
Impact of TNF and IL-33 Cytokines on Mast Cells in Neuroinflammation

 ,
, 
 ,
,  , , , and
, , , and
Abstract
1. Introduction
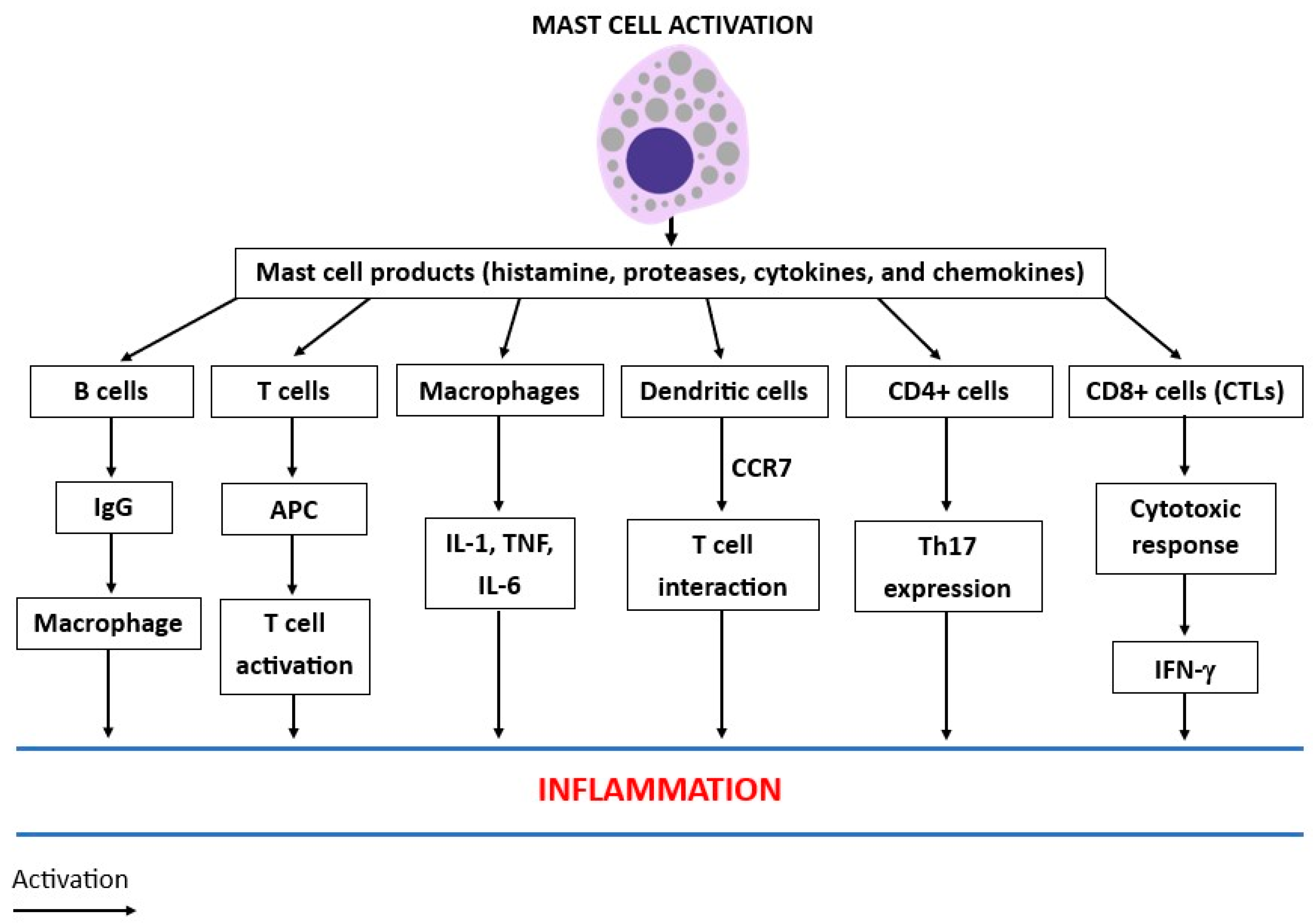
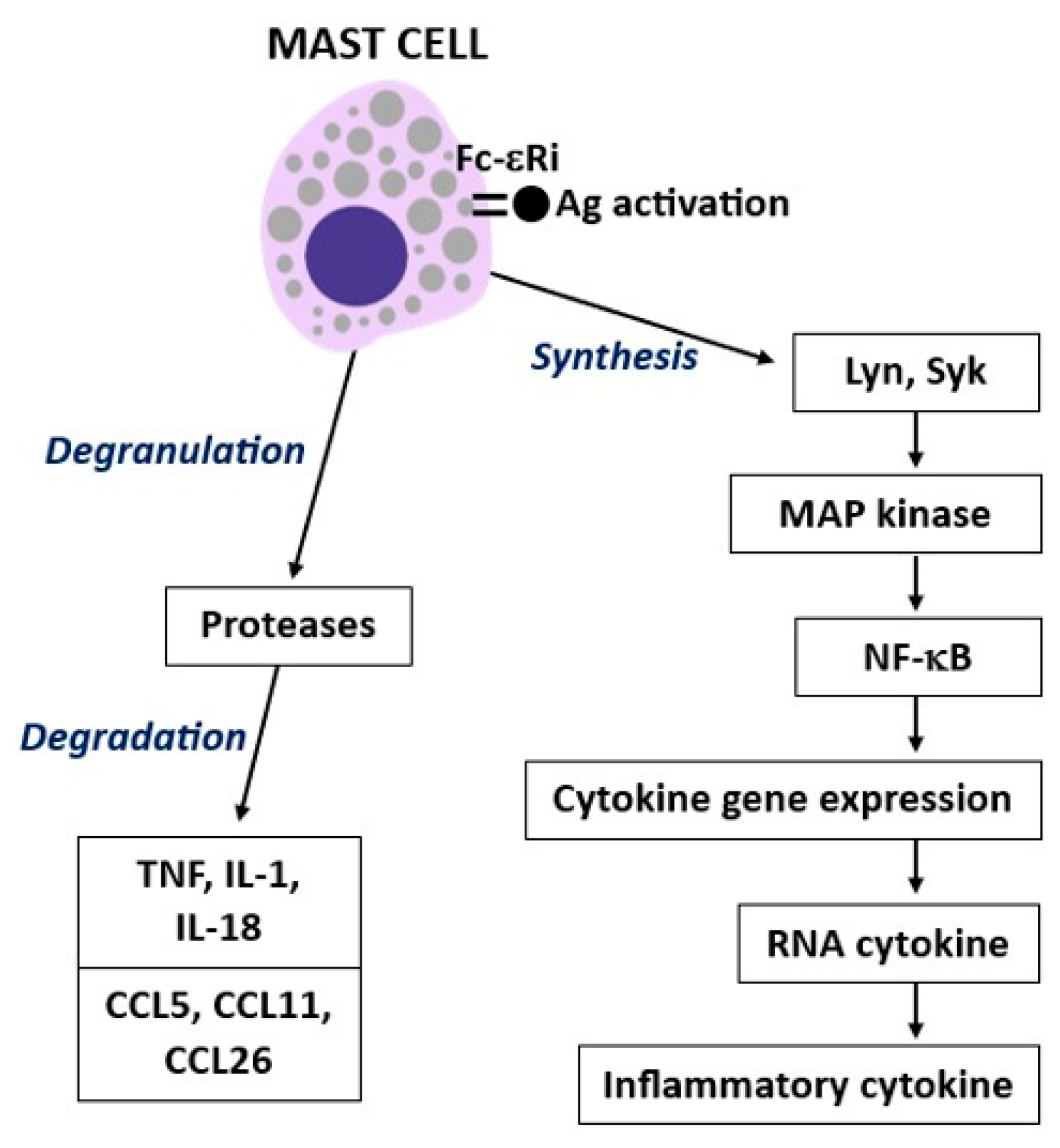
2. Mast Cell Activity
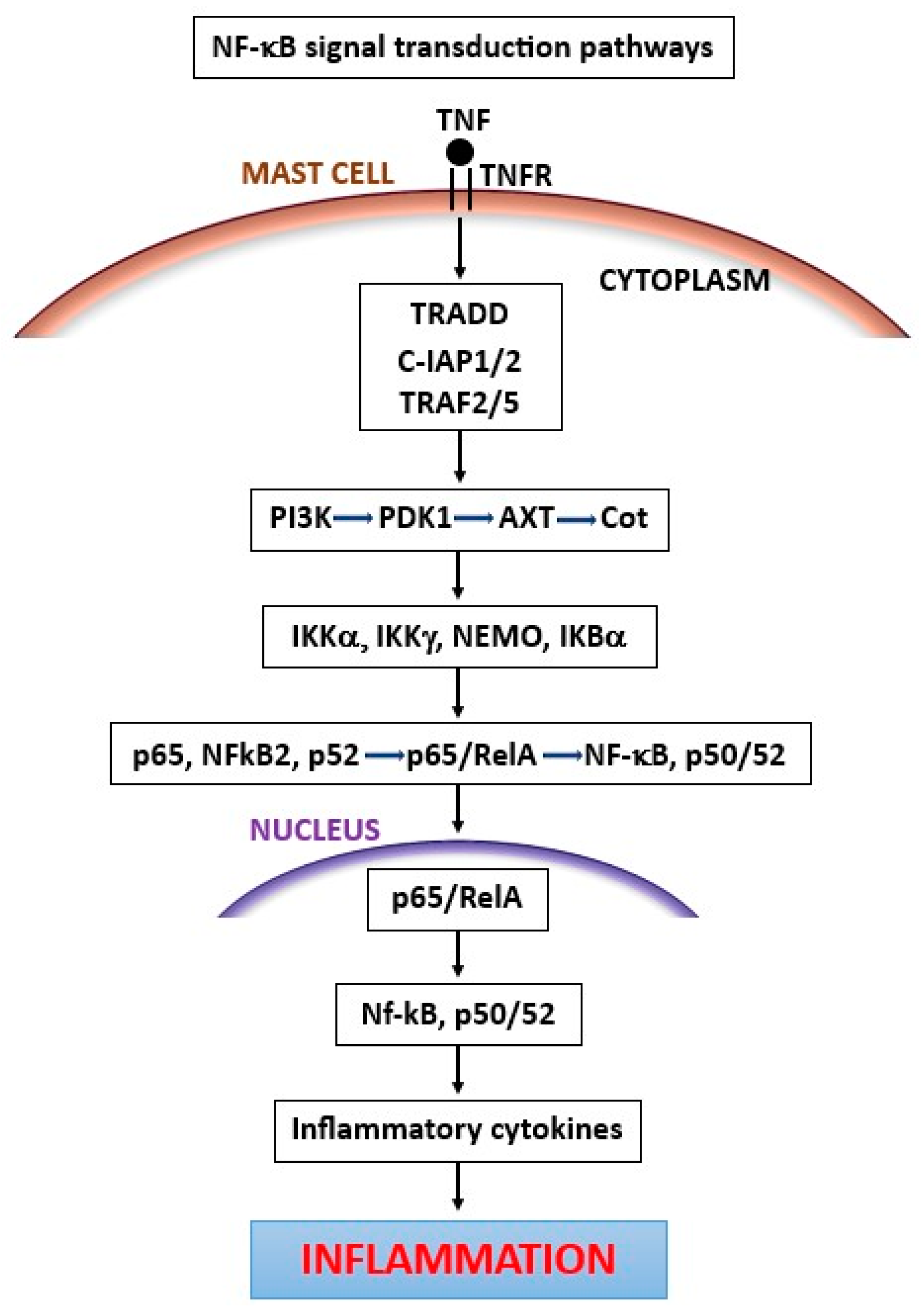
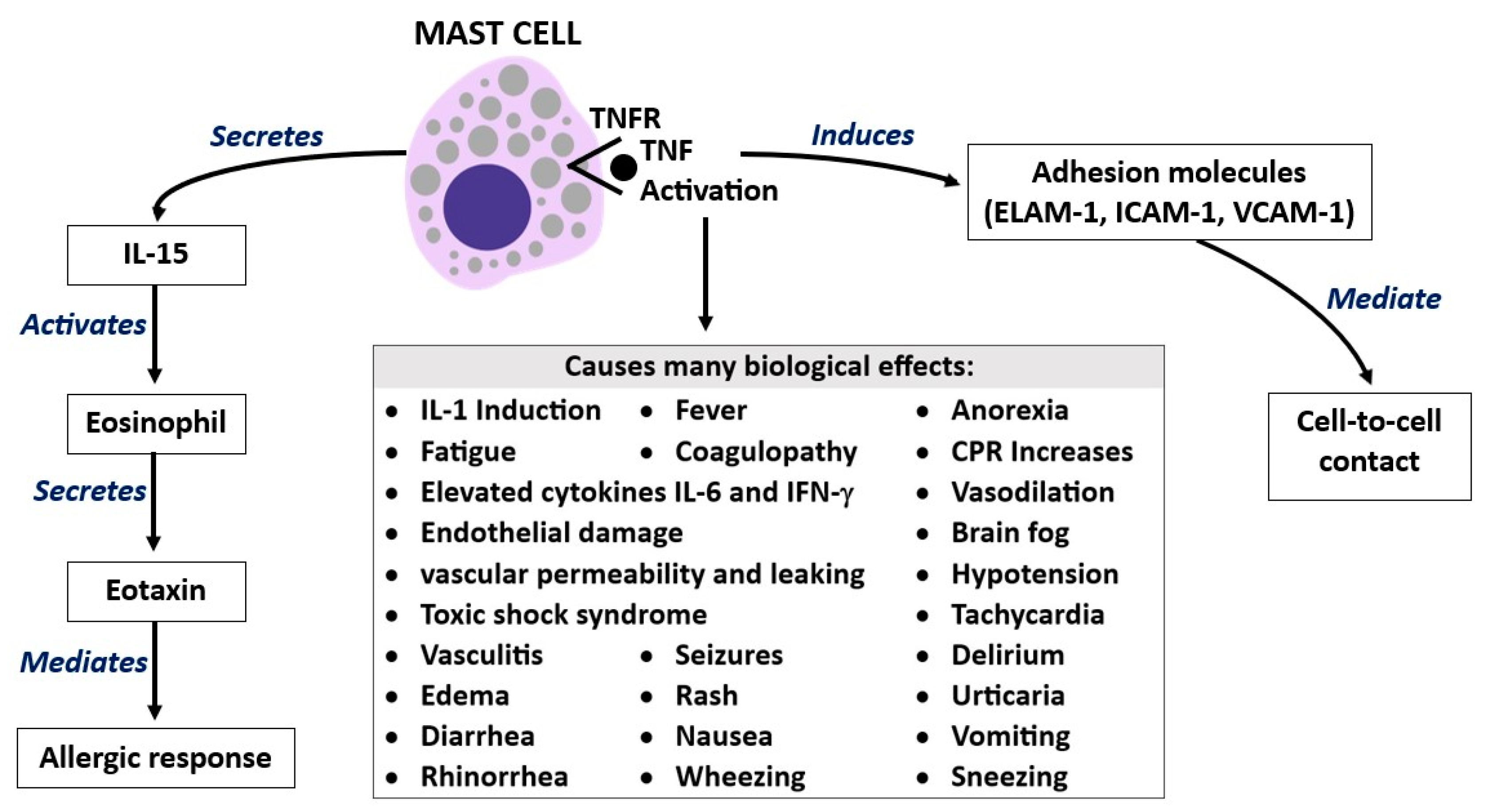
3. Tumor Necrosis Factor (TNF)
4. IL-33
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tyler, K.L. Acute Viral Encephalitis. N. Engl. J. Med. 2018, 379, 557–566. [Google Scholar] [CrossRef]
- An, Y.; Li, S.; Huang, X.; Chen, X.; Shan, H.; Zhang, M. The Role of Copper Homeostasis in Brain Disease. Int. J. Mol. Sci. 2022, 23, 13850. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Li, Y. Pyroptosis and Respiratory Diseases: A Review of Current Knowledge. Front. Immunol. 2022, 13, 920464. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Twahir, A.; Kempuraj, D. Mast Cells in the Autonomic Nervous System and Potential Role in Disorders with Dysautonomia and Neuroinflammation. Ann. Allergy Asthma Immunol. 2023, 23, S1081–S1206. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate Immunity in the Central Nervous System. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Suto, H.; Nakae, S.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast Cell-Associated TNF Promotes Dendritic Cell Migration. J. Immunol. 2006, 176, 4102–4112. [Google Scholar] [CrossRef]
- Chen, C.-C.; Grimbaldeston, M.A.; Tsai, M.; Weissman, I.L.; Galli, S.J. From the Cover: Identification of Mast Cell Progenitors in Adult Mice. Proc. Natl. Acad. Sci. USA 2005, 102, 11408–11413. [Google Scholar] [CrossRef]
- Maślińiska, D.; Dambska, M.; Kaliszek, A.; Maśliński, S. Accumulation, Distribution and Phenotype Heterogeneity of Mast Cells (MC) in Human Brains with Neurocysticercosis. Folia Neuropathol. 2001, 39, 7–13. [Google Scholar]
- Kuehn, H.S.; Gilfillan, A.M. G Protein-Coupled Receptors and the Modification of FcεRI-Mediated Mast Cell Activation. Immunol. Lett. 2007, 113, 59–69. [Google Scholar] [CrossRef]
- Zhou, T.; Du, X.; Zhang, L.; Zheng, Y.; Jia, T.; Song, X.; Che, D.; Geng, S. Suprabasin-Derived Polypeptides: SBSN(50-63) Induces Inflammatory Response via TLR4-Mediated Mast Cell Activation. Inflammopharmacology 2023, 31, 1329–1339. [Google Scholar] [CrossRef]
- Hart, P.H. Regulation of the Inflammatory Response in Asthma by Mast Cell Products. Immunol. Cell Biol. 2001, 79, 149–153. [Google Scholar] [CrossRef]
- Conti, P.; Pang, X.; Boucher, W.; Letourneau, R.; Reale, M.; Barbacane, R.C.; Thibault, J.; Theoharides, T.C. Impact of Rantes and MCP-1 Chemokines on in Vivo Basophilic Cell Recruitment in Rat Skin Injection Model and Their Role in Modifying the Protein and MRNA Levels for Histidine Decarboxylase. Blood 1997, 89, 4120–4127. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Tsilioni, I.; Conti, P. Mast Cells May Regulate the Anti-Inflammatory Activity of IL-37. Int. J. Mol. Sci. 2019, 20, 3701. [Google Scholar] [CrossRef]
- Conti, P.; Lessiani, G.; Kritas, S.K.; Ronconi, G.; Caraffa, A.; Theoharides, T.C. Mast Cells Emerge as Mediators of Atherosclerosis: Special Emphasis on IL-37 Inhibition. Tissue Cell 2017, 49, 393–400. [Google Scholar] [CrossRef]
- Abdollahiyan, S.; Nabavi-Rad, A.; Raftar, S.K.A.; Monnoye, M.; Salarieh, N.; Farahanie, A.; Aghdaei, H.A.; Zali, M.R.; Hatami, B.; Gérard, P.; et al. Characterization of Gut Microbiome Composition in Iranian Patients with Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Sci. Rep. 2023, 13, 20584. [Google Scholar] [CrossRef]
