
Not Only Editing: A Cas-Cade of CRISPR/Cas-Based Tools for Functional Genomics in Plants and Animals
 , ,
, ,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Overview of the Main CRISPR/Cas Systems That Are Exploitable for Both Editing and Non-Editing Uses
2.1. CRISPRCas9 and dCas9
2.2. CRISPR–Cas12a
2.3. CRISPR/Cas13 and dCas13
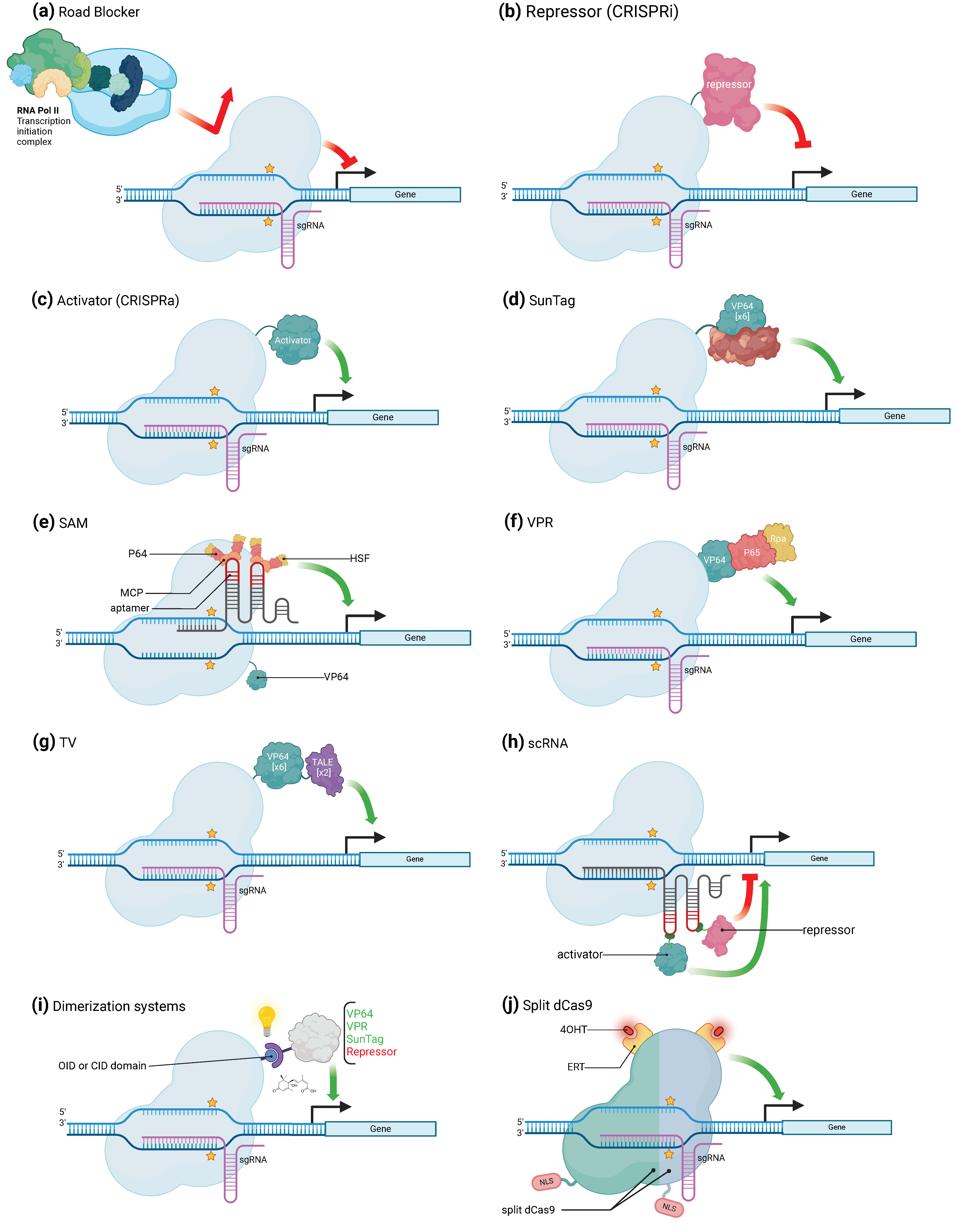
3. CRISPR/Cas-Mediated Transcriptional Regulation
3.1. CRISPR/Cas Mediated Gene Activation
3.2. CRISPR/Cas-Mediated Gene Repression
3.3. Alternative Uses of CRISPR/Cas for Transcriptional Regulation
4. CRISPR/Cas-Mediated in-Depth Study of Gene Regulation
4.1. Study of Gene Regulation at the Transcriptional Level

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Organism | CRISPR/Cas System | Type of Regulation | Performance | References |
|---|---|---|---|---|---|---|
| VP64 | Single activator (VP16 or p65) | Mammalian cells and budding yeast | dCas9 | CRISPRa | Between 2- and 5-fold | [3,26] |
| SunTag | Tandem array of peptides, which recruits several copies of VP64 | HEK293 and U2OS cells. Arabidopsis thaliana | dCas9 | CRISPRa | Up to 50-fold | [28,30] |
| VPR | Tripartite peptide composed by the VP64, p65, and Rta activators placed in a specific order to maximize gene activation | HEK293T and Neuro-2A cells. Nicotiana benthamiana | dCas9 | CRISPRa | Up to 300-fold | [32,33] |
| SAM | VP64 and sgRNA with two MS2 on turn fused to p65 and HSF1 | HEK293FT and Neuro-2a cells | dCas9 | CRISPRa | Variable | [34] |
| TV | Six copies of TAD motif and two copies of the VP64 activator | HEK293T cells. Arabidopsis thaliana and Oryza sativa | dCas9 | CRISPRa | Variable | [35] |
| Road blocker | Steric hamper due to simple bound of dCas9 | E. coli and mammalian | dCas9 | CRISPRi | Depends on organism | [17] |
| Transcriptional repressors | KRAB, CS, WPRW, SID4X, 3xSRDX, and SRDX domains | Mammalian cells. Arabidopsis thaliana and Nicotiana benthamiana | dCas9 | CRISPRi | Between 40 and 99% | [36,37,38,39,40] |
| scRNA | Differential regulation (both activation and repression) of a set of gene targets simultaneously | Human cells | dCas9 | Both | NA | [29] |
| Dimerization systems | Spatial and temporal control of gene function through sense input signals and generate functional outputs | HEK293T cells, mice and Avena sativa | dCas9 | Both | NA | [2,44,46] |
| Split dCas9 | Fusing ligand-binding domains of nuclear receptors to split Cas9 protein fragments can provide chemical control over split Cas9 activity | HEK293T cells. | dCas9- | Both | NA | [50] |
4.2. Study of Gene Regulation at the Post-Transcriptional Level
5. CRISPR/Cas System to Image Specific Portions of Nucleic Acids in Plants and Animals
| Name | Description | Organism | Type(s) of Cas Protein | Advantages | Disadvantages | Performances | References |
|---|---|---|---|---|---|---|---|
| dCas9∷eGFP fusion protein | Imaging of DNA loci with a GFP-dCas9, expressed in situ along with the gRNA from transfected vectors. | Human | dCas9 | The use of an sgRNA guide with a custom scaffold reduces non-specific binding of Cas9. Possibility to label heterochromatin regions. | Labeling of repetitive sequences as well as single loci. Tracking of telomere dynamics in live cells Labeling of different positions of the same gene. Gene copy-number identification | [73] | |
| Cas9-mediated fluorescence in situ hybridization (CASFISH) | dCas9 harbors a HaloTag flag and can be bound by fluorophores that are linked to HaloTag ligand. | Human | dCas9 | Highly stable sgRNA-dCas9-fluorophore complex High specificity Several loci can be stained at the same time: multiplexed imaging. Very quick protocol (15 min) Performed at room temperature | Imaging of repetitive sequences in detection of the allele of a certain sequence in the genome of cells at a tissue scale. Dual color Genetic diagnosis | [74] | |
| LiveFISH | One Cas9 harbors a labeled sgRNA with a short protospacer to disable cutting and one other Cas9 harbors a normal unlabeled sgRNA | Human | Cas9, dCas9, Cas13, dCas13 | One type of Cas9 is used because the RNP complex is preassembled. Live imaging is possible. Combinable with other CRISPR/Cas system-based techniques. |
| [72] | |
| Labeling sgRNA scaffolds in animals | sgRNA carries a long 3′ scaffold that harbors aptamers, to which fluorescently labeled proteins bind. | Mouse | dCas9 | Fewer background compared to labeling with GFP-fused dCas9 because non-specific binding of dCas9 is not visible. A single vector encodes every component of the system. Live imaging is possible. Multiplex labeling | Non-specific binding is not visible, i.e., off target cannot be characterized. | Labeling of nuclear structures, repetitive sequences, and single loci. Study of chromatin dynamics during cell division. Labeling of two loci in different colors. | [75] |
| Labeling sgRNA scaffolds in plants | sgRNA carries a long 3′ scaffold that harbors aptamers, to which fluorescently labeled proteins bind. | Tobacco | dCas9 | Fewer background compared to labeling with GFP-fused dCas9 because non-specific binding of dCas9 is not visible. A single construct encodes every component of the system. This construct is inserted with A. tumefaciens-mediated transformation. Up to 2 simultaneous labeling.
