
Repurposed Drugs Celecoxib and Fmoc-L-Leucine Alone and in Combination as Temozolomide-Resistant Antiglioma Agents—Comparative Studies on Normal and Immortalized Cell Lines, and on C. elegans

 ,
,
Abstract
1. Introduction
2. Results and Discussion
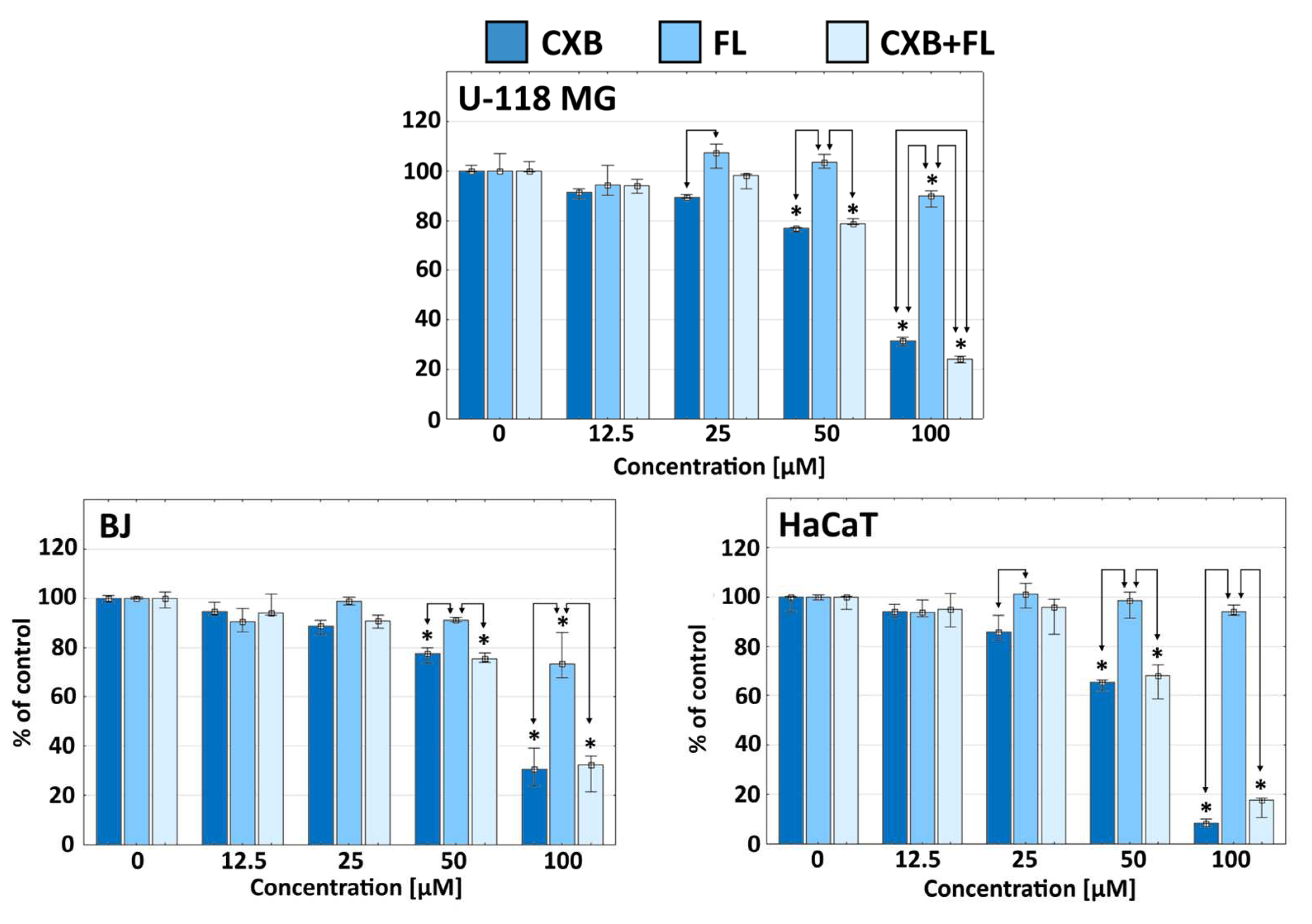
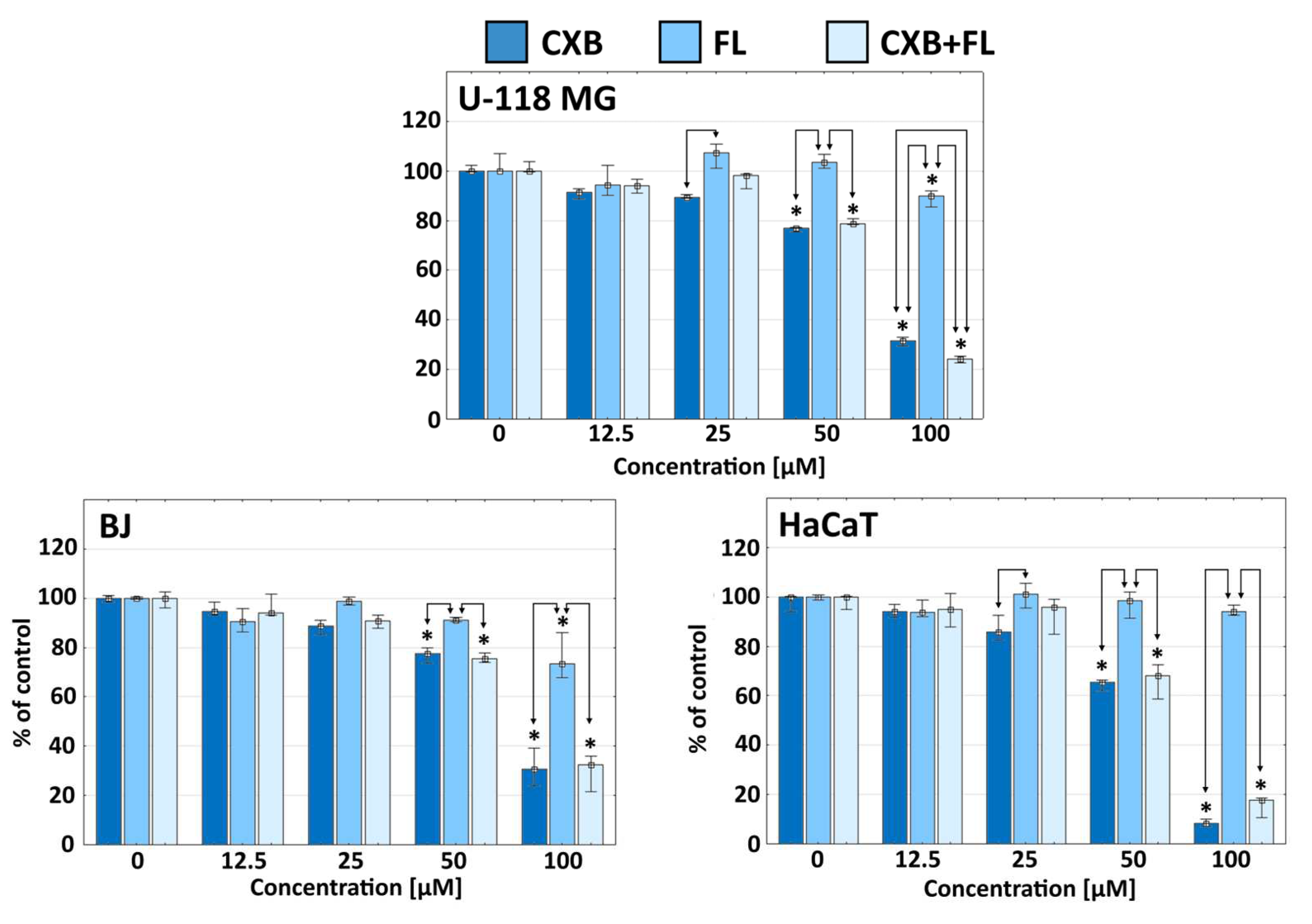
