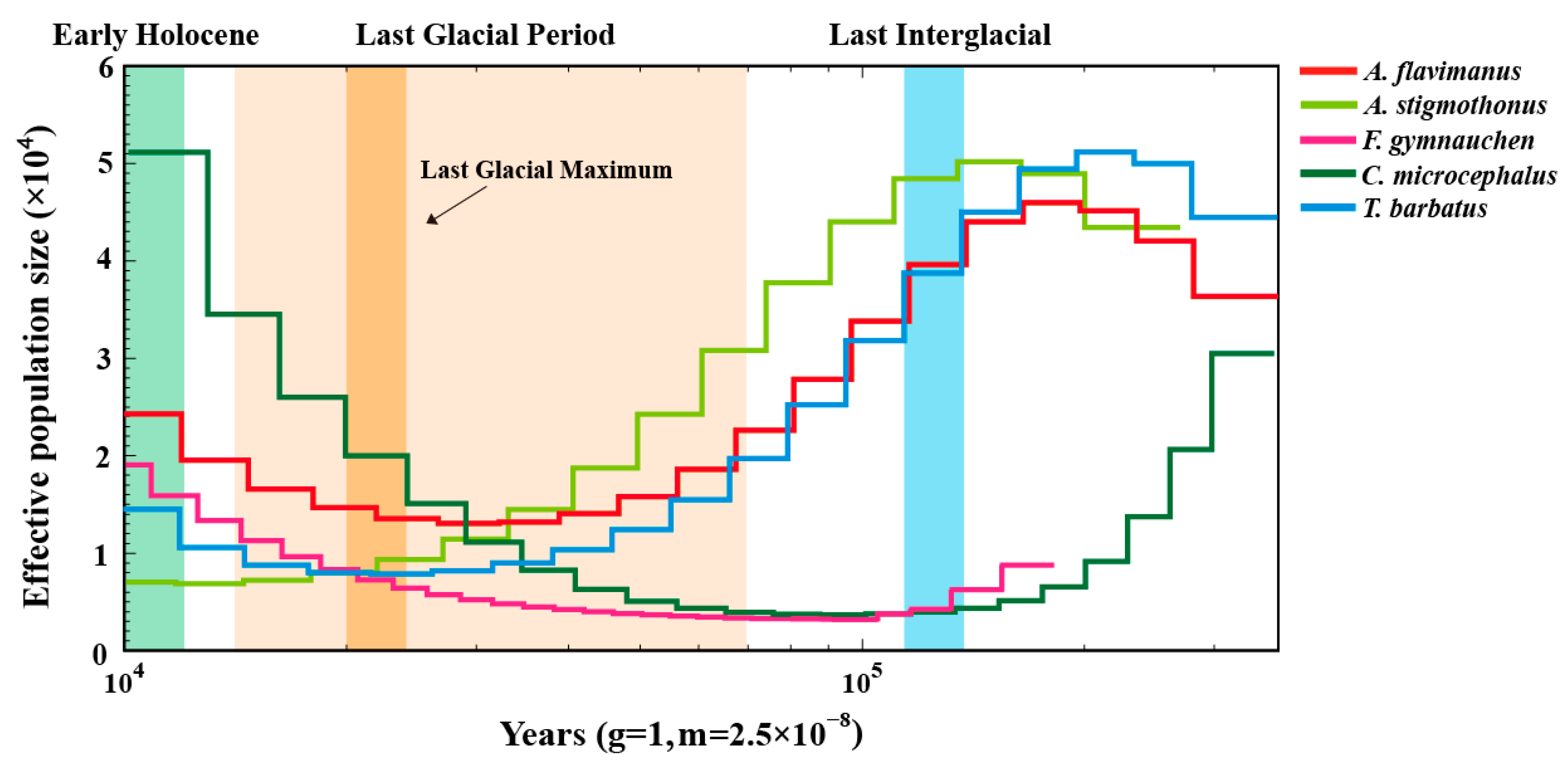
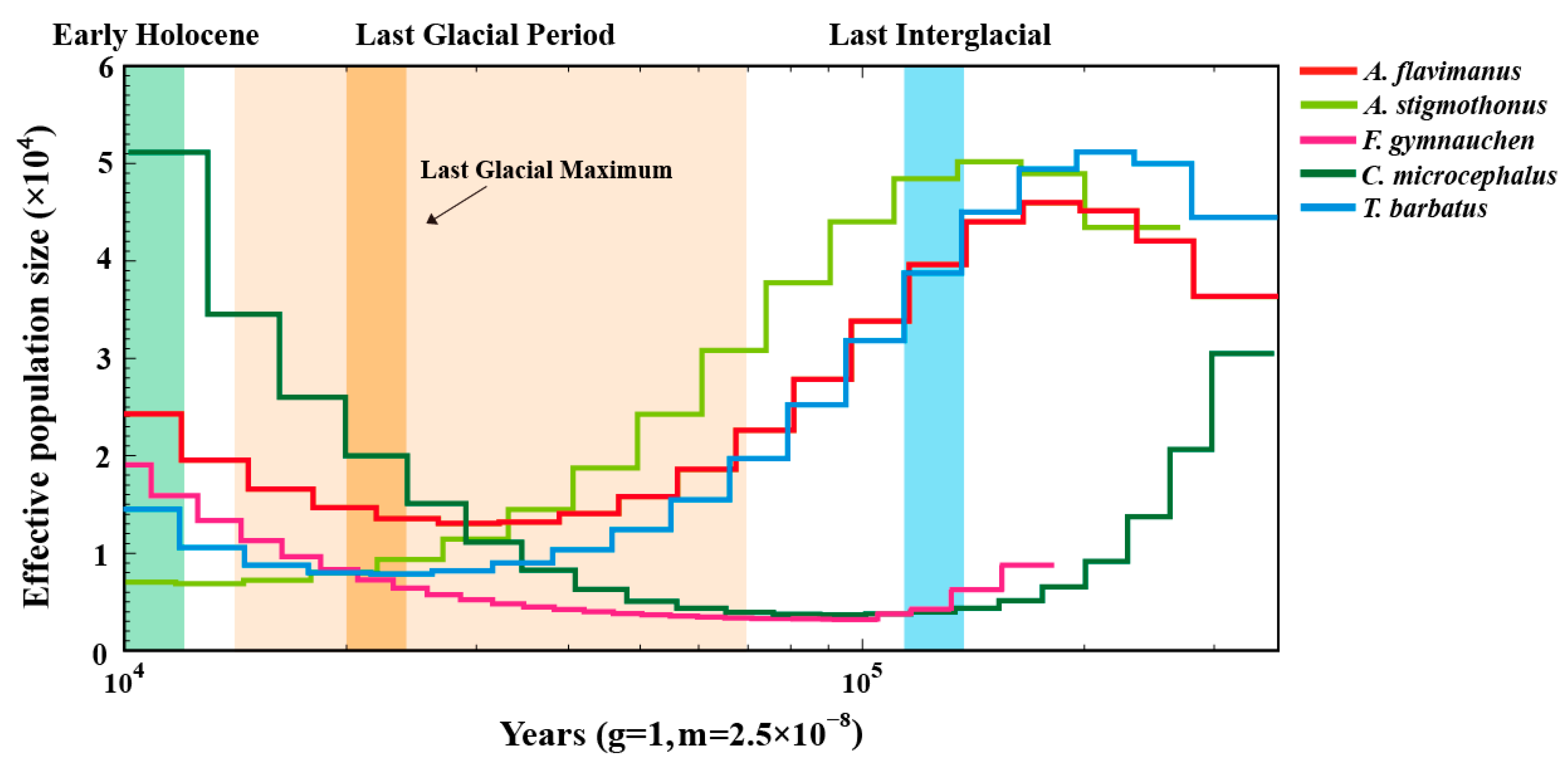
2.7. Demographic History
3. Discussion
3.1. Genome Sequencing and Characteristics
3.2. Profile of Repeat Sequences in 5 Gobiidae Fish
3.3. Genome Annotation
3.4. Overview of Mitochondrial Genome and Phylogenetic Relationship
3.5. Demographic Analysis
4. Materials and Methods
4.1. Ethics Statement
4.2. Sample Preparation and Genome Sequencing
4.3. Analysis of K-mer and GC Content
4.4. Microsatellite Identification
4.5. Search of Single-Copy Homologous Genes and Functional Annotation
4.6. Mitochondrial DNA Assembly and Phylogenetic Research
4.7. Demography History Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agorreta, A.; Mauro, D.S.; Schliewen, U.; Van Tassell, J.L.; Kovačić, M.; Zardoya, R.; Rüber, L. Molecular phylogenetics of Gobioidei and phylogenetic placement of European gobies. Mol. Phylogenet. Evol. 2013, 69, 619–633. [Google Scholar] [CrossRef]
- Winterbottom, R.; Emery, A.R. A new genus and two new species of gobiid fishes (Perciformes) from the Chagos Archipelago, Central Indian Ocean. Environ. Biol. Fishes 1981, 6, 139–149. [Google Scholar] [CrossRef]
- Froese, R. FishBase. World Wide Web Electronic Publication. 2005. Available online: http://www.fishbase.org (accessed on 2 January 2024).