| Non-specific binding is not visible, i.e., off target cannot be characterized. Lack of telomeric foci compared to FISH because of the working temperature in plants. Cannot be improved by modification of the RNA scaffold. Only repetitive sequences have yet been targeted.
| Live imaging of telomeric repeats in plant cells. | [76] |
| RGEN-ISL/CRISPR-FISH | Imaging of loci in purified fixed nuclei using a preassembled ribonucleoprotein that contains the dCas9 and its sgRNA, for which the tracrRNA part is fused to a fluorophore for labeling. | Soybean, mouse, wheat, rye, maize, and tobacco | dCas9 | No plasmid construct. No in vitro RNA synthesis. Theoretically available in any species Non disruptive technique Simple and fast Usable for repetitive sequences and single loci. | Fixation of nuclei is required. ATT550-labeled tracrRNA and the crRNA that can bind to it must be ordered and are costly. | Labeling of centromeric and telomeric repeats in diverse species. Optimization of sample fixation to increase labeling yield. Time-lapse-mediated study of the binding dynamics of dCas9-sgRNA complex to DNA. | [78,79,82] |
| mRNA imaging | dLwaCas13 is fused to GFP expressed along with the specific sgRNA using a transient vector. | Rice, mammals | dCas13 | Specific targeting of mRNA Applicable to live or fixed samples. | Imaging of a specific gene’s mRNA to track its localization at stress-granules. | [13] |
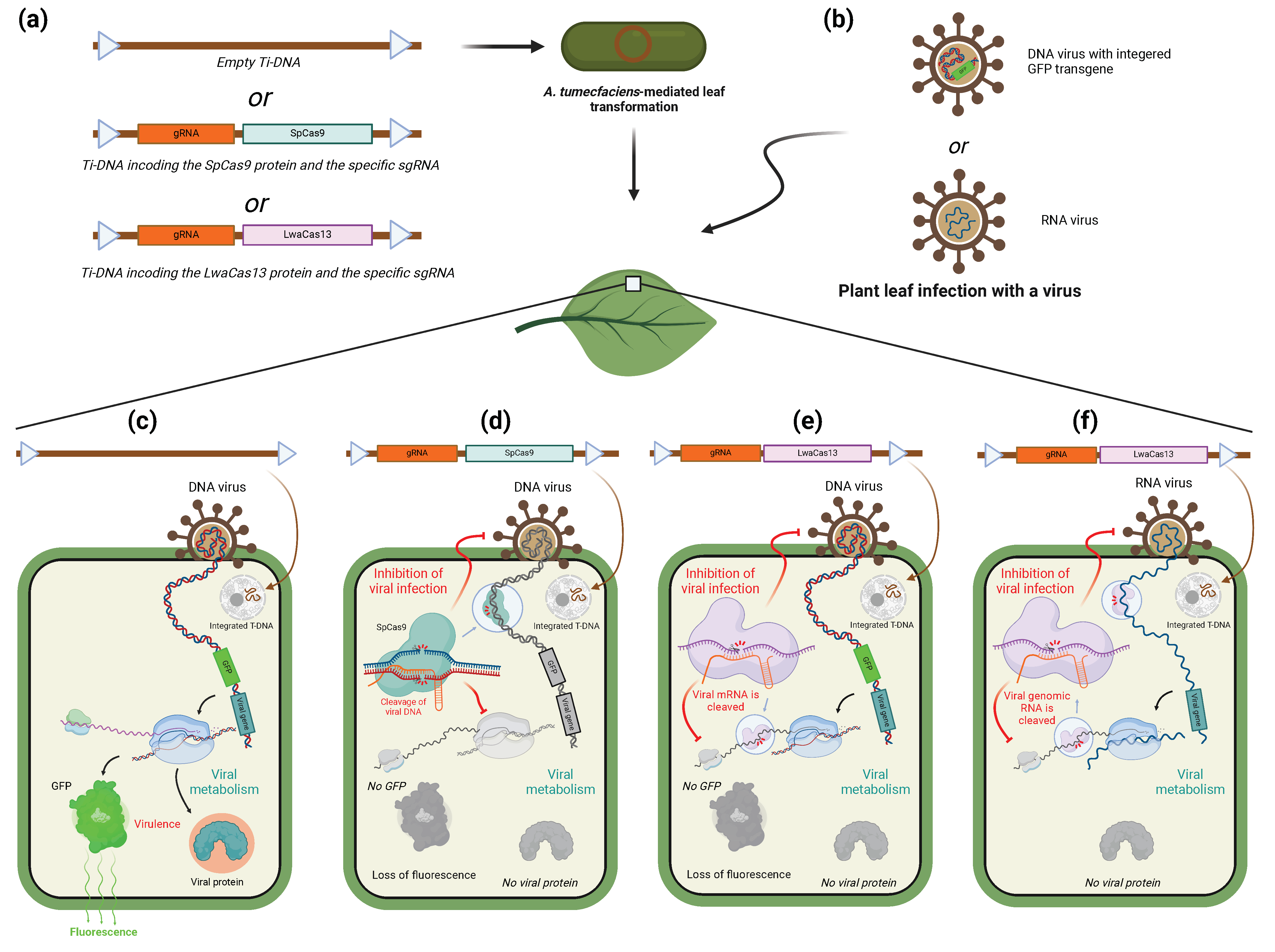
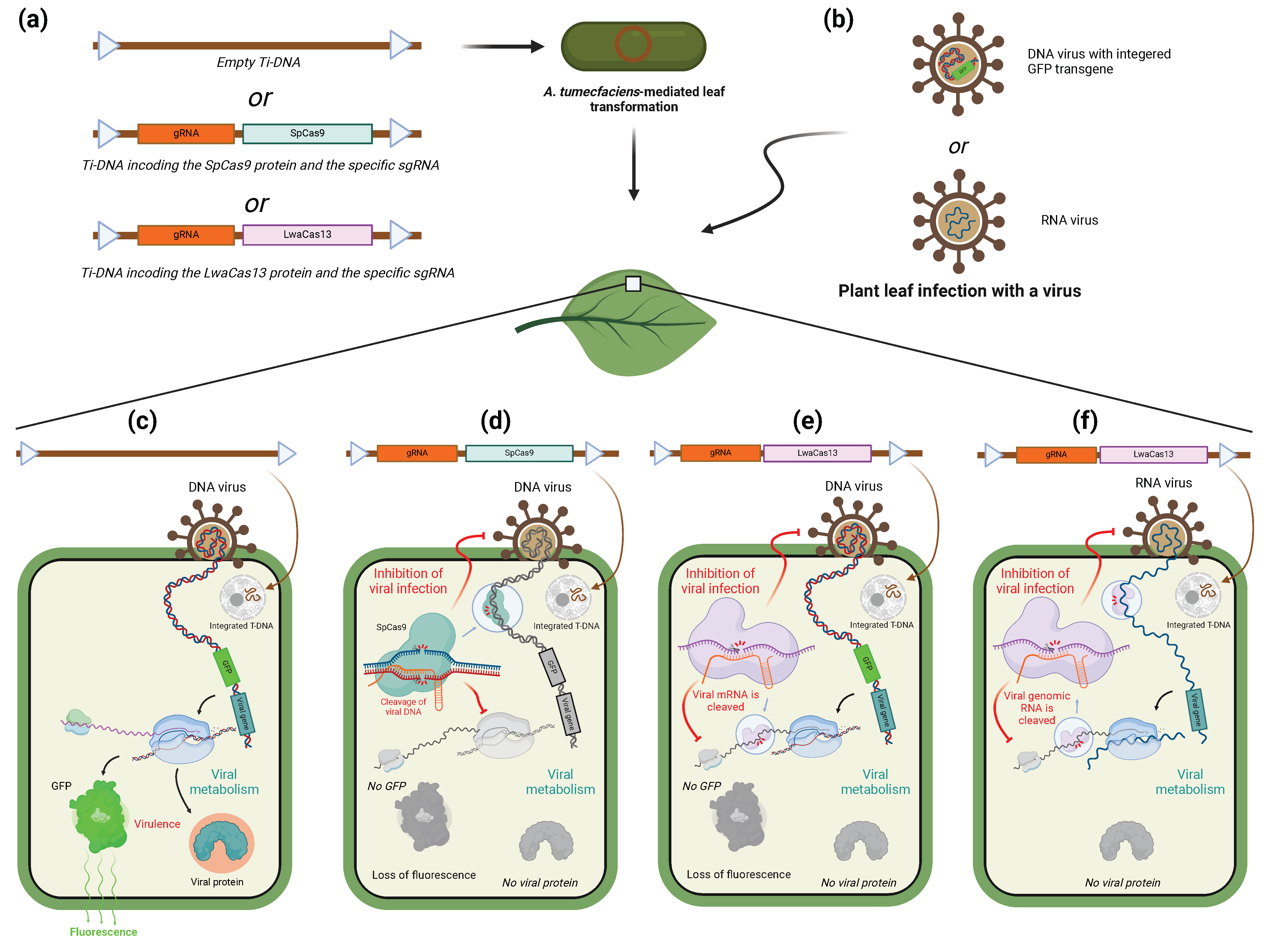
6. CRISPR/Cas-System as a Tool to Target Viruses
6.1. CRISPR/Cas-System for Inhibition of Viral Infection in Plants
6.2. Use of CRISPR/Cas System to Target Viruses in Animals
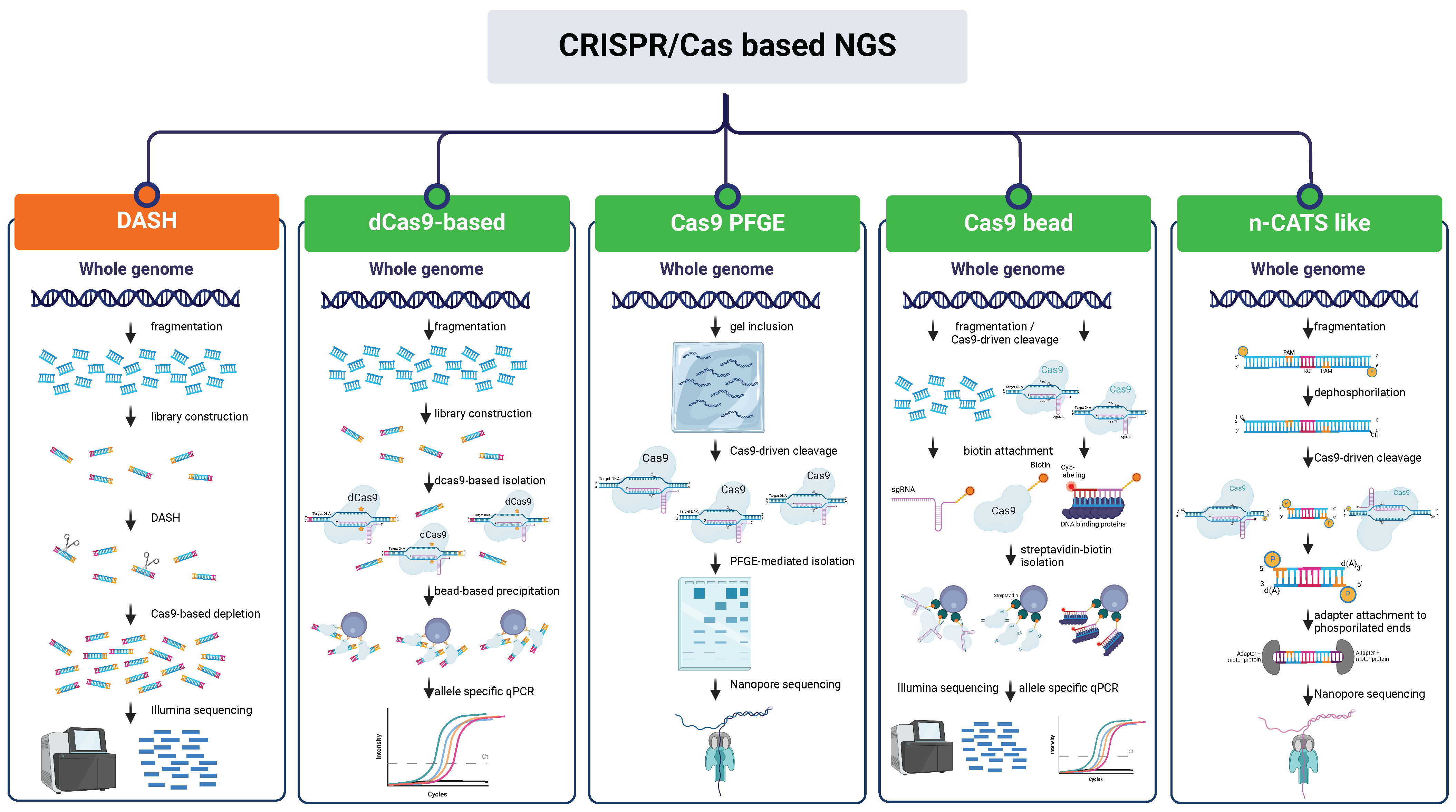
7. CRISPR/Cas as an Enrichment Tool for Next-Generation Sequencing
7.1. CRISPR/Cas NGS Approaches Based on Depletion of Undesired Region
7.2. CRISPR/Cas NGS Approaches Based on Enrichment of Regions of Interest (ROI)
| Name | Organism | CRISPR/Cas System | Description | Pros | Cons | Desired Sequences Fold Enrichment/ Undesired Sequences Fold Depletion | Target Sequence Length | References |
|---|---|---|---|---|---|---|---|---|
| DASH (Depletion of Abundant Sequences by Hybridization) | Human | Cas9 | DNA is fragmented, adapter ligated, sgRNA-driven cleavage, and sequencing of adapters-ligated fragments. | Highly efficient for background noise reduction | Background sequences must be repetitive; high amount of sgRNAs to be provided; amplification step needed | 3–10-fold enrichment/30–105-fold depletion | 2.5 kb | [101] |
| Cas-16S-seq | Rice | Cas9 | 16S rRNA amplification and adapter ligation, host’s 16S sgRNA-driven cleavage, second amplification of remaining microbial 16S, and sequencing. | 8.6–22-fold depletion | [103] | |||
| Lens culinaris | Cas9 | Custom gRNAs-driven cleavage of DNA repetitive regions, PCR amplification, purification, and sequencing. | 2.6-fold enrichment | 2.9 Gb of repeats | [104] | |||
| CUT-PCR (CRISPR-mediated, Ultrasensitive detection of Target DNA by PCR) | Human | Cas9 | sgRNA-driven cleavage of WT DNA with intact PAM site, PCR enrichment of mutants, cleavage step repeated, and sequencing. | Detected mutations must be in the PAM site; several amplification steps needed | 30–600-fold enrichment | 1 kb | [102] | |
| CRISDA (CRISPR–Cas9-triggered nicking endonuclease mediated Strand Displacement Amplification) | Human | Cas9 | Two Cas9 RNPs recognize each border of the target DNA nicking non-target strands, primers hybridize to the exposed non-target strands, SDA linearly replaces single -for and -rev strands, exponential SDA of target sequence, and amplicon quantification with biotin and Cy5-labeled PNA probes via magnetic pull-down and fluorescence measurements. | Highly specific and sensitive | Complex procedure; amplification step needed | 200 bp | [107] | |
| CRISPR-Cap | Escherichia coli | Cas9 | For cleavage of target regions, a biotinylated sgRNA library and genomic DNA are mixed and incubated, cleaved CRISPR-DNA complexes are bound to streptavidin magnetic beads, and target DNA is released and sequenced. | Easy procedure; no mandatory amplification step | Reduced sequence length; high amount of sgRNAs needed | 183.6-fold enrichment | 13 kb | [108] |
| RGEN-R and RGEN-TdT | Mammalians | Cas9 | Random fragmentation of genomic DNA (20–30 kb) and ddNTP treatment, Cas9-driven cleavage, and reparation with biotinylated adapters (RGEN-R) or with biotin-dUTP tail (RGEN-TdT), straptavidin-mediated isolation, PCR-free enrichment, and sequencing. | No amplification step needed; long sequences; low amount of sgRNAs needed | Highly dependent on the streptavidin purification step | 5–60-fold enrichment | 15 kb | [109] |
| Human | Cas9 | DNA digestion with EcoRI-HF and BamHI-HF and a sgRNA is designed adjacent to the region of interest. Digestion with Cas9 enables ligation with a capture adapter and SMRTbell molecules that contain the capture adapter are enriched on magnetic beads and prepared for SMRT PacBio sequencing. | No amplification step needed; long sequences | High amount of input DNA | up to 64,000-fold enrichment | 1 kb | [110] | |
| CISMR (CRISPR mediated isolation of specific megabase-sized regions of the genome | Yeast, Mouse | Cas9 | CRISPR-mediated site-specific cleavage, isolation of megabase-sized DNA segment via PFGE, random fragmentation of DNA to 20 kb for Oxford Nanopore/PacBio long-read sequencing, or to 40 kb for fosmid clone library. | Long sequences | Complex procedure; not multiplexable; risk of yield loss during gel extraction | 39–174-fold enrichment | up to 2.3 Mb | [111] |
| CATCH (Cas9-assisted targeting of chromosome segments) | Human | Cas9 | Cells are embedded in an agarose gel-plug and lysed. Genomic DNA is Cas9-cleaved in the plug and the target DNA is separated by PFGE. The desired band is then excised from the gel, DNA is isolated, purified, and sequenced. | Long sequences | Complex procedure; not multiplexable; risk of yield loss during gel extraction; amplification step needed | 237-fold enrichment | 200 kb | [113] |