2.1. Cytotoxicity
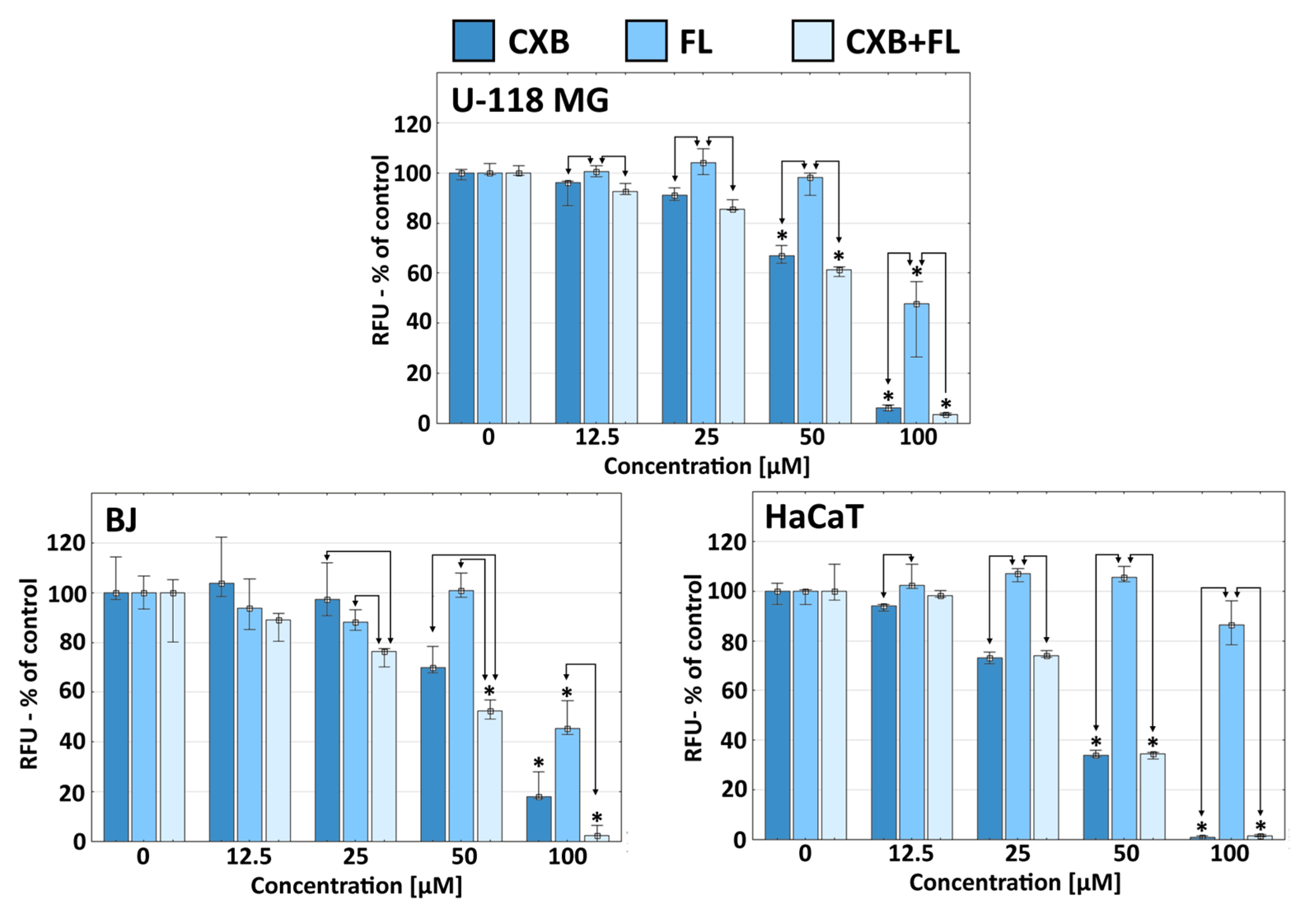
2.2. Proliferation