- Han, D.; Xue, Y.; Ji, Y.; Xu, B.; Liu, H.; Ma, Q. Trophic and spatial niche of five gobiid fishes in Jiaozhou Bay. J. Fish. Sci. China 2013, 20, 148–156. [Google Scholar] [CrossRef]
- Koonin, E.V.; Wolf, Y.I. Constraints and plasticity in genome and molecular-phenome evolution. Nat. Rev. Genet. 2010, 11, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Roche, K.; Janáč, M.; Šlapanský, L.; Mikl, L.; Kopeček, L.; Jurajda, P. A newly established round goby (Neogobius melanostomus) population in the upper stretch of the river Elbe. Knowl. Manag. Aquat. Ecosyst. 2015, 416, 33. [Google Scholar] [CrossRef]
- Zhou, W. A preliminary study on endangerment mechanism of freshwater fish species in wetland ecosystem of Yunnan. Biodivers. Sci. 2000, 8, 163. [Google Scholar] [CrossRef]
- Aguilar-Medrano, R.; Durand, J.R.; Cruz-Escalona, V.H.; Moyle, P.B. Fish functional groups in the San Francisco Estuary: Understanding new fish assemblages in a highly altered estuarine ecosystem. Estuar. Coast. Shelf Sci. 2019, 227, 106331. [Google Scholar] [CrossRef]
- O’grady, K.; Cowx, I. Nonindigenous fishes introduced into inland waters of the United States. Fish. Manag. Ecol. 2000, 2, 172–175. [Google Scholar] [CrossRef]
- Liu, J.; Du, J.C. Progress on the Application of Bioinformatics in Digital Agriculture. Chin. Agric. Sci. Bull. 2014, 30, 279–284. [Google Scholar]
- Van Tassell, J.L.; Kovacic, M. The Biology of Gobies; Patzner, R.A., Kapoor, B.G., Eds.; Science Publishers: Enfield, UK, 2011; pp. 3–138. [Google Scholar]
- Bai, C.; Liu, S.; Zhuang, C. Distribution characteristics of microsatellite DNA in the genome of Gobiidae. Prog. Fish. Sci. 2016, 5, 9–15. [Google Scholar]
- Shang, Y.; Wang, X.; Liu, G.; Wu, X.; Wei, Q.; Sun, G.; Mei, X.; Dong, Y.; Sha, W.; Zhang, H. Adaptability and Evolution of Gobiidae: A Genetic Exploration. Animals 2022, 12, 1741. [Google Scholar] [CrossRef]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef]
- Lu, M.; An, H.; Li, L. Genome Survey Sequencing for the Characterization of the Genetic Background of Rosa roxburghii Tratt and Leaf Ascorbate Metabolism Genes. PLoS ONE 2016, 11, e0147530. [Google Scholar] [CrossRef]
- Sarropoulou, E.; Sundaram, M.Y.A.; Kaitetzidou, E.; Kotoulas, G.; Gilfillan, D.G.; Papandroulakis, N.; Mylonas, C.C.; Magoulas, A. Full genome survey and dynamics of gene expression in the greater amberjack Seriola dumerili. Gigascience 2017, 6, 12. [Google Scholar] [CrossRef]
- Shi, L.; Yi, S.; Li, Y. Genome survey sequencing of red swamp crayfish Procambarus clarkii. Mol. Biol. Rep. 2018, 45, 799–806. [Google Scholar] [CrossRef]
- Song, H.; Zhang, Y.; Yang, M.; Sun, J.; Zhang, T.; Wang, H. Genome survey on invasive veined rapa whelk (Rapana venosa) and development of microsatellite loci on large scale. J. Genet. 2018, 97, 79–86. [Google Scholar] [CrossRef]
- Gilbert, P.S.; Chang, J.; Pan, C.; Sobel, E.M.; Sinsheimer, J.S.; Faircloth, B.C.; Alfaro, M.E. Genome-wide ultraconserved elements exhibit higher phylogenetic informativeness than traditional gene markers in percomorph fishes. Mol. Phylogenet. Evol. 2015, 92, 140–146. [Google Scholar] [CrossRef]
- Koprek, T.; Rangel, S.; McElroy, D.; Louwerse, J.D.; Williams-Carrier, R.E.; Lemaux, P.G. Transposon-Mediated Single-Copy Gene Delivery Leads to Increased Transgene Expression Stability in Barley. Plant Physiol. 2001, 125, 1354–1362. [Google Scholar] [CrossRef]