| nCATS (nanopore Cas9 Targeted-Sequencing) | Human | Cas9 | DNA is fragmented, ends are dephosphorylated, and Cas9 -driven cleavage is performed. Nanopore sequencing adapters are ligated to new ends and fragments are sequenced. | Low amount of sgRNAs needed; no amplification step needed | Shorter sequences compared to other nCATS-like methods | 10–300-fold enrichment | 12.3–24.3 kb | [114] |
| Mouse | Cas9 | nCATS-like procedure with custom sequencing data analysis pipeline | Long sequences; low amount of sgRNAs needed; no amplification step needed | High amount of input DNA | 400–700-fold enrichment | 200 kb | [115] | |
| FUDGE (FUsion Detection from Gene Enrichment) | Human | Cas9 | nCATS-like procedure with custom sequencing data analysis pipeline | Low amount of sgRNAs needed; no amplification step needed | Shorter sequences compared to other nCATS-like methods | 300–700-fold enrichment | up to 9.9 kb | [116] |
| ACME (Affinity-based Cas9 Mediated Enrichment) | Human | Cas9 | nCATS-like procedure with a background reduction step | Long sequences; on-target to off-target ratio increase; no amplification step needed | Inability to sequence deeply targets >100 kb in size without coverage dropouts in the center | 3–60-fold enrichment | 200 kb | [117] |
| Malus domestica | Cas9 | nCATS-like procedure with custom sequencing data analysis pipeline | No amplification step needed; possible multiplexing | High quality input DNA needed | 180× and 196× read depths | 9.78 and 9.89 kb | [127] | |
| Prunus salicina | Cas9 | nCATS-like procedure with custom sequencing data analysis pipeline | Long sequences; easy procedure; no previous knowledge of the DNA sequence is needed; no amplification step needed | Manual curation needed in case of de novo sequencing | 11.9× read depth | 194 Mb | [128] | |
| Triticale | Cas9 | nCATS-like procedure with custom sequencing data analysis pipeline | Low sequencing depth for small loci in big genomes; no amplification step needed | No cost-effective if studying one or a few loci | 3.4 kb, 5.1 kb and 3.6 kb | [118] | ||
| Cas9-tiling | Human; Rhesus macaques | Cas9 | nCATS-like procedure on shorter sequences | Sequencing of small overlapping regions to avoid inaccuracies in polymorphism detections; no amplification step needed; further replacement of poor-quality assemblies in the reference genome | sgRNA design preferably on coding regions; high amount of sgRNAs needed depending on the entire sequence length | 215–394-fold enrichment (human); 128–637-fold enrichment (macaques) | 176 kb (human); 280 kb (macaques) | [124] |
| Human | Cas9 | 19× to 238× target depth | 28–100 kb | [125] | ||||
| Phaseolus vulgaris | Cas9 | 113.16-fold enrichment | 250 kb | [126] | ||||
| FLASH (Finding Low Abundance Sequences by Hybridization) | Pneumonia-causing gram+ bacteria; Plasmodium falciparum | Cas9 | nCATS-like procedure with custom sequencing data analysis pipeline | Low amount of sgRNAs needed; low amount of input DNA | Amplification step needed; shorter sequences compared to other methods | >5000-fold enrichment | [120] | |
| CANS (CRISPR-Associated Nanopore Sequencing)-NanoCasTE | Arabidopsis | Cas9 | nCATS-like procedure with custom sequencing data analysis pipeline | Rapid procedure; no amplification step needed; high mappability of the reads | Shorter sequences compared to other methods | 0.2× to 40× target depth | 14–35 kb | [122] |
| s | Human | Cas9 | nCATS-like procedure with gRNA specifically targeting 3’ end of the MEI sequences. | Low amount of sgRNAs needed; no amplification step needed; high sensitivity | Shorter sequences compared to other methods; possible recalcitrance of some chromosome locations | 13.4× to 54× enrichment | 14.9–32.3 kb | [121] |
| Negative enrichment | Human | Cas9 | Cas9-driven cleavage of sequences flanking target region, treatment with exonucleases while Cas9 remains bound, protecting ends of target, purification from exonucleases and digested DNA, library preparation, and sequencing. | Easy procedure; low amount of sgRNAs needed; long sequences | Long persistence of Cas9 on the target | 127 to 197-fold enrichment | 36 kb | [97] |
| CAMP (CRISPR Associated Multiplexed PCR) | Human | Cas9 | Cas9-driven cleavage at either side of the target locus. Universal UPS adapters are ligated for amplification. CAMP uses primers that have complementarity to the UPS adapter only, cCAMP uses chimeric primers that have complementarity to the UPS adapter and several bases of target DNA and cTRACE uses chimeric primers that have complementarity to the UPS adapter, several bases of target DNA, and specificity for a mutation. | High specificity; short DNA targeting; significant enrichment; multiplexable | Amplification step needed | 2.6 × 104 to 3.1 × 106-fold enrichment | ~100 kb | [119] |
| cCAMP (chimeric CRISPR Associated Multiplexed PCR) | Cas9 | High specificity; significant enrichment; multiplexable | Amplification step needed | 4.7 × 106 to 2.1 × 108-fold enrichment | ~100 kb | |||
| cTRACE (chimeric Targeting Rare Alleles with CRISPR-based Enrichment) | Cas9 | Significant enrichment; flexibility in the location of the mutation in reference to a PAM site | Amplification step needed | |||||
| TRACE (Targeting Rare Alleles with CRISPR-based Enrichment) | Cas9 | TRACE uses Cas9/sgRNA to protect targeted DNA from exonuclease, which digests off-target sequences; the protection provided by the Cas9 complex confers single base discrimination to protect a single base mutation while digesting the normal variant. | High specificity; No amplification step needed | Mutations must be in proximity to a PAM site; short sequences | up to 820 bp | |||
| CRISPR-DS (CRISPR-Duplex Sequencing) | Human | Cas9 | Cas9-driven cleavage of target sequence, targeted cutting of fragments containing coding exons using sgRNAs. Fragment size selection with beads. Double-stranded DNA fragmented and ligated with double-stranded DS adapters. Creation of single-strand consensus sequence (SSCS) reads and comparison to create a double-strand consensus sequence (DCS). Only mutations found in both SSCS reads are counted as true mutations in DCS reads. | Low amount of input DNA | Complex procedure; high quality input DNA needed; previous knowledge of the DNA target sequences needed | 49,000-fold enrichment | [123] | |