- Brzezińska-Błaszczyk, E.; Wierzbicki, M. Mast Cell Toll-like Receptors (TLRs). Postep. Hig. Med. Dosw. 2010, 64, 11–21. [Google Scholar]
- Carta, S.; Tassi, S.; Pettinati, I.; Delfino, L.; Dinarello, C.A.; Rubartelli, A. The Rate of Interleukin-1β Secretion in Different Myeloid Cells Varies with the Extent of Redox Response to Toll-like Receptor Triggering. J. Biol. Chem. 2011, 286, 27069–27080. [Google Scholar] [CrossRef]
- Akira, S. TLR Signaling. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2006; Volume 311, pp. 1–16. [Google Scholar] [CrossRef]
- Zhang, G.; Ghosh, S. Toll-like receptor-mediated NF-κB activation: A phylogenetically conserved paradigm in innate immunity. J. Clin. Investig. 2001, 107, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Saunders, S.P.; Barlow, J.L.; Walsh, C.M.; Bellsoi, A.; Smith, P.; McKenzie, A.N.J.; Fallon, P.G. C-Type Lectin SIGN-R1 Has a Role in Experimental Colitis and Responsiveness to Lipopolysaccharide. J. Immunol. 2010, 184, 2627–2637. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Power, M.R.; Li, B.; Lin, T.-J. Inhibition of IKK Down-Regulates Antigen + IgE-Induced TNF Production by Mast Cells: A Role for the IKK-IκB-NF-ΚB Pathway in IgE-Dependent Mast Cell Activation. J. Leukoc. Biol. 2005, 77, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Kalesnikoff, J.; Grimbaldeston, M.A.; Piliponsky, A.M.; Williams, C.M.M.; Tsai, M. Mast Cells as “TUNABLE” Effector and Immunoregulatory Cells: Recent Advances. Annu. Rev. Immunol. 2005, 23, 749–786. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Valent, P.; Akin, C. Mast Cells, Mastocytosis, and Related Disorders. N. Engl. J. Med. 2015, 373, 163–172. [Google Scholar] [CrossRef]
- Lee, Y.S.; Saxena, V.; Bromberg, J.S.; Scalea, J.R. G-CSF Promotes Alloregulatory Function of MDSCs through a C-Kit Dependent Mechanism. Cell. Immunol. 2021, 364, 104346. [Google Scholar] [CrossRef] [PubMed]
- Marichal, T.; Starkl, P.; Reber, L.L.; Kalesnikoff, J.; Oettgen, H.C.; Tsai, M.; Metz, M.; Galli, S.J. A Beneficial Role for Immunoglobulin E in Host Defense against Honeybee Venom. Immunity 2013, 39, 963–975. [Google Scholar] [CrossRef]
- Kanagaratham, C.; El Ansari, Y.S.; Lewis, O.L.; Oettgen, H.C. IgE and IgG Antibodies as Regulators of Mast Cell and Basophil Functions in Food Allergy. Front. Immunol. 2020, 11, 3000. [Google Scholar] [CrossRef]
- Marone, G.; de Crescenzo, G.; Florio, G.; Granata, F.; Dente, V.; Genovese, A. Immunological Modulation of Human Cardiac Mast Cells. Neurochem. Res. 1999, 24, 1195–1202. [Google Scholar] [CrossRef]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 Families of Cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef]
- Conti, P.; Stellin, L.; Caraffa, A.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G. Advances in Mast Cell Activation by IL-1 and IL-33 in Sjögren’s Syndrome: Promising Inhibitory Effect of IL-37. Int. J. Mol. Sci. 2020, 21, 4297. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.O.; Pullen, N.A. Beyond IgE: Alternative Mast Cell Activation across Different Disease States. Int. J. Mol. Sci. 2020, 21, 1498. [Google Scholar] [CrossRef]
- Gri, G.; Frossi, B.; D’Inca, F.; Danelli, L.; Betto, E.; Mion, F.; Sibilano, R.; Pucillo, C. Mast cell: An emerging partner in immune interaction. Front. Immunol. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Kambayashi, T.; Allenspach, E.J.; Chang, J.T.; Zou, T.; Shoag, J.E.; Reiner, S.L.; Caton, A.J.; Koretzky, G.A. Inducible MHC Class II Expression by Mast Cells Supports Effector and Regulatory T Cell Activation. J. Immunol. 2009, 182, 4686–4695. [Google Scholar] [CrossRef]