- Ma, Y.; Lou, F.; Yin, X.; Cong, B.; Liu, S.; Zhao, L.; Zheng, L. Whole-genome survey and phylogenetic analysis of Gadus macrocephalus. Biosci. Rep. 2022, 42, 7. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Y.; Du, X.; Ma, S.; Song, N.; Zhao, L. Whole-Genome Survey Analyses Provide a New Perspective for the Evolutionary Biology of Shimofuri Goby, Tridentiger bifasciatus. Animals 2022, 12, 1914. [Google Scholar] [CrossRef]
- Baeza, J.A.; Molina-Quirós, J.L.; Hernández-Muñoz, S. Genome survey sequencing of an iconic ‘Trophy’sportfish, the Roosterfish Nematistius pectoralis: Genome size, repetitive elements, nuclear RNA gene operon, and microsatellite discovery. Genes 2021, 12, 1710. [Google Scholar] [CrossRef]
- Shibukawa, K.; Iwata, A. Review of the East Asian Gobiid genus Chaeturichthys (Teleostei: Perciformes: Gobioidei), with description of a new species. Bull. Natl. Mus. Nat. Sci. 2013, 4, 31–51. [Google Scholar]
- Das, S.; Hirano, M. Editorial [Hot Topic: Comparative Genomics and Genome Evolution (Guest Editors: Sabyasachi Das and Masayuki Hirano)]. Curr. Genom. 2012, 13, 85. [Google Scholar] [CrossRef]
- Pan, Y.; Sun, Z.; Gao, T.; Zhao, L.; Song, N. Chromosome-level genome assembly of Acanthogobius ommaturus provides insights into evolution and lipid metabolism. Front. Mar. Sci. 2022, 9, 894821. [Google Scholar] [CrossRef]
- Adrian-Kalchhauser, I.; Blomberg, A.; Larsson, T.; Musilova, Z.; Peart, C.R.; Pippel, M.; Solbakken, M.H.; Suurväli, J.; Walser, J.-C.; Wilson, J.Y.; et al. The round goby genome provides insights into mechanisms that may facilitate biological invasions. BMC Biol. 2020, 18, 11. [Google Scholar] [CrossRef]
- Qiu, B.; Fang, S.; Ikhwanuddin, M.; Wong, L.; Ma, H. Genome survey and development of polymorphic microsatellite loci for Sillago sihama based on Illumina sequencing technology. Mol. Biol. Rep. 2020, 47, 3011–3017. [Google Scholar] [CrossRef]
- Cao, S.; Yang, Y.; Bi, G.; Nelson, D.; Hu, S.; Makunga, N.P.; Yu, B.; Liu, X.; Li, X.; Hu, X. Genomic and Transcriptomic Insight of Giant Sclerotium Formation of Wood-Decay Fungi. Front. Microbiol. 2021, 12, 746121. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Sternberg, R. Why repetitive DNA is essential to genome function. Biol. Rev. Camb. Philos. Soc. 2005, 80, 227–250. [Google Scholar] [CrossRef]
- Xu, S.; Song, N.; Xiao, S.; Gao, T. Whole genome survey analysis and microsatellite motif identification of Sebastiscus marmoratus. Biosci. Rep. 2020, 40, 2. [Google Scholar] [CrossRef]
- Lei, Y.; Zhou, Y.; Price, M.; Song, Z. Genome-wide characterization of microsatellite DNA in fishes: Survey and analysis of their abundance and frequency in genome-specific regions. BMC Genom. 2021, 22, 421. [Google Scholar] [CrossRef]
- Fraimout, A.; Loiseau, A.; Price, D.K.; Xuéreb, A.; Jean-François, M.; Vitalis, R.; Fellous, S.; Debat, V.; Estoup, A. New set of microsatellite markers for the spotted-wing Drosophila suzukii (Diptera: Drosophilidae): A promising molecular tool for inferring the invasion history of this major insect pest. Eur. J. Entomol. 2015, 112, 855. [Google Scholar] [CrossRef]