| Human | dCas9 | Target-bound dCas9 isolated through immunomagnetic precipitation, purification, and allele-specific qPCR. | Highly sensitive | Mutations must be in proximity to a PAM site | 21-fold enrichment | [105] | ||
| CATE-seq (CRISPR-assisted targeted enrichment-sequencing) | Human | dCas9 | DNA fragmentation, adapter ligation, target dCas9 binding, purification, and allele-specific PCR. | Highly specific and sensitive; low amount of sgRNAs needed | Complex procedure; short sequences | 3700-fold enrichment | 235 bp | [106] |
8. Conclusions and Future Perspectives
| Category of Tools | Animals | Plants | Notes |
|---|---|---|---|
| Gene expression modulation | Most reported examples were developed on animals including humans. In plants, most of them are still lacking, presumably due to the time that it takes to import those approaches in vegetal systems. The inherent differences in the genome and cell structure between the two kingdoms could be an obstacle to the delivery and the functioning of the Cas system. | ||
| Study of transcription regulation | |||
| Imaging/structural genomics | |||
| Immunization against viruses and other pathogen | In the case of the techniques that aim at conferring immunity against viruses, it seems that the plant system is more in advance due to the high economic impact that such bioengineering could have on agriculture. Plus, in this case, the target of the Cas-mediated cleavage is no longer the endogenous genome of the plant cell but one of the viruses, whose structure is simpler. Application in animals must face stronger ethical considerations here, which may explain why there is more to find in vegetal systems when it comes to this field. On the contrary, the advances in medical care that would bring a better detection of pathogens account for the plethora of techniques that were developed in animals to assess the presence of pathogens; however, they are still mainly viral. | ||
| Detection of viruses and other pathogen | |||
| NGS | The applicability of NGS-mediated techniques can be considered as being on the same level in both biological systems. Indeed, they are based on nucleic acids from cell extracts, which makes them independent from the biological properties of the system. |
Author Contributions
Funding
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next Generation of CRISPR-Cas Technologies and Applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary Classification of CRISPR-Cas Systems: A Burst of Class 2 and Derived Variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Evolutionary Classification of CRISPR-Cas Systems. In Crispr; John Wiley & Sons, Ltd.: Bognor Regis, UK, 2022; pp. 13–38. ISBN 978-1-68367-379-8. [Google Scholar]
- Hryhorowicz, M.; Lipiński, D.; Zeyland, J.; Słomski, R. CRISPR/Cas9 Immune System as a Tool for Genome Engineering. Arch. Immunol. Ther. Exp. 2017, 65, 233–240. [Google Scholar] [CrossRef]
- Kozovska, Z.; Rajcaniova, S.; Munteanu, P.; Dzacovska, S.; Demkova, L. CRISPR: History and Perspectives to the Future. Biomed. Pharmacother. 2021, 141, 111917. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ma, J.; Wu, Z.; Wang, Z.; Zhang, H.; Fu, W.; Pan, D.; Shi, J.; Ji, Q. Cas12n Nucleases, Early Evolutionary Intermediates of Type V CRISPR, Comprise a Distinct Family of Miniature Genome Editors. Mol. Cell 2023, 83, 2768–2780.e6. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Young, J.K.; Karvelis, T.; Kazlauskas, D.; Urbaitis, T.; Jasnauskaite, M.; Grusyte, M.M.; Paulraj, S.; Wang, P.-H.; Hou, Z.; et al. A Catalogue of Biochemically Diverse CRISPR-Cas9 Orthologs. Nat. Commun. 2020, 11, 5512. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, R.; Ishiguro, S.; Okazaki, S.; Mori, H.; Tanaka, M.; Aburatani, H.; Yachie, N.; Nishimasu, H.; Nureki, O. Engineered Campylobacter Jejuni Cas9 Variant with Enhanced Activity and Broader Targeting Range. Commun. Biol. 2022, 5, 211. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA Recognition and Cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed Activation of Endogenous Genes by CRISPR-on, an RNA-Guided Transcriptional Activator System. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA Editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA Targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic Acid Detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a Target Binding Unleashes Indiscriminate Single-Stranded DNase Activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- Cao, H.; Wang, Y.; Zhang, N.; Xia, S.; Tian, P.; Lu, L.; Du, J.; Du, Y. Progress of CRISPR-Cas13 Mediated Live-Cell RNA Imaging and Detection of RNA-Protein Interactions. Front. Cell Dev. Biol. 2022, 10, 866820. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Pan, Z.; Wang, X.; Bian, X.; Wang, W.; Liang, Q.; Kou, M.; Ji, H.; Li, Y.; Ma, D.; et al. Targeting of SPCSV-RNase3 via CRISPR-Cas13 Confers Resistance against Sweet Potato Virus Disease. Mol. Plant Pathol. 2022, 23, 104–117. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Grotewold, E.; Stam, M. Cis-Regulatory Sequences in Plants: Their Importance, Discovery, and Future Challenges. Plant Cell 2022, 34, 718–741. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and Validation of Promoters and Cis-Acting Regulatory Elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable Repression and Activation of Bacterial Gene Expression Using an Engineered CRISPR-Cas System. Nucleic Acids Res. 2013, 41, 7429–7437. [Google Scholar] [CrossRef]
- La Russa, M.F.; Qi, L.S. The New State of the Art: Cas9 for Gene Activation and Repression. Mol. Cell. Biol. 2015, 35, 3800–3809. [Google Scholar] [CrossRef]