- Mekori, Y.A.; Baram, D. Heterotypic Adhesion-Induced Mast Cell Activation: Biologic Relevance in the Inflammatory Context. Mol. Immunol. 2002, 38, 1363–1367. [Google Scholar] [CrossRef]
- Cornejo-García, J.A.; Perkins, J.R.; Jurado-Escobar, R.; García-Martín, E.; Agúndez, J.A.; Viguera, E.; Pérez-Sánchez, N.; Blanca-López, N. Pharmacogenomics of Prostaglandin and Leukotriene Receptors. Front. Pharmacol. 2016, 7, 316. [Google Scholar] [CrossRef]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.-P. Regulation of Endothelial Cell Adhesion Molecule Expression by Mast Cells, Macrophages, and Neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef]
- Lauritano, D.; Mastrangelo, F.; D’Ovidio, C.; Ronconi, G.; Caraffa, A.; Gallenga, C.E.; Frydas, I.; Kritas, S.K.; Trimarchi, M.; Carinci, F.; et al. Activation of Mast Cells by Neuropeptides: The Role of Pro-Inflammatory and Anti-Inflammatory Cytokines. Int. J. Mol. Sci. 2023, 24, 4811. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.V.; Bailey, D.; Kashyap, M.; Kepley, C.L.; Drutskaya, M.S.; Nedospasov, S.A.; Ryan, J.J. IL-3-Mediated TNF Production Is Necessary for Mast Cell Development. J. Immunol. 2006, 176, 2114–2121. [Google Scholar] [CrossRef]
- Nishida, S.; Yoshioka, S.; Kinoshita-Kimoto, S.; Kotani, M.; Tsubaki, M.; Fujii, Y.; Tomura, T.T.; Irimajiri, K. Pretreatment with PKC Inhibitor Triggers TNF-α Induced Apoptosis in TNF-α–Resistant B16 Melanoma BL6 Cells. Life Sci. 2003, 74, 781–792. [Google Scholar] [CrossRef]
- Gordon, J.R.; Galli, S.J. Mast Cells as a Source of Both Preformed and Immunologically Inducible TNF-Alpha/Cachectin. Nature 1990, 346, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Taracanova, A.; Alevizos, M.; Karagkouni, A.; Weng, Z.; Norwitz, E.; Conti, P.; Leeman, S.E.; Theoharides, T.C. SP and IL-33 Together Markedly Enhance TNF Synthesis and Secretion from Human Mast Cells Mediated by the Interaction of Their Receptors. Proc. Natl. Acad. Sci. USA 2017, 114, E4002–E4009. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Kuroda, E.; Yoshida, Y.; Yamashita, U.; Suzumura, A.; Tsuji, S. Effects of Interferon-β on the Cytokine Production of Astrocytes. J. Neuroimmunol. 2005, 159, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R. TNF-Mediated Inflammatory Disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and Adaptive Immunity in Inflammatory Bowel Disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-W.; Chang, Y.-C.; Chen, S.-J.; Tseng, C.-H.; Tu, Y.-F.; Liao, N.-S.; Huang, C.-C.; Ho, C.-J. TNFR1-JNK Signaling Is the Shared Pathway of Neuroinflammation and Neurovascular Damage after LPS-Sensitized Hypoxic-Ischemic Injury in the Immature Brain. J. Neuroinflamm. 2014, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Caraffa, A.; Mastrangelo, F.; Tettamanti, L.; Ronconi, G.; Frydas, I.; Kritas, S.K.; Theoharides, T.C. Critical Role of Inflammatory Mast Cell in Fibrosis: Potential Therapeutic Effect of IL-37. Cell Prolif. 2018, 51, e12475. [Google Scholar] [CrossRef]
- Nakae, S.; Suto, H.; Berry, G.J.; Galli, S.J. Mast Cell–Derived TNF Can Promote Th17 Cell–Dependent Neutrophil Recruitment in Ovalbumin-Challenged OTII Mice. Blood 2006, 109, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Sibilano, R.; Gaudenzio, N.; DeGorter, M.K.; Reber, L.L.; Hernandez, J.D.; Starkl, P.M.; Zurek, O.W.; Tsai, M.; Zahner, S.; Montgomery, S.B.; et al. A TNFRSF14-FcɛRI-Mast Cell Pathway Contributes to Development of Multiple Features of Asthma Pathology in Mice. Nat. Commun. 2016, 7, 13696. [Google Scholar] [CrossRef]