- Huang, X.; Jiang, Y.; Jiang, X.; Yang, T. Analysis of Microsatellite Markers in Harpadon Nehereus Based on Transcriptome Sequencing Illumina Hiseq tm 2500. J. Zhejiang Ocean. Univ. Nat. Sci. 2021, 40, 189–197. [Google Scholar]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, J.; Han, R.; Zhang, F.; Mao, A.; Luo, J.; Dong, B.; Liu, H.; Tang, H.; Zhang, J.; et al. Target SSR-Seq: A novel SSR genotyping technology associate with perfect SSRs in genetic analysis of cucumber varieties. Front. Plant Sci. 2019, 10, 531. [Google Scholar] [CrossRef]
- Song, N.; Zhao, X.; Cai, C.; Gao, T. Profile of the genomic characteristics and comparative studies of five Trichiuridae species by genome survey sequencing. Front. Microbiol. 2022, 9, 962307. [Google Scholar] [CrossRef]
- Fitch, M.W. Distinguishing homologous from analogous proteins. Syst. Zool. 1970, 19, 99–113. [Google Scholar] [CrossRef]
- Huang, Y.; Feulner, P.G.; Eizaguirre, C.; Lenz, T.L.; Bornberg-Bauer, E.; Milinski, M.; Reusch, H.B.T.; Chain, J.J.F. Genome-wide genotype-expression relationships reveal both copy number and single nucleotide differentiation contribute to differential gene expression between stickleback ecotypes. Genome Biol. Evol. 2019, 11, 2344–2359. [Google Scholar] [CrossRef]
- Ištvánek, J.; Dluhošová, J.; Dluhoš, P.; Pátková, L.; Nedělník, J.; Řepková, J. Classification and Mining of Molecular Markers Useful in Red Clover (Trifolium pratense) Breeding. Front. Plant Sci. 2017, 8, 367. [Google Scholar] [CrossRef]
- Borgonove, C.M.; Cavallari, C.B.; Santos, M.H.; Rossetti, R.; Hartfelder, K.; Manfrin, M.H. Identification of differentially expressed genes in female Drosophila antonietae and Drosophila meridionalis in response to host cactus odor. BMC Evol. Biol. 2014, 14, 191. [Google Scholar] [CrossRef]
- Moran, E.V.; Alexander, J.M. Evolutionary responses to global change: Lessons from invasive species. Ecol. Lett. 2014, 17, 637–649. [Google Scholar] [CrossRef]
- Carroll, S.P. Natives adapting to invasive species: Ecology, genes, and the sustainability of conservation. Ecol. Res. 2007, 22, 892–901. [Google Scholar] [CrossRef]
- Ahmar, S.; Gruszka, D. In-silico study of brassinosteroid signaling genes in rice provides insight into mechanisms which regulate their expression. Front. Genet. 2022, 13, 953458. [Google Scholar] [CrossRef] [PubMed]
- Barthélémy, C.; de Baulny, H.; Lombès, A. D-loop mutations in mitochondrial DNA: Link with mitochondrial DNA depletion? Hum. Genet. 2002, 110, 479–487. [Google Scholar] [CrossRef]
- Allio, R.; Schomaker-Bastos, A.; Romiguier, J.; Prosdocimi, F.; Nabholz, B.; Delsuc, F. MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 2020, 20, 892–905. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. NAR 2019, 47, e63. [Google Scholar] [CrossRef]
- Thacker, E.C.; Roje, M.D. Phylogeny of cardinalfishes (Teleostei: Gobiiformes: Apogonidae) and the evolution of visceral bioluminescence. Mol. Phylogenet. Evol. 2009, 52, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.S. Fish of the World; John Wiley Sons: New York, NY, USA, 2006; Volume 4, p. 601. [Google Scholar]
- Zheng, J.; Gao, W.; Sun, Z.; Gao, T.; Song, N. The complete mitochondrial genome of Acanthogobius stigmothonus (Perciformes, Gobiidae). Mitochondrial DNA Part B 2020, 5, 2888–2889. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.B.; Jun, J.; Choi, S.H.; Suk, H.Y. Diversification and colonization processes in Gobioidei predicted based on mitochondrial 12S rRNA with focusing on Oxudercidae. Mitochondrial DNA Part B 2021, 6, 1166–1172. [Google Scholar] [CrossRef]