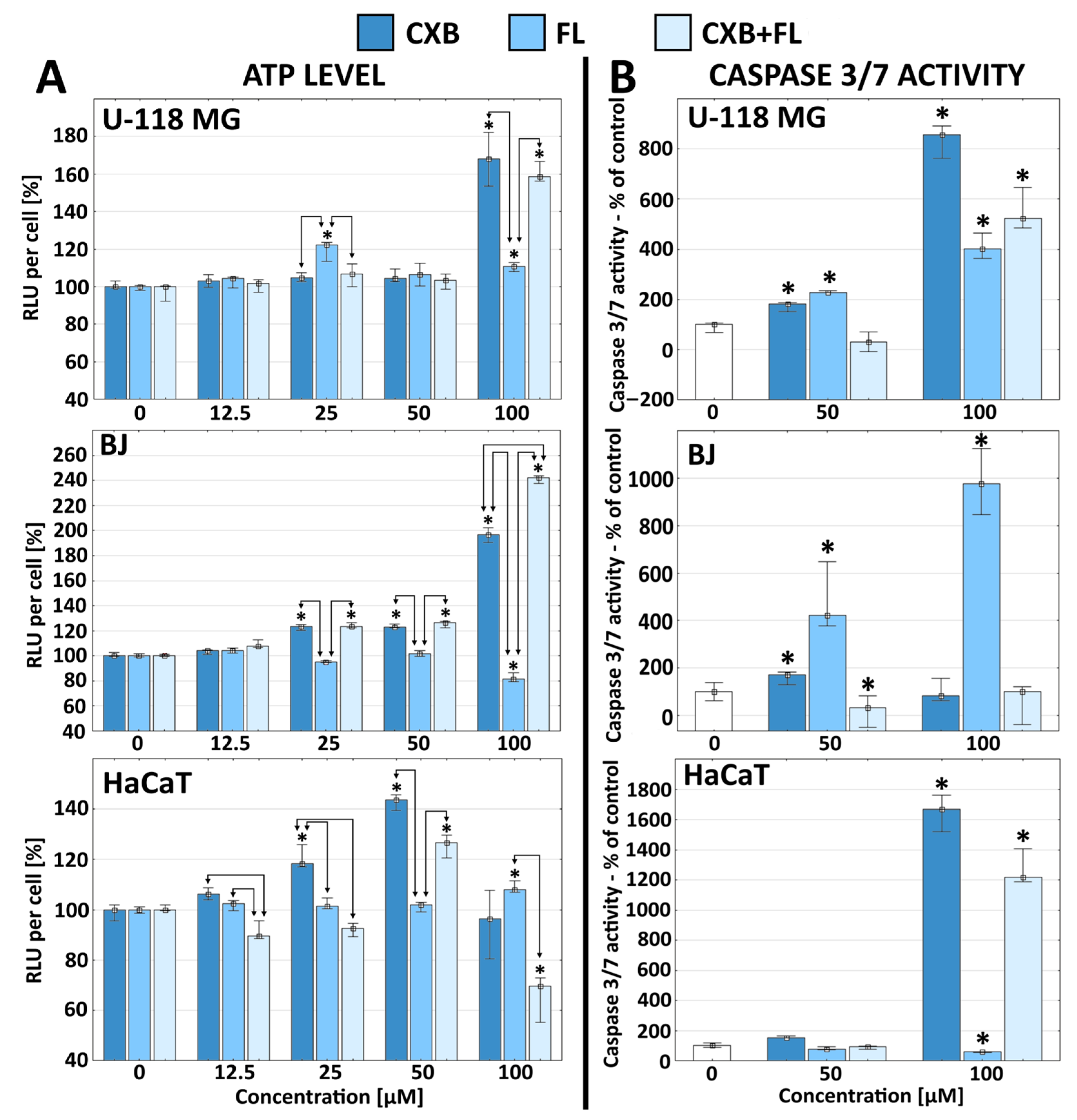
2.3. Apoptosis and ATP Level
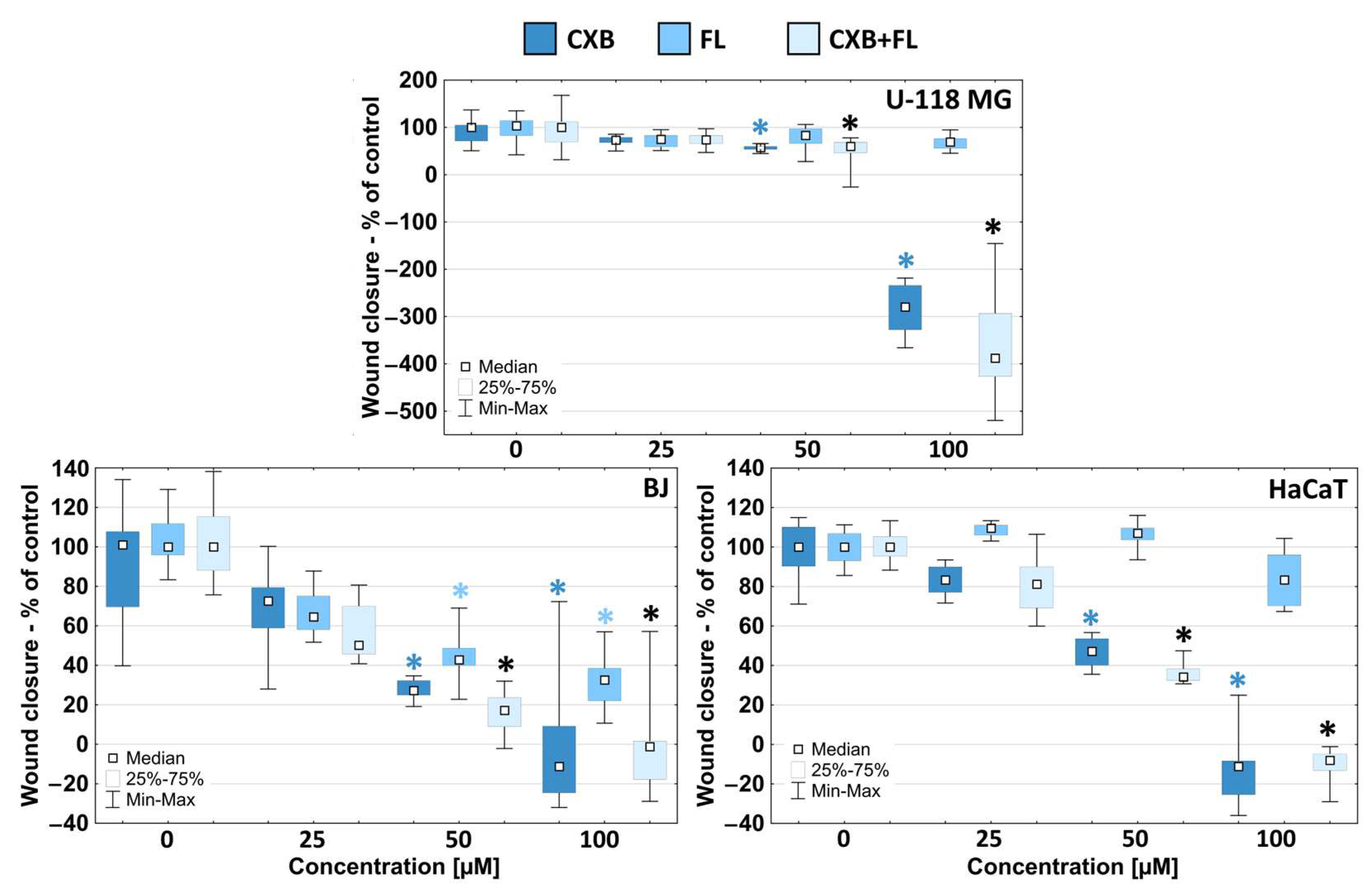