- Haaster, V.; Derhaag, J.G.; Engels, W.; Lemmens, R.; Gijsen, A.P.; Hornstra, G.; Der, V.; Duijvestijn, A.M. Mast Cell-Mediated Induction of ICAM-1, VCAM-1 and E-Selectin in Endothelial Cells in Vitro: Constitutive Release of Inducing Mediators but No Effect of Degranulation. Pflügers Arch. Eur. J. Physiol. 1997, 435, 137–144. [Google Scholar] [CrossRef]
- Fu, Z.; Akula, S.; Thorpe, M.; Hellman, L. Highly Selective Cleavage of TH2-Promoting Cytokines by the Human and the Mouse Mast Cell Tryptases, Indicating a Potent Negative Feedback Loop on TH2 Immunity. Int. J. Mol. Sci. 2019, 20, 5147. [Google Scholar] [CrossRef]
- Brzezińska-Błaszczyk, E.; Pietrzak, A.; Misiak-Tłoczek, A. Tumor Necrosis Factor (TNF) Is a Potent Rat Mast Cell Chemoattractant. J. Interferon Cytokine Res. 2007, 27, 911–920. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Tsilioni, I.; Bawazeer, M. Mast Cells, Neuroinflammation and Pain in Fibromyalgia Syndrome. Front. Cell. Neurosci. 2019, 13, 353. [Google Scholar] [CrossRef]
- Mittal, A.; Sagi, V.; Gupta, M.; Gupta, K. Mast Cell Neural Interactions in Health and Disease. Front. Cell. Neurosci. 2019, 13, 110. [Google Scholar] [CrossRef]
- Abraham, S.N.; St John, A.L. Mast Cell-Orchestrated Immunity to Pathogens. Nat. Rev. Immunol. 2010, 10, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Tweedie, D.; Sambamurti, K.; Greig, N. TNF-α Inhibition as a Treatment Strategy for Neurodegenerative Disorders: New Drug Candidates and Targets. Curr. Alzheimer Res. 2007, 4, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Kraft, A.D.; McPherson, C.A.; Harry, G.J. Heterogeneity of Microglia and TNF Signaling as Determinants for Neuronal Death or Survival. NeuroToxicology 2009, 30, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Papazian, I.; Tsoukala, E.; Boutou, A.; Karamita, M.; Kambas, K.; Iliopoulou, L.; Fischer, R.; Kontermann, R.E.; Denis, M.C.; Kollias, G.; et al. Fundamentally Different Roles of Neuronal TNF Receptors in CNS Pathology: TNFR1 and IKKβ Promote Microglial Responses and Tissue Injury in Demyelination While TNFR2 Protects against Excitotoxicity in Mice. J. Neuroinflamm. 2021, 18, 222. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Conti, P. Mast Cells: The JEKYLL and HYDE of Tumor Growth. Trends Immunol. 2004, 25, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Okayama, Y. Mast Cell-Derived Cytokine Expression Induced via Fc Receptors and Toll-like Receptors. Nephrology 2005, 87, 101–110. [Google Scholar] [CrossRef]
- Yang, D.; Chen, Q.; Chertov, O.; Oppenheim, J.J. Human Neutrophil Defensins Selectively Chemoattract Naive T and Immature Dendritic Cells. J. Leukoc. Biol. 2000, 68, 9–14. [Google Scholar] [CrossRef]
- Hill, G.R.; Teshima, T.; Rebel, V.I.; Krijanovski, O.I.; Cooke, K.R.; Brinson, Y.S.; Ferrara, J.L.M. The P55 TNF-α Receptor Plays a Critical Role in T Cell Alloreactivity. J. Immunol. 2000, 164, 656–663. [Google Scholar] [CrossRef]
- Namba, S.; Nakano, R.; Kitanaka, T.; Kitanaka, N.; Nakayama, T.; Sugiya, H. ERK2 and JNK1 Contribute to TNF-α-Induced IL-8 Expression in Synovial Fibroblasts. PLoS ONE 2017, 12, e0182923. [Google Scholar] [CrossRef]
- Matsushima, K.; Yang, D.; Oppenheim, J.J. Interleukin-8: An Evolving Chemokine. Cytokine 2022, 153, 155828. [Google Scholar] [CrossRef] [PubMed]