- Thacker, C. Phylogeny of Gobioidei and Placement within Acanthomorpha, with a New Classification and Investigation of Diversification and Character Evolution. Copeia 2009, 2009, 93–104. [Google Scholar] [CrossRef]
- Cowman, P.F.; Bellwood, D.R. Coral reefs as drivers of cladogenesis: Expanding coral reefs, cryptic extinction events, and the development of biodiversity hotspots. J. Evol. Biol. 2011, 24, 2543–2562. [Google Scholar] [CrossRef]
- Chamala, S.; Feng, G.; Chavarro, C.; Barbazuk, W.B. Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants. Front. Bioeng. Biotechnol. 2015, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, A.A.; Skrbinšek, T.; Amato, G.; Dendrinos, P.; Gaughran, S.; Kasapidis, P.; Kopatz, A.; Stronen, V.A. Genetic and demographic history define a conservation strategy for earth’s most endangered pinniped, the Mediterranean monk seal Monachus Monachus. Sci. Rep. 2021, 11, 373. [Google Scholar] [CrossRef]
- Otto-Bliesner, B.L.; Marshall, S.J.; Overpeck, J.T.; Miller, G.H.; Hu, A. CAPE Last Interglacial Project members. Simulating Arctic climate warmth and icefield retreat in the last interglaciation. Science 2006, 311, 1751–1753. [Google Scholar] [CrossRef]
- Abbott, R.J.; Brochmann, C. History and evolution of the arctic flora: In the footsteps of Eric Hultén. Mol. Ecol. 2003, 12, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Gao, T.X.; Wu, S.F.; Zhang, Y.P. Pleistocene isolation in the Northwestern Pacific marginal seas and limited dispersal in a marine fish, Chelon haematocheilus (Temminck & Schlegel, 1845). Mol. Ecol. 2007, 16, 275–288. [Google Scholar] [CrossRef]
- Wu, H.; Zhong, J. Osteichthys, Perciformes, Gobioidei. In Fauna Sinica, 1st ed.; Science Press: Beijing, China, 2008; Volume 5, pp. 211–215. [Google Scholar]
- Delrieu-Trottin, E.; Maynard, J.; Planes, S. Endemic and widespread coral reef fishes have similar mitochondrial genetic diversity. Proc. Biol. Sci. 2014, 281, 1797. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Varshney, R.K.; Chen, W.; Li, Y.; Bharti, A.K.; Saxena, R.K.; Schlueter, J.A.; Donoghue, M.T.A.; Azam, S.; Fan, G.; Whaley, A.M.; et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 2012, 30, 83–89. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Baldwin, R.M.; Owzar, K.; Zembutsu, H.; Chhibber, A.; Kubo, M.; Jiang, C.; Watson, D.; Eclov, R.J.; Mefford, J.; McLeod, H.L.; et al. A Genome-Wide Association Study Identifies Novel Loci for Paclitaxel-Induced Sensory Peripheral Neuropathy in CALGB 40101. Clin. Cancer Res. 2012, 18, 5099–5109. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Emms, M.D.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Z. PAMLX: A graphical user interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Cantalapiedra, P.C.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Raw Base (Gb) | Effective Rate (%) | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|
| A. flavimanus | 52.48 | 99.51 | 0.04 | 96.31 | 91.18 | 41.04 |
| A. stigmothonus | 54.54 | 99.54 | 0.04 | 96.78 | 92.22 | 40.80 |
| F. gymnauchen | 53.77 | 99.18 | 0.05 | 96.83 | 91.19 | 40.05 |
| C. microcephalus | 53.55 | 96.87 | 0.03 | 96.43 | 91.42 | 41.69 |
| T. barbatus | 52.22 | 98.52 | 0.03 | 96.58 | 91.61 | 39.91 |
| Species | K-mer Number | Genome Size (Mb) | Heterozygous Ratio (%) | Repeat Ratio (%) |
|---|---|---|---|---|
| A. flavimanus | 38,806,743,595 | 978.96 | 0.21 | 50.94 |
| A. stigmothonus | 39,715,938,956 | 998.27 | 0.24 | 50.52 |