- Morita, S.; Horii, T.; Hatada, I. Regulation of Gene Expression Using dCas9-SunTag Platforms. In Epigenomics: Methods and Protocols; Hatada, I., Horii, T., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2023; pp. 189–195. ISBN 978-1-07-162724-2. [Google Scholar]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- Morita, S.; Horii, T.; Kimura, M.; Hatada, I. Synergistic Upregulation of Target Genes by TET1 and VP64 in the dCas9–SunTag Platform. Int. J. Mol. Sci. 2020, 21, 1574. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Selma, S.; Bernabé-Orts, J.M.; Vazquez-Vilar, M.; Diego-Martin, B.; Ajenjo, M.; Garcia-Carpintero, V.; Granell, A.; Orzaez, D. Strong Gene Activation in Plants with Genome-Wide Specificity Using a New Orthogonal CRISPR/Cas9-Based Programmable Transcriptional Activator. Plant Biotechnol. J. 2019, 17, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, D.; Xiong, X.; Yan, B.; Xie, W.; Sheen, J.; Li, J.-F. A Potent Cas9-Derived Gene Activator for Plant and Mammalian Cells. Nat. Plants 2017, 3, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef]
- Lowder, L.G.; Zhang, D.; Baltes, N.J.; Paul, J.W., III; Tang, X.; Zheng, X.; Voytas, D.F.; Hsieh, T.-F.; Zhang, Y.; Qi, Y. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiol. 2015, 169, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-Guided Transcriptional Regulation in Planta via Synthetic dCas9-Based Transcription Factors. Plant Biotechnol. J. 2015, 13, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Sretenovic, S.; Qi, Y. CRISPR/dCas-Mediated Transcriptional and Epigenetic Regulation in Plants. Curr. Opin. Plant Biol. 2021, 60, 101980. [Google Scholar] [CrossRef]
- Cai, R.; Lv, R.; Shi, X.; Yang, G.; Jin, J. CRISPR/dCas9 Tools: Epigenetic Mechanism and Application in Gene Transcriptional Regulation. Int. J. Mol. Sci. 2023, 24, 14865. [Google Scholar] [CrossRef]
- Zetsche, B.; Volz, S.E.; Zhang, F. A Split-Cas9 Architecture for Inducible Genome Editing and Transcription Modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef]
- Xu, X.; Qi, L.S. A CRISPR–dCas Toolbox for Genetic Engineering and Synthetic Biology. J. Mol. Biol. 2019, 431, 34–47. [Google Scholar] [CrossRef]
- Bao, Z.; Jain, S.; Jaroenpuntaruk, V.; Zhao, H. Orthogonal Genetic Regulation in Human Cells Using Chemically Induced CRISPR/Cas9 Activators. ACS Synth. Biol. 2017, 6, 686–693. [Google Scholar] [CrossRef]
- Chen, F.; Hu, Y.; Vannozzi, A.; Wu, K.; Cai, H.; Qin, Y.; Mullis, A.; Lin, Z.; Zhang, L. The WRKY Transcription Factor Family in Model Plants and Crops. Crit. Rev. Plant Sci. 2017, 36, 311–335. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, X.; Wong, S.; Charles, E.J.; Lim, W.A.; Qi, L.S. Complex Transcriptional Modulation with Orthogonal and Inducible dCas9 Regulators. Nat. Methods 2016, 13, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal Control of Cell Signalling Using a Light-Switchable Protein Interaction. Nature 2009, 461, 997–1001. [Google Scholar] [CrossRef]
- Nihongaki, Y.; Yamamoto, S.; Kawano, F.; Suzuki, H.; Sato, M. CRISPR-Cas9-Based Photoactivatable Transcription System. Chem. Biol. 2015, 22, 169–174. [Google Scholar] [CrossRef]
- Polstein, L.R.; Gersbach, C.A. A Light-Inducible CRISPR-Cas9 System for Control of Endogenous Gene Activation. Nat. Chem. Biol. 2015, 11, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Miyaoka, Y.; Gilbert, L.A.; Mayerl, S.J.; Lee, B.H.; Weissman, J.S.; Conklin, B.R.; Wells, J.A. Ligand-Binding Domains of Nuclear Receptors Facilitate Tight Control of Split CRISPR Activity. Nat. Commun. 2016, 7, 12009. [Google Scholar] [CrossRef] [PubMed]
- Tarasava, K.; Oh, E.J.; Eckert, C.A.; Gill, R.T. CRISPR-Enabled Tools for Engineering Microbial Genomes and Phenotypes. Biotechnol. J. 2018, 13, e1700586. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Xie, B.; Chen, L.; Zhang, N.; Jiang, Y.; Qin, H.; Wang, H.; Hao, P.; Yang, S.; Li, X. Implementation of the CRISPR-Cas13a System in Fission Yeast and Its Repurposing for Precise RNA Editing. Nucleic Acids Res. 2018, 46, e90. [Google Scholar] [CrossRef]
- Kushawah, G.; Hernandez-Huertas, L.; Abugattas-Nuñez del Prado, J.; Martinez-Morales, J.R.; DeVore, M.L.; Hassan, H.; Moreno-Sanchez, I.; Tomas-Gallardo, L.; Diaz-Moscoso, A.; Monges, D.E.; et al. CRISPR-Cas13d Induces Efficient mRNA Knockdown in Animal Embryos. Dev. Cell 2020, 54, 805–817.e7. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Qi, Y. Plant Gene Knockout and Knockdown by CRISPR-Cpf1 (Cas12a) Systems. In Plant Genome Editing with CRISPR Systems: Methods and Protocols; Qi, Y., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 245–256. ISBN 978-1-4939-8991-1. [Google Scholar]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Chavez, M.; Chen, X.; Finn, P.B.; Qi, L.S. Advances in CRISPR Therapeutics. Nat. Rev. Nephrol. 2023, 19, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Gao, Y.; Dominguez, A.A.; Qi, L.S. CRISPR Technologies for Precise Epigenome Editing. Nat. Cell Biol. 2021, 23, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef] [PubMed]
- Stepper, P.; Kungulovski, G.; Jurkowska, R.Z.; Chandra, T.; Krueger, F.; Reinhardt, R.; Reik, W.; Jeltsch, A.; Jurkowski, T.P. Efficient Targeted DNA Methylation with Chimeric dCas9-Dnmt3a-Dnmt3L Methyltransferase. Nucleic Acids Res. 2017, 45, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.-L.; Hu, R. A CRISPR-Based Approach for Targeted DNA Demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA Demethylation in Vivo Using dCas9-Peptide Repeat and scFv-TET1 Catalytic Domain Fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome Editing by a CRISPR-Cas9-Based Acetyltransferase Activates Genes from Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Y.; Li, M.; Zhou, F.; Li, K.; Cao, H.; Ni, M.; Liu, Y.; Gu, Z.; et al. In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell 2017, 170, 1028–1043.e19. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Y.; Li, M.; Shao, Z.; Zhang, M.Q.; Xu, J. CAPTURE: In Situ Analysis of Chromatin Composition of Endogenous Genomic Loci by Biotinylated dCas9. Curr. Protoc. Mol. Biol. 2018, 123, e64. [Google Scholar] [CrossRef]
- Wang, Z.; He, Z.; Liu, Z.; Qu, M.; Gao, C.; Wang, C.; Wang, Y. A Reverse Chromatin Immunoprecipitation Technique Based on the CRISPR–dCas9 System. Plant Physiol. 2023, 191, 1505–1519. [Google Scholar] [CrossRef]