- Dri, P.; Haas, E.; Cramer, R.; Menegazzi, R.; Gasparini, C.; Martinelli, R.; Scheurich, P.; Patriarca, P. Role of the 75-KDa TNF Receptor in TNF-Induced Activation of Neutrophil Respiratory Burst. J. Immunol. 1999, 162, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Boyce, J.A.; Mellor, E.A.; Perkins, B.; Lim, Y.-C.; Luscinskas, F.W. Human Mast Cell Progenitors Use α4-Integrin, VCAM-1, and PSGL-1 E-Selectin for Adhesive Interactions with Human Vascular Endothelium under Flow Conditions. Blood 2002, 99, 2890–2896. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sheibani, N. Expression Pattern of Alternatively Spliced PECAM-1 Isoforms in Hematopoietic Cells and Platelets. J. Cell. Biochem. 2002, 87, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.E. The Unfolding Tale of PECAM-1. FEBS Lett. 2003, 540, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Flores, A.; Rodríguez, G.M.; Soria-Castro, R.; López-Santiago, R.; Rodríguez-Cortés, O.; Pérez-Tapia, S.M.; Chávez-Blanco, A.D.; Estrada-Parra, S.; Flores-Mejía, R.; Chacón-Salinas, R. Brucella Abortus Induces Mast Cell Activation through TLR-2 and TLR-4. Microb. Pathog. 2023, 176, 106005. [Google Scholar] [CrossRef]
- Portales-Cervantes, L.; Haidl, I.D.; Lee, P.W.; Marshall, J.S. Virus-Infected Human Mast Cells Enhance Natural Killer Cell Functions. J. Innate Immun. 2016, 9, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Ortega, P.; Ragu Varman, D.; Rodríguez, V.M.; Reyes-Haro, D. Anorexia Induces a Microglial Associated Pro-Inflammatory Environment and Correlates with Neurodegeneration in the Prefrontal Cortex of Young Female Rats. Behav. Brain Res. 2020, 392, 112606. [Google Scholar] [CrossRef]
- Bussolino, F.; Camussi, G. Platelet-Activating Factor Produced by Endothelial Cells. A Molecule with Autocrine and Paracrine Properties. Eur. J. Biochem. 1995, 229, 327–337. [Google Scholar] [CrossRef]
- Dickel, H.; Gambichler, T.; Kamphowe, J.; Altmeyer, P.; Skrygan, M. Standardized Tape Stripping prior to Patch Testing Induces Upregulation of Hsp90, Hsp70, IL-33, TNF-α and IL-8/CXCL8 MRNA: New Insights into the Involvement of “Alarmins”. Contact Dermat. 2010, 63, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Mege, J.-L.; Gomez-Cambronero, J.; Molski, T.F.; Becker, E.L.; Sha’afi, R.I. Effect of Granulocyte-Macrophage Colony-Stimulating Factor on Superoxide Production in Cytoplasts and Intact Human Neutrophils: Role of Protein Kinase and G-Proteins. J. Leukoc. Biol. 1989, 46, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Miller, S.D.; Koh, S. Immune Mechanisms in Epileptogenesis. Front. Cell. Neurosci. 2013, 7, 195. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-Z.; Nygård, M.; Kristensson, K.; Bentivoglio, M. Regulation of Cytokine Signaling and T-Cell Recruitment in the Aging Mouse Brain in Response to Central Inflammatory Challenge. Brain Behav. Immun. 2010, 24, 138–152. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, M.J.; Ibarra-Sánchez, A.; Román-Figueroa, A.; Pérez-Severiano, F.; González-Espinosa, C. Mutant Huntingtin Affects Toll-like Receptor 4 Intracellular Trafficking and Cytokine Production in Mast Cells. J. Neuroinflamm. 2020, 17, 95. [Google Scholar] [CrossRef]
- Askenase, P.W.; Kawikova, I.; Paliwal, V.; Akahira-Azuma, M.; Gerard, C.; Hugli, T.; Tsuji, R. A New Paradigm of T Cell Allergy: Requirement for the B–1 Cell Subset. Int. Arch. Allergy Immunol. 1999, 118, 145–149. [Google Scholar] [CrossRef]
- Breil, I.; Koch, T.; Belz, M.; Van Ackern, K.; Neuhof, H. Effects of Bradykinin, Histamine and Serotonin on Pulmonary Vascular Resistance and Permeability. Acta Physiol. Scand. 1997, 159, 189–198. [Google Scholar] [CrossRef]