| F. gymnauchen | 44,941,714,426 | 1601.98 | 1.56 | 59.83 |
| C. microcephalus | 47,124,301,675 | 812.88 | 0.64 | 52.89 |
| T. barbatus | 45,948,893,647 | 808.55 | 0.56 | 46.90 |
| Species | Assembly Level | Total Length (bp) | Total Number | Max Length (bp) | N50 Length (bp) | N90 Length (bp) |
|---|---|---|---|---|---|---|
| A. flavimanus | Contig | 750,612,336 | 766,615 | 72,830 | 3318 | 382 |
| Scaffold | 764,365,216 | 612,519 | 78,798 | 4596 | 542 | |
| A. stigmothonus | Contig | 801,994,264 | 783,027 | 142,602 | 6066 | 301 |
| Scaffold | 817,012,736 | 631,430 | 230,933 | 12,605 | 508 | |
| F. gymnauchen | Contig | 1,538,128,271 | 7,370,457 | 75,210 | 2120 | 122 |
| Scaffold | 1,558,061,154 | 6,001,135 | 90,310 | 3120 | 129 | |
| C. microcephalus | Contig | 789,064,495 | 2,896,033 | 22,967 | 4431 | 150 |
| Scaffold | 782,790,775 | 2,117,910 | 24,126 | 8719 | 335 | |
| T. barbatus | Contig | 811,858,718 | 1,790,899 | 35,267 | 2116 | 245 |
| Scaffold | 805,390,951 | 1,216,192 | 35,267 | 2990 | 307 |
| A. flavimanus | A. stigmothonus | F. gymnauchen | C. microcephalus | T. barbatus | |
|---|---|---|---|---|---|
| Total amount of sequences assessed | 612,519 | 631,430 | 6,510,510 | 2,117,910 | 1,216,192 |
| Total size of assessed sequences (bp) | 764,365,216 | 817,012,736 | 1,653,145,701 | 806,337,820 | 817,882,156 |
| Total identified SSRs | 340,592 | 351,606 | 362,191 | 494,515 | 349,412 |
| Number of sequences containing SSRs | 163,241 | 106,981 | 337,951 | 371,928 | 226,596 |
| Number of sequences with multiple SSRs | 72,809 | 55,926 | 21,999 | 81,266 | 72,335 |
| Number of SSRs involved in compound synthesis | 57,550 | 62,289 | 15,217 | 73,846 | 41,068 |
| Genus | Species | Sampling Time | Sampling Site | Sample Quantity Obtained | Sample Quantity Used |
|---|---|---|---|---|---|
| Acanthogobius | Acanthogobius flavimanus | 26 November 2018 | Qingdao, Yellow Sea of China | 24 | 1 |
| Acanthogobius stigmothonus | 2 September 2020 | Beihai, South China Sea | 18 | 1 | |
| Favonigobius | Favonigobius gymnauchen | 20 November 2019 | Qingdao, Yellow Sea of China | 1 | 1 |
| Ctenotrypauchen | Ctenotrypauchen microcephalus | 12 April 2022 | Rizhao, Yellow Sea of China | 12 | 1 |
| Tridentiger | Tridentiger barbatus | 20 November 2021 | Yantai, Bohai Sea of China | 20 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Zhao, X.; Song, N. Whole-Genome Survey Analyses of Five Goby Species Provide Insights into Their Genetic Evolution and Invasion-Related Genes. Int. J. Mol. Sci. 2024, 25, 3293. https://doi.org/10.3390/ijms25063293
Ma S, Zhao X, Song N. Whole-Genome Survey Analyses of Five Goby Species Provide Insights into Their Genetic Evolution and Invasion-Related Genes. International Journal of Molecular Sciences. 2024; 25(6):3293. https://doi.org/10.3390/ijms25063293
Chicago/Turabian StyleMa, Siyu, Xiang Zhao, and Na Song. 2024. "Whole-Genome Survey Analyses of Five Goby Species Provide Insights into Their Genetic Evolution and Invasion-Related Genes" International Journal of Molecular Sciences 25, no. 6: 3293. https://doi.org/10.3390/ijms25063293
APA StyleMa, S., Zhao, X., & Song, N. (2024). Whole-Genome Survey Analyses of Five Goby Species Provide Insights into Their Genetic Evolution and Invasion-Related Genes. International Journal of Molecular Sciences, 25(6), 3293. https://doi.org/10.3390/ijms25063293