- Orphanides, G.; Reinberg, D. A Unified Theory of Gene Expression. Cell 2002, 108, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Chen, S.; Jia, G. Detection, Regulation, and Functions of RNA N6-Methyladenosine Modification in Plants. Plant Commun. 2023, 4, 100546. [Google Scholar] [CrossRef]
- Maslova, A.; Krasikova, A. FISH Going Meso-Scale: A Microscopic Search for Chromatin Domains. Front. Cell Dev. Biol. 2021, 9, 753097. [Google Scholar] [CrossRef]
- Khosravi, S.; Ishii, T.; Dreissig, S.; Houben, A. Application and Prospects of CRISPR/Cas9-Based Methods to Trace Defined Genomic Sequences in Living and Fixed Plant Cells. Chromosome Res. 2020, 28, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, S.; Dreissig, S.; Schindele, P.; Wolter, F.; Rutten, T.; Puchta, H.; Houben, A. Live-Cell CRISPR Imaging in Plant Cells with a Telomere-Specific Guide RNA. In RNA Tagging: Methods and Protocols; Heinlein, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; pp. 343–356. ISBN 978-1-07-160712-1. [Google Scholar]
- Wu, X.; Mao, S.; Ying, Y.; Krueger, C.J.; Chen, A.K. Progress and Challenges for Live-Cell Imaging of Genomic Loci Using CRISPR-Based Platforms. Genom. Proteom. Bioinform. 2019, 17, 119–128. [Google Scholar] [CrossRef]
- Wang, H.; Nakamura, M.; Abbott, T.R.; Zhao, D.; Luo, K.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-Mediated Live Imaging of Genome Editing and Transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef]
- Deng, W.; Shi, X.; Tjian, R.; Lionnet, T.; Singer, R.H. CASFISH: CRISPR/Cas9-Mediated in Situ Labeling of Genomic Loci in Fixed Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 11870–11875. [Google Scholar] [CrossRef]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA Scaffolds Enable Dual-Colour Live Imaging of Satellite Sequences and Repeat-Enriched Individual Loci. Nat. Commun. 2016, 7, 11707. [Google Scholar] [CrossRef]
- Khosravi, S.; Schindele, P.; Gladilin, E.; Dunemann, F.; Rutten, T.; Puchta, H.; Houben, A. Application of Aptamers Improves CRISPR-Based Live Imaging of Plant Telomeres. Front. Plant Sci. 2020, 11, 1254. [Google Scholar] [CrossRef]
- Dreissig, S.; Schiml, S.; Schindele, P.; Weiss, O.; Rutten, T.; Schubert, V.; Gladilin, E.; Mette, M.F.; Puchta, H.; Houben, A. Live-Cell CRISPR Imaging in Plants Reveals Dynamic Telomere Movements. Plant J. 2017, 91, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Schubert, V.; Khosravi, S.; Dreissig, S.; Metje-Sprink, J.; Sprink, T.; Fuchs, J.; Meister, A.; Houben, A. RNA-Guided Endonuclease—In Situ Labelling (RGEN-ISL): A Fast CRISPR/Cas9-Based Method to Label Genomic Sequences in Various Species. New Phytol. 2019, 222, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Potlapalli, B.P.; Schubert, V.; Metje-Sprink, J.; Liehr, T.; Houben, A. Application of Tris-HCl Allows the Specific Labeling of Regularly Prepared Chromosomes by CRISPR-FISH. Cytogenet. Genome Res. 2020, 160, 156–165. [Google Scholar] [CrossRef]
- Jacobi, A.M.; Rettig, G.R.; Turk, R.; Collingwood, M.A.; Zeiner, S.A.; Quadros, R.M.; Harms, D.W.; Bonthuis, P.J.; Gregg, C.; Ohtsuka, M.; et al. Simplified CRISPR Tools for Efficient Genome Editing and Streamlined Protocols for Their Delivery into Mammalian Cells and Mouse Zygotes. Methods 2017, 121–122, 16–28. [Google Scholar] [CrossRef]
- Nagaki, K.; Yamaji, N. Decrosslinking Enables Visualization of RNA-Guided Endonuclease–in Situ Labeling Signals for DNA Sequences in Plant Tissues. J. Exp. Bot. 2020, 71, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Potlapalli, B.P.; Ishii, T.; Nagaki, K.; Somasundaram, S.; Houben, A. CRISPR-FISH: A CRISPR/Cas9-Based In Situ Labeling Method. In Plant Cytogenetics and Cytogenomics: Methods and Protocols; Heitkam, T., Garcia, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2023; pp. 315–335. ISBN 978-1-07-163226-0. [Google Scholar]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and Classification of the CRISPR–Cas Systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef]
- Manjunatha, L.; Rajashekara, H.; Uppala, L.S.; Ambika, D.S.; Patil, B.; Shankarappa, K.S.; Nath, V.S.; Kavitha, T.R.; Mishra, A.K. Mechanisms of Microbial Plant Protection and Control of Plant Viruses. Plants 2022, 11, 3449. [Google Scholar] [CrossRef]
- Ali, Z.; Abulfaraj, A.; Idris, A.; Ali, S.; Tashkandi, M.; Mahfouz, M.M. CRISPR/Cas9-Mediated Viral Interference in Plants. Genome Biol. 2015, 16, 238. [Google Scholar] [CrossRef]
- Kalinina, N.O.; Khromov, A.; Love, A.J.; Taliansky, M.E. CRISPR Applications in Plant Virology: Virus Resistance and Beyond. Phytopathology 2020, 110, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Taliansky, M.; Samarskaya, V.; Zavriev, S.K.; Fesenko, I.; Kalinina, N.O.; Love, A.J. RNA-Based Technologies for Engineering Plant Virus Resistance. Plants 2021, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Spencer, K.P.; Burger, J.T.; Campa, M. CRISPR-Based Resistance to Grapevine Virus A. Front. Plant Sci. 2023, 14, 1296251. [Google Scholar] [CrossRef] [PubMed]
- Gobena, D.; Shimels, M.; Rich, P.J.; Ruyter-Spira, C.; Bouwmeester, H.; Kanuganti, S.; Mengiste, T.; Ejeta, G. Mutation in Sorghum LOW GERMINATION STIMULANT 1 Alters Strigolactones and Causes Striga Resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 4471–4476. [Google Scholar] [CrossRef]
- Shahriari, Z.; Su, X.; Zheng, K.; Zhang, Z. Advances and Prospects of Virus-Resistant Breeding in Tomatoes. Int. J. Mol. Sci. 2023, 24, 15448. [Google Scholar] [CrossRef] [PubMed]
- Karpov, D.S.; Demidova, N.A.; Kulagin, K.A.; Shuvalova, A.I.; Kovalev, M.A.; Simonov, R.A.; Karpov, V.L.; Snezhkina, A.V.; Kudryavtseva, A.V.; Klimova, R.R.; et al. Complete and Prolonged Inhibition of Herpes Simplex Virus Type 1 Infection In Vitro by CRISPR/Cas9 and CRISPR/CasX Systems. Int. J. Mol. Sci. 2022, 23, 14847. [Google Scholar] [CrossRef] [PubMed]