- Kanashiro, A.; Hiroki, C.H.; da Fonseca, D.M.; Birbrair, A.; Ferreira, R.G.; Bassi, G.S.; Fonseca, M.D.; Kusuda, R.; Cebinelli, G.C.M.; da Silva, K.P.; et al. The Role of Neutrophils in Neuro-Immune Modulation. Pharmacol. Res. 2020, 151, 104580. [Google Scholar] [CrossRef]
- DiStasi, M.R.; Ley, K. Opening the Flood-Gates: How Neutrophil-Endothelial Interactions Regulate Permeability. Trends Immunol. 2009, 30, 547–556. [Google Scholar] [CrossRef]
- Takaoka, K.; Shirai, Y.; Saito, N. Inflammatory Cytokine Tumor Necrosis Factor-α Enhances Nerve Growth Factor Production in Human Keratinocytes, HaCaT Cells. J. Pharmacol. Sci. 2009, 111, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Piliponsky, A.M.; Chen, C.-C.; Grimbaldeston, M.A.; Burns-Guydish, S.M.; Hardy, J.; Kalesnikoff, J.; Contag, C.H.; Tsai, M.; Galli, S.J. Mast Cell-Derived TNF Can Exacerbate Mortality during Severe Bacterial Infections in C57BL/6-Kit Mice. Am. J. Pathol. 2010, 176, 926–938. [Google Scholar] [CrossRef]
- Reber, L.L.; Hernandez, J.D.; Galli, S.J. The Pathophysiology of Anaphylaxis. J. Allergy Clin. Immunol. 2018, 140, 335–348. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast Cells as Sources of Cytokines, Chemokines, and Growth Factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Huang, X.; Zhao, Y.; Chen, F.; Sun, M.; Yang, W.; Chen, L.; Yao, S.; Peniche, A.; Dann, S.M.; et al. Interleukin-33 Promotes REG3γ Expression in Intestinal Epithelial Cells and Regulates Gut Microbiota. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Jeong, A.P.; Kim, J.; Seung-Sik, R.; Park, H.; Young-Myeong, K.; Young-Guen, K. Nuclear IL-33 Is a Transcriptional Regulator of NF-ΚB P65 and Induces Endothelial Cell Activation. Biochem. Biophys. Res. Commun. 2012, 421, 305–311. [Google Scholar] [CrossRef]
- Gadani, S.P.; Walsh, J.T.; Smirnov, I.; Zheng, J.; Kipnis, J. The Glia-Derived Alarmin IL-33 Orchestrates the Immune Response and Promotes Recovery Following CNS Injury. Neuron 2015, 85, 703–709. [Google Scholar] [CrossRef]
- Zharichenko, N.; Njoku, D.B. The Role of Pro-Inflammatory and Regulatory Signaling by IL-33 in the Brain and Liver: A Focused Systematic Review of Mouse and Human Data and Risk of Bias Assessment of the Literature. Int. J. Mol. Sci. 2020, 21, 3933. [Google Scholar] [CrossRef]
- Chan, B.C.L.; Lam, C.W.K.; Tam, L.-S.; Wong, C.K. IL33: Roles in Allergic Inflammation and Therapeutic Perspectives. Front. Immunol. 2019, 10, 364. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in Health and Disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Yasuoka, S.; Kawanokuchi, J.; Parajuli, B.; Jin, S.; Doi, Y.; Noda, M.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Production and Functions of IL-33 in the Central Nervous System. Brain Res. 2011, 1385, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A.; et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef]
- Zhu, J. T Helper 2 (Th2) Cell Differentiation, Type 2 Innate Lymphoid Cell (ILC2) Development and Regulation of Interleukin-4 (IL-4) and IL-13 Production. Cytokine 2015, 75, 14–24. [Google Scholar] [CrossRef]
- Iwaszko, M.; Biały, S.; Bogunia-Kubik, K. Significance of Interleukin (IL)-4 and IL-13 in Inflammatory Arthritis. Cells 2021, 10, 3000. [Google Scholar] [CrossRef]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Cardamone, C.; Parente, R.; Feo, G.D.; Triggiani, M. Mast Cells as Effector Cells of Innate Immunity and Regulators of Adaptive Immunity. Immunol. Lett. 2016, 178, 10–14. [Google Scholar] [CrossRef]
- Larsen, K.; Minaya, M.; Vaish, V.; Peña, M. The Role of IL-33/ST2 Pathway in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef]