- Bayoumi, M.; Munir, M. Potential Use of CRISPR/Cas13 Machinery in Understanding Virus-Host Interaction. Front. Microbiol. 2021, 12, 743580. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, R.; Li, J. CRISPR-Cas-Based Techniques for Pathogen Detection: Retrospect, Recent Advances, and Future Perspectives. J. Adv. Res. 2023, 50, 69–82. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, T.; Liu, C.; Xu, Q.; Liu, Q. Pathogen Detection Strategy Based on CRISPR. Microchem. J. 2022, 174, 107036. [Google Scholar] [CrossRef]
- Zhou, J.; Yin, L.; Dong, Y.; Peng, L.; Liu, G.; Man, S.; Ma, L. CRISPR-Cas13a Based Bacterial Detection Platform: Sensing Pathogen Staphylococcus Aureus in Food Samples. Anal. Chim. Acta 2020, 1127, 225–233. [Google Scholar] [CrossRef]
- Schultzhaus, Z.; Wang, Z.; Stenger, D. CRISPR-Based Enrichment Strategies for Targeted Sequencing. Biotechnol. Adv. 2021, 46, 107672. [Google Scholar] [CrossRef]
- Stevens, R.C.; Steele, J.L.; Glover, W.R.; Sanchez-Garcia, J.F.; Simpson, S.D.; O’Rourke, D.; Ramsdell, J.S.; MacManes, M.D.; Thomas, W.K.; Shuber, A.P. A Novel CRISPR/Cas9 Associated Technology for Sequence-Specific Nucleic Acid Enrichment. PLoS ONE 2019, 14, e0215441. [Google Scholar] [CrossRef]
- Paul, W. Hook & Winston Timp Beyond Assembly: The Increasing Flexibility of Single-Molecule Sequencing Technology. Nat. Rev. Genet. 2023, 24, 627–641. [Google Scholar] [CrossRef]
- Malekshoar, M.; Azimi, S.A.; Kaki, A.; Mousazadeh, L.; Motaei, J.; Vatankhah, M. CRISPR-Cas9 Targeted Enrichment and Next-Generation Sequencing for Mutation Detection. J. Mol. Diagn. 2023, 25, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.; Shendure, J. Smash and DASH with Cas9. Genome Biol. 2016, 17, 42. [Google Scholar] [CrossRef]
- Gu, W.; Crawford, E.D.; O’Donovan, B.D.; Wilson, M.R.; Chow, E.D.; Retallack, H.; DeRisi, J.L. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to Remove Unwanted High-Abundance Species in Sequencing Libraries and Molecular Counting Applications. Genome Biol. 2016, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Yu, J.; Hwang, G.-H.; Kim, S.; Kim, H.S.; Ye, S.; Kim, K.; Park, J.; Park, D.Y.; Cho, Y.-K.; et al. CUT-PCR: CRISPR-Mediated, Ultrasensitive Detection of Target DNA Using PCR. Oncogene 2017, 36, 6823–6829. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Xie, K. Engineering CRISPR/Cas9 to Mitigate Abundant Host Contamination for 16S rRNA Gene-Based Amplicon Sequencing. Microbiome 2020, 8, 80. [Google Scholar] [CrossRef]
- Rossato, M.; Marcolungo, L.; De Antoni, L.; Lopatriello, G.; Bellucci, E.; Cortinovis, G.; Frascarelli, G.; Nanni, L.; Bitocchi, E.; Di Vittori, V.; et al. CRISPR/Cas9-Based Repeat Depletion for the High-Throughput Genotyping of Complex Plant Genomes. Genomics Res. 2023, 33, 787–797. [Google Scholar] [CrossRef]
- Aalipour, A.; Dudley, J.C.; Park, S.; Murty, S.; Chabon, J.J.; Boyle, E.A.; Diehn, M.; Gambhir, S.S. Deactivated CRISPR Associated Protein 9 for Minor-Allele Enrichment in Cell-Free DNA. Clin. Chem. 2018, 64, 307–316. [Google Scholar] [CrossRef]
- Xu, X.; Xia, Q.; Zhang, S.; Gao, J.; Dai, W.; Wu, J.; Wang, J. CRISPR-Assisted Targeted Enrichment-Sequencing (CATE-Seq). bioRxiv 2019. [Google Scholar] [CrossRef]
- Zhou, W.; Hu, L.; Ying, L.; Zhao, Z.; Chu, P.K.; Yu, X.-F. A CRISPR–Cas9-Triggered Strand Displacement Amplification Method for Ultrasensitive DNA Detection. Nat. Commun. 2018, 9, 5012. [Google Scholar] [CrossRef]
- Lee, J.; Lim, H.; Jang, H.; Hwang, B.; Lee, J.H.; Cho, J.; Lee, J.H.; Bang, D. CRISPR-Cap: Multiplexed Double-Stranded DNA Enrichment Based on the CRISPR System. Nucleic Acids Res. 2019, 47, e1. [Google Scholar] [CrossRef]
- Slesarev, A.; Viswanathan, L.; Tang, Y.; Borgschulte, T.; Achtien, K.; Razafsky, D.; Onions, D.; Chang, A.; Cote, C. CRISPR/Cas9 Targeted CAPTURE of Mammalian Genomic Regions for Characterization by NGS. Sci. Rep. 2019, 9, 3587. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Greenberg, D.; Powell, J.; Höijer, I.; Ameur, A.; Strahl, M.; Ellis, E.; Jonasson, I.; Mouro Pinto, R.; Wheeler, V.C.; et al. Amplification-free, CRISPR-Cas9 targeted enrichment and SMRT sequencing of repeat-expansion disease causative genomic regions. bioRxiv 2017. [Google Scholar] [CrossRef]
- Bennett-Baker, P.E.; Mueller, J.L. CRISPR-Mediated Isolation of Specific Megabase Segments of Genomic DNA. Nucleic Acids Res. 2017, 45, e165. [Google Scholar] [CrossRef]
- Li, T.; Du, D.; Zhang, D.; Lin, Y.; Ma, J.; Zhou, M.; Meng, W.; Jin, Z.; Chen, Z.; Yuan, H.; et al. CRISPR-Based Targeted Haplotype-Resolved Assembly of a Megabase Region. Nat. Commun. 2023, 14, 22. [Google Scholar] [CrossRef]
- Gabrieli, T.; Sharim, H.; Fridman, D.; Arbib, N.; Michaeli, Y.; Ebenstein, Y. Selective Nanopore Sequencing of Human BRCA1 by Cas9-Assisted Targeting of Chromosome Segments (CATCH). Nucleic Acids Res. 2018, 46, e87. [Google Scholar] [CrossRef]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted Nanopore Sequencing with Cas9-Guided Adapter Ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef]
- Watson, C.M.; Crinnion, L.A.; Hewitt, S.; Bates, J.; Robinson, R.; Carr, I.M.; Sheridan, E.; Adlard, J.; Bonthron, D.T. Cas9-Based Enrichment and Single-Molecule Sequencing for Precise Characterization of Genomic Duplications. Lab. Investig. 2020, 100, 135–146. [Google Scholar] [CrossRef]
- Stangl, C.; de Blank, S.; Renkens, I.; Westera, L.; Verbeek, T.; Valle-Inclan, J.E.; González, R.C.; Henssen, A.G.; van Roosmalen, M.J.; Stam, R.W.; et al. Partner Independent Fusion Gene Detection by Multiplexed CRISPR-Cas9 Enrichment and Long Read Nanopore Sequencing. Nat. Commun. 2020, 11, 2861. [Google Scholar] [CrossRef]
- Iyer, S.V.; Kramer, M.; Goodwin, S.; McCombie, W.R. ACME: An Affinity-Based Cas9 Mediated Enrichment Method for Targeted Nanopore Sequencing. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kirov, I.; Merkulov, P.; Gvaramiya, S.; Komakhin, R.; Omarov, M.; Dudnikov, M.; Kocheshkova, A.; Soloviev, A.; Karlov, G.; Divashuk, M. Illuminating the Transposon Insertion Landscape in Plants Using Cas9-Targeted Nanopore Sequencing and a Novel Pipeline. bioRxiv 2021. [Google Scholar] [CrossRef]