- Pelaia, C.; Paoletti, G.; Puggioni, F.; Racca, F.; Pelaia, G.; Canonica, G.W.; Heffler, E. Interleukin-5 in the Pathophysiology of Severe Asthma. Front. Physiol. 2019, 10, 1514. [Google Scholar] [CrossRef] [PubMed]
- Foster, P.S.; Hogan, S.P.; Ramsay, A.J.; Matthaei, K.I.; Young, I.G. Interleukin 5 Deficiency Abolishes Eosinophilia, Airways Hyperreactivity, and Lung Damage in a Mouse Asthma Model. J. Exp. Med. 1996, 183, 195–201. [Google Scholar] [CrossRef]
- Kusano, S.; Kukimoto-Niino, M.; Hino, N.; Ohsawa, N.; Ikutani, M.; Takaki, S.; Sakamoto, K.; Hara-Yokoyama, M.; Shirouzu, M.; Takatsu, K.; et al. Structural Basis of Interleukin-5 Dimer Recognition by Its α Receptor. Protein Sci. 2012, 21, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Nishida, T.; Fukushima, A. Synergistic Induction of Eotaxin and VCAM-1 Expression in Human Corneal Fibroblasts by Staphylococcal Peptidoglycan and Either IL-4 or IL-13. Allergol. Int. 2011, 60, 355–363. [Google Scholar] [CrossRef]
- Guseh, J.S.; Bores, S.A.; Stanger, B.Z.; Zhou, Q.; Anderson, W.J.; Melton, D.A.; Rajagopal, J. Notch Signaling Promotes Airway Mucous Metaplasia and Inhibits Alveolar Development. Development 2009, 136, 1751–1759. [Google Scholar] [CrossRef]
- Hueber, A.J.; Asquith, D.L.; Miller, A.M.; Reilly, J.; Kerr, S.; Leipe, J.; Melendez, A.J.; McInnes, I.B. Cutting Edge: Mast Cells Express IL-17A in Rheumatoid Arthritis Synovium. J. Immunol. 2010, 184, 3336–3340. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.G.; Lavelle, E.C. A Crucial Role for Interleukin (IL)-1 in the Induction of IL-17–Producing T Cells That Mediate Autoimmune Encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 Cells in Human Disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Gao, Y.; Guo, W. Th17 Immunity in Patients with Allergic Asthma. Int. Arch. Allergy Immunol. 2009, 151, 297–307. [Google Scholar] [CrossRef]
- Schuijs, M.J.; Hammad, H.; Lambrecht, B.N. Professional and “Amateur” Antigen-Presenting Cells in Type 2 Immunity. Trends Immunol. 2019, 40, 22–34. [Google Scholar] [CrossRef]
- Brown, M.A.; Weinberg, R.B. Mast Cells and Innate Lymphoid Cells: Underappreciated Players in CNS Autoimmune Demyelinating Disease. Front. Immunol. 2018, 9, 514. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.S.-H.; Ho, Y.-S.; Chang, R.C.-C. The Role of Meningeal Populations of Type II Innate Lymphoid Cells in Modulating Neuroinflammation in Neurodegenerative Diseases. Exp. Mol. Med. 2021, 53, 1251–1267. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.Y.; Hung, K.-W.; Yuen, M.Y.F.; Zhou, X.; Mak, D.S.Y.; Chan, I.C.W.; Cheung, T.H.; Zhang, B.; Fu, W.-Y.; Liew, F.Y.; et al. IL-33 Ameliorates Alzheimer’s Disease-like Pathology and Cognitive Decline. Proc. Natl. Acad. Sci. USA 2016, 113, E2705–E2713. [Google Scholar] [CrossRef]
- Rao, X.; Hua, F.; Zhang, L.; Lin, Y.; Fang, P.; Chen, S.; Ying, J.; Wang, X. Dual Roles of Interleukin-33 in Cognitive Function by Regulating Central Nervous System Inflammation. J. Transl. Med. 2022, 20, 369. [Google Scholar] [CrossRef]
- Jiang, H.-R.; Milovanović, M.; Allan, D.; Niedbala, W.; Besnard, A.-G.; Fukada, S.Y.; Alves-Filho, J.C.; Togbe, D.; Goodyear, C.S.; Linington, C.; et al. IL-33 Attenuates EAE by Suppressing IL-17 and IFN-γ Production and Inducing Alternatively Activated Macrophages. Eur. J. Immunol. 2012, 42, 1804–1814. [Google Scholar] [CrossRef]