- Steele, J.L.; Stevens, R.C.; Cabrera, O.A.; Bassill, G.J.; Cramer, S.M.; Guzman, F.; Shuber, A.P. Novel CRISPR-Based Sequence Specific Enrichment Methods for Target Loci and Single Base Mutations. PLoS ONE 2020, 15, e0243781. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Langelier, C.; Kuchta, A.; Batson, J.; Teyssier, N.; Lyden, A.; Caldera, S.; McGeever, A.; Dimitrov, B.; King, R.; et al. FLASH: A next-Generation CRISPR Diagnostic for Multiplexed Detection of Antimicrobial Resistance Sequences. Nucleic Acids Res. 2019, 47, e83. [Google Scholar] [CrossRef]
- McDonald, T.L.; Zhou, W.; Castro, C.P.; Mumm, C.; Switzenberg, J.A.; Mills, R.E.; Boyle, A.P. Cas9 Targeted Enrichment of Mobile Elements Using Nanopore Sequencing. Nat. Commun. 2021, 12, 3586. [Google Scholar] [CrossRef] [PubMed]
- Merkulov, P.; Gvaramiya, S.; Dudnikov, M.; Komakhin, R.; Omarov, M.; Kocheshkova, A.; Konstantinov, Z.; Soloviev, A.; Karlov, G.; Divashuk, M.; et al. Cas9-Targeted Nanopore Sequencing Rapidly Elucidates the Transposition Preferences and DNA Methylation Profiles of Mobile Elements in Plants. J. Integr. Plant Biol. 2023, 65, 2242–2261. [Google Scholar] [CrossRef] [PubMed]
- Nachmanson, D.; Lian, S.; Schmidt, E.K.; Hipp, M.J.; Baker, K.T.; Zhang, Y.; Tretiakova, M.; Loubet-Senear, K.; Kohrn, B.F.; Salk, J.J.; et al. Targeted Genome Fragmentation with CRISPR/Cas9 Enables Fast and Efficient Enrichment of Small Genomic Regions and Ultra-Accurate Sequencing with Low DNA Input (CRISPR-DS). Genome Res. 2018, 28, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Bruijnesteijn, J.; van der Wiel, M.; de Groot, N.G.; Bontrop, R.E. Rapid Characterization of Complex Killer Cell Immunoglobulin-Like Receptor (KIR) Regions Using Cas9 Enrichment and Nanopore Sequencing. Front. Immunol. 2021, 12, 722181. [Google Scholar] [CrossRef] [PubMed]
- Rubben, K.; Tilleman, L.; Deserranno, K.; Tytgat, O.; Deforce, D.; Nieuwerburgh, F.V. Cas9 Targeted Nanopore Sequencing with Enhanced Variant Calling Improves CYP2D6-CYP2D7 Hybrid Allele Genotyping. PLoS Genet. 2022, 18, e1010176. [Google Scholar] [CrossRef]
- Lopatriello, G.; Maestri, S.; Alfano, M.; Papa, R.; Di Vittori, V.; De Antoni, L.; Bellucci, E.; Pieri, A.; Bitocchi, E.; Delledonne, M.; et al. CRISPR/Cas9-Mediated Enrichment Coupled to Nanopore Sequencing Provides a Valuable Tool for the Precise Reconstruction of Large Genomic Target Regions. Int. J. Mol. Sci. 2023, 24, 1076. [Google Scholar] [CrossRef] [PubMed]
- López-Girona, E.; Davy, M.W.; Albert, N.W.; Hilario, E.; Smart, M.E.M.; Kirk, C.; Thomson, S.J.; Chagné, D. CRISPR-Cas9 Enrichment and Long Read Sequencing for Fine Mapping in Plants. Plant Methods 2020, 16, 121. [Google Scholar] [CrossRef] [PubMed]
- Fiol, A.; Jurado-Ruiz, F.; López-Girona, E.; Aranzana, M.J. An Efficient CRISPR-Cas9 Enrichment Sequencing Strategy for Characterizing Complex and Highly Duplicated Genomic Regions. A Case Study in the Prunus Salicina LG3-MYB10 Genes Cluster. Plant Methods 2022, 18, 105. [Google Scholar] [CrossRef] [PubMed]
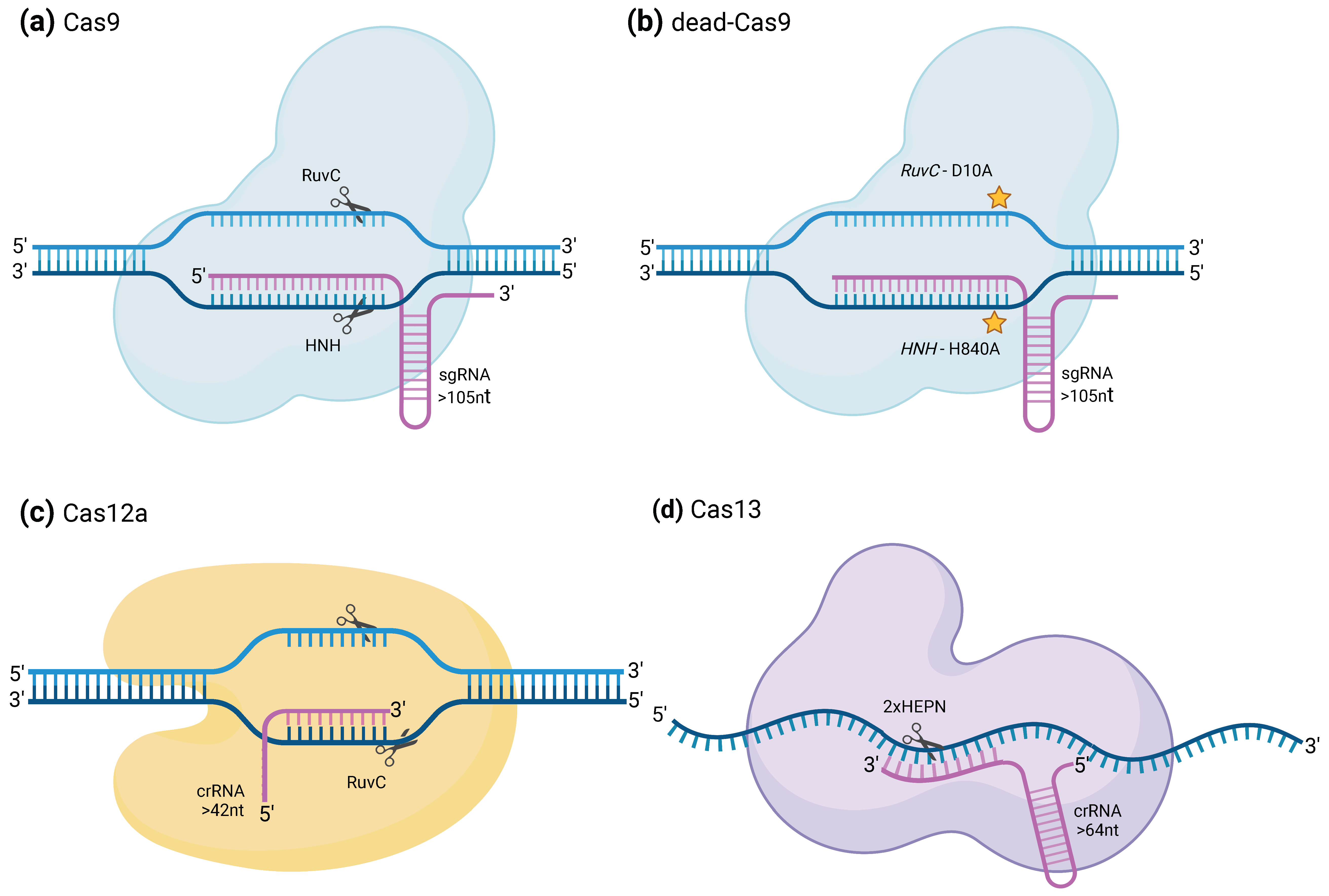

| Features | Cas9 | Cas12a | Cas13 |
|---|---|---|---|
| Other names | Cpf1 | C2c2 | |
| Type of CRISPR/Cas system | Class 2 type II | Class 2 type V | Class 2 type VI |
| Size (amino acids) | 1368 (SpCas9) | 1307 (AaCas12a) | 1389 (LshCas13a) |
| Nuclease domain | RuvC and HNH | RuvC | HEPN (×2) |
| Mutations inducing loss of function in nuclease domain. | D10A and H840A | - | D474A and D1046A |
| sgRNA components | crRNA and tracrRNA | crRNA | crRNA |
| sgRNA crRNA processing | tracrRNA-dependent | tracrRNA-independent | - |
| sgRNA protospacer length (nucleotides) | 20 (minimum ensure DNA cleavage) | 20 | 22–28 |
| sgRNA total length (nucleotides) | >105 | >42 | >140 |
| Targeted nuclei acid | dsDNA (can be induced to cleave ssRNA) | dsDNA, ssDNA (non-specific cleavage) | ssRNA |
| PAM sequence (5′-3′) | NGG (SpCas9) | T-rich, e.g., TTTV (AsCas12a, LbCas12a) | None |
| Cleavage | Blunt ended double-stranded break, 3 nucleotides before the PAM sequence. Each nuclease domain cleaves one strand. | PAM-distal dsDNA break with staggered 5′ and 3′ ends | Cleavage patterns depend on features of the target sequence (like accessibility) rather than the distance from the binding site. Single mismatches may be tolerated. |
| Other properties | - | Non-targeted ssDNA cleaving activity upon recognition of target sequence | Non-targeted ssRNA cleaving activity upon recognition of target sequence |
| Non-editing applications presented in this review | Modulation of gene expression and regulation Viral DNA targeting In situ DNA imaging New sequencing techniques | Modulation of gene expression and regulation Viral DNA targeting | In situ RNA imaging Viral RNA targeting Gene post-transcriptional regulation RNA detection techniques |
| References | [2,5,9,10] | [2,5,11] | [5,12,13,14] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devillars, A.; Magon, G.; Pirrello, C.; Palumbo, F.; Farinati, S.; Barcaccia, G.; Lucchin, M.; Vannozzi, A. Not Only Editing: A Cas-Cade of CRISPR/Cas-Based Tools for Functional Genomics in Plants and Animals. Int. J. Mol. Sci. 2024, 25, 3271. https://doi.org/10.3390/ijms25063271
Devillars A, Magon G, Pirrello C, Palumbo F, Farinati S, Barcaccia G, Lucchin M, Vannozzi A. Not Only Editing: A Cas-Cade of CRISPR/Cas-Based Tools for Functional Genomics in Plants and Animals. International Journal of Molecular Sciences. 2024; 25(6):3271. https://doi.org/10.3390/ijms25063271
Chicago/Turabian StyleDevillars, Aurélien, Gabriele Magon, Carlotta Pirrello, Fabio Palumbo, Silvia Farinati, Gianni Barcaccia, Margherita Lucchin, and Alessandro Vannozzi. 2024. "Not Only Editing: A Cas-Cade of CRISPR/Cas-Based Tools for Functional Genomics in Plants and Animals" International Journal of Molecular Sciences 25, no. 6: 3271. https://doi.org/10.3390/ijms25063271
APA StyleDevillars, A., Magon, G., Pirrello, C., Palumbo, F., Farinati, S., Barcaccia, G., Lucchin, M., & Vannozzi, A. (2024). Not Only Editing: A Cas-Cade of CRISPR/Cas-Based Tools for Functional Genomics in Plants and Animals. International Journal of Molecular Sciences, 25(6), 3271. https://doi.org/10.3390/ijms25063271