- Chapuis, J.; Hot, D.; Hansmannel, F.; Kerdraon, O.; Ferreira, S.; Hubans, C.; Maurage, C.A.; Huot, L.; Bensemain, F.; Laumet, G.; et al. Transcriptomic and Genetic Studies Identify IL-33 as a Candidate Gene for Alzheimer’s Disease. Mol. Psychiatry 2009, 14, 1004–1016. [Google Scholar] [CrossRef]
- Hudson, C.A.; Christophi, G.P.; Gruber, R.C.; Wilmore, J.R.; Lawrence, D.A.; Massa, P.T. Induction of IL-33 Expression and Activity in Central Nervous System Glia. J. Leukoc. Biol. 2008, 84, 631–643. [Google Scholar] [CrossRef]
- Christophi, G.P.; Gruber, R.C.; Panos, M.; Christophi, R.L.; Jubelt, B.; Massa, P.T. Interleukin-33 Upregulation in Peripheral Leukocytes and CNS of Multiple Sclerosis Patients. Clin. Immunol. 2012, 142, 308–319. [Google Scholar] [CrossRef]
- Allan, D.; Fairlie-Clarke, K.J.; Elliott, C.; Schuh, C.; Barnett, S.C.; Lassmann, H.; Linnington, C.; Jiang, H.-R. Role of IL-33 and ST2 Signalling Pathway in Multiple Sclerosis: Expression by Oligodendrocytes and Inhibition of Myelination in Central Nervous System. Acta Neuropathol. Commun. 2016, 4, 75. [Google Scholar] [CrossRef]
- Marashian, S.M.; Mortaz, E.; Jamaati, H.R.; Alavi-Moghaddam, M.; Kiani, A.; Abedini, A.; Garssen, J.; Adcock, I.M.; Velayati, A.A. Role of Innate Lymphoid Cells in Lung Disease. Iran. J. Allergy Asthma Immunol. 2015, 14, 346–360. [Google Scholar]
- Fung, I.T.H.; Sankar, P.; Zhang, Y.; Robison, L.S.; Zhao, X.; D’Souza, S.S.; Salinero, A.E.; Wang, Y.; Qian, J.; Kuentzel, M.L.; et al. Activation of Group 2 Innate Lymphoid Cells Alleviates Aging-Associated Cognitive Decline. J. Exp. Med. 2020, 217, e20190915. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
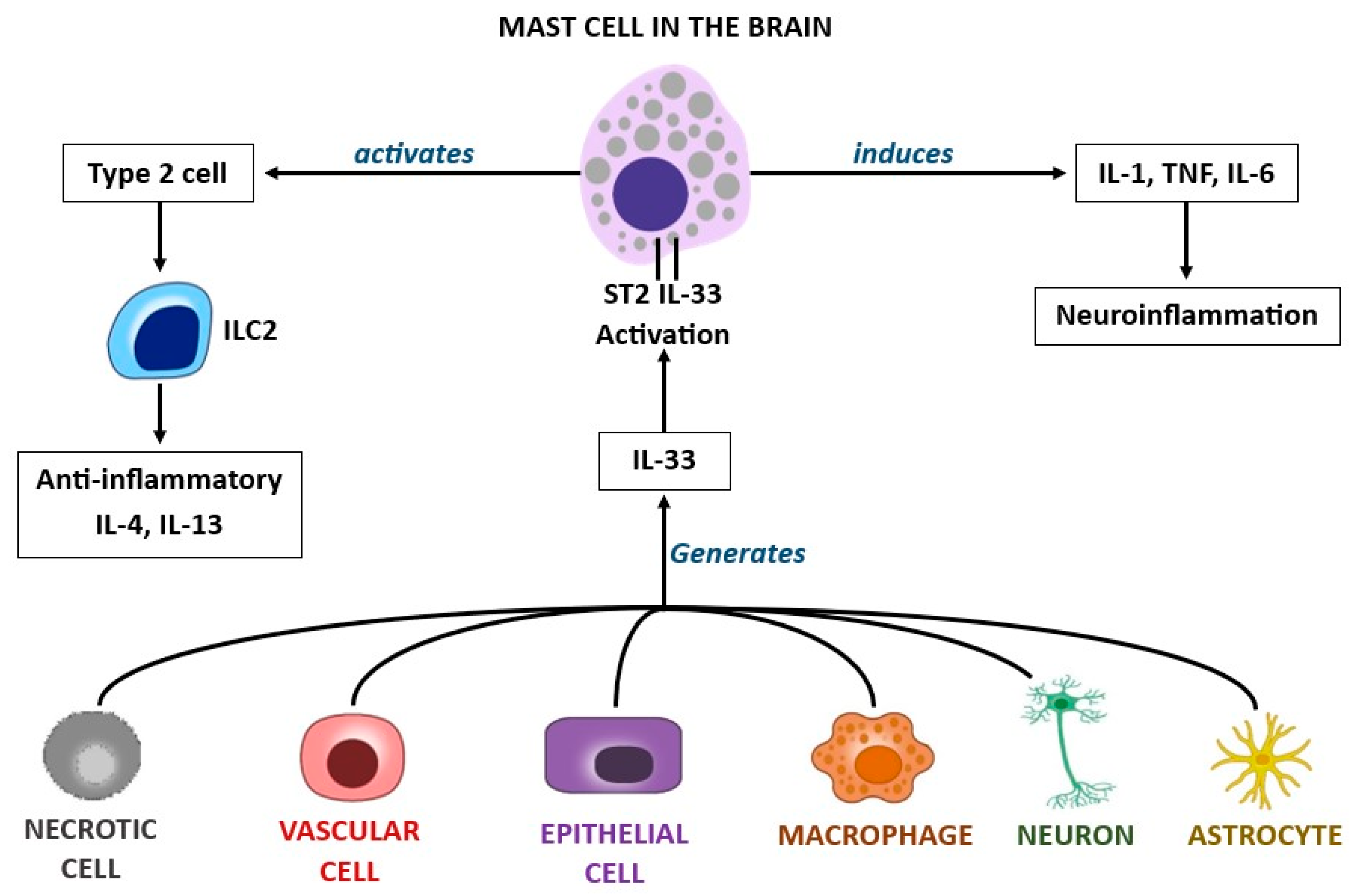{kind=link}
| Stored in the cytoplasmatic granules: Histamine, serotonin (in mice), tryptase, chymase, peroxidase, heparin, chondroitin Sulphates, hydrolases, carboxypeptidases. |
| Cytokines secreted after appropriate activation: TNF, IL-1, IL-3, IL-4, IL-5, IL-6, IL-8, IL-10, IL-9, IL-11, IL-12, IL-13, IL-15, IL-16, IL-18, IL-25, TGF-β, VEGF, IFN-γ, GM-CSF, (and probably many more). |
| Chemokines: CCL1, CCL2, CCL3, CCL4, CCL7, CCL8, CCL9, CCL17, CCL20, CCL22, CXCL2, CXCL8, Eotaxin 1 and 3. |
| Arachidonic acid mediators after activation: PGD2, PGE2, LTB4, LTC4 |
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conti, P.; Ronconi, G.; Lauritano, D.; Mastrangelo, F.; Caraffa, A.; Gallenga, C.E.; Frydas, I.; Kritas, S.K.; Carinci, F.; Gaudelli, F.; et al. Impact of TNF and IL-33 Cytokines on Mast Cells in Neuroinflammation. Int. J. Mol. Sci. 2024, 25, 3248. https://doi.org/10.3390/ijms25063248
Conti P, Ronconi G, Lauritano D, Mastrangelo F, Caraffa A, Gallenga CE, Frydas I, Kritas SK, Carinci F, Gaudelli F, et al. Impact of TNF and IL-33 Cytokines on Mast Cells in Neuroinflammation. International Journal of Molecular Sciences. 2024; 25(6):3248. https://doi.org/10.3390/ijms25063248
Chicago/Turabian StyleConti, Pio, Gianpaolo Ronconi, Dorina Lauritano, Filiberto Mastrangelo, Alessandro Caraffa, Carla E. Gallenga, Ilias Frydas, Spyridon K. Kritas, Francesco Carinci, Federico Gaudelli, and et al. 2024. "Impact of TNF and IL-33 Cytokines on Mast Cells in Neuroinflammation" International Journal of Molecular Sciences 25, no. 6: 3248. https://doi.org/10.3390/ijms25063248
APA StyleConti, P., Ronconi, G., Lauritano, D., Mastrangelo, F., Caraffa, A., Gallenga, C. E., Frydas, I., Kritas, S. K., Carinci, F., Gaudelli, F., Annicchiarico, C., & D’Ovidio, C. (2024). Impact of TNF and IL-33 Cytokines on Mast Cells in Neuroinflammation. International Journal of Molecular Sciences, 25(6), 3248. https://doi.org/10.3390/ijms25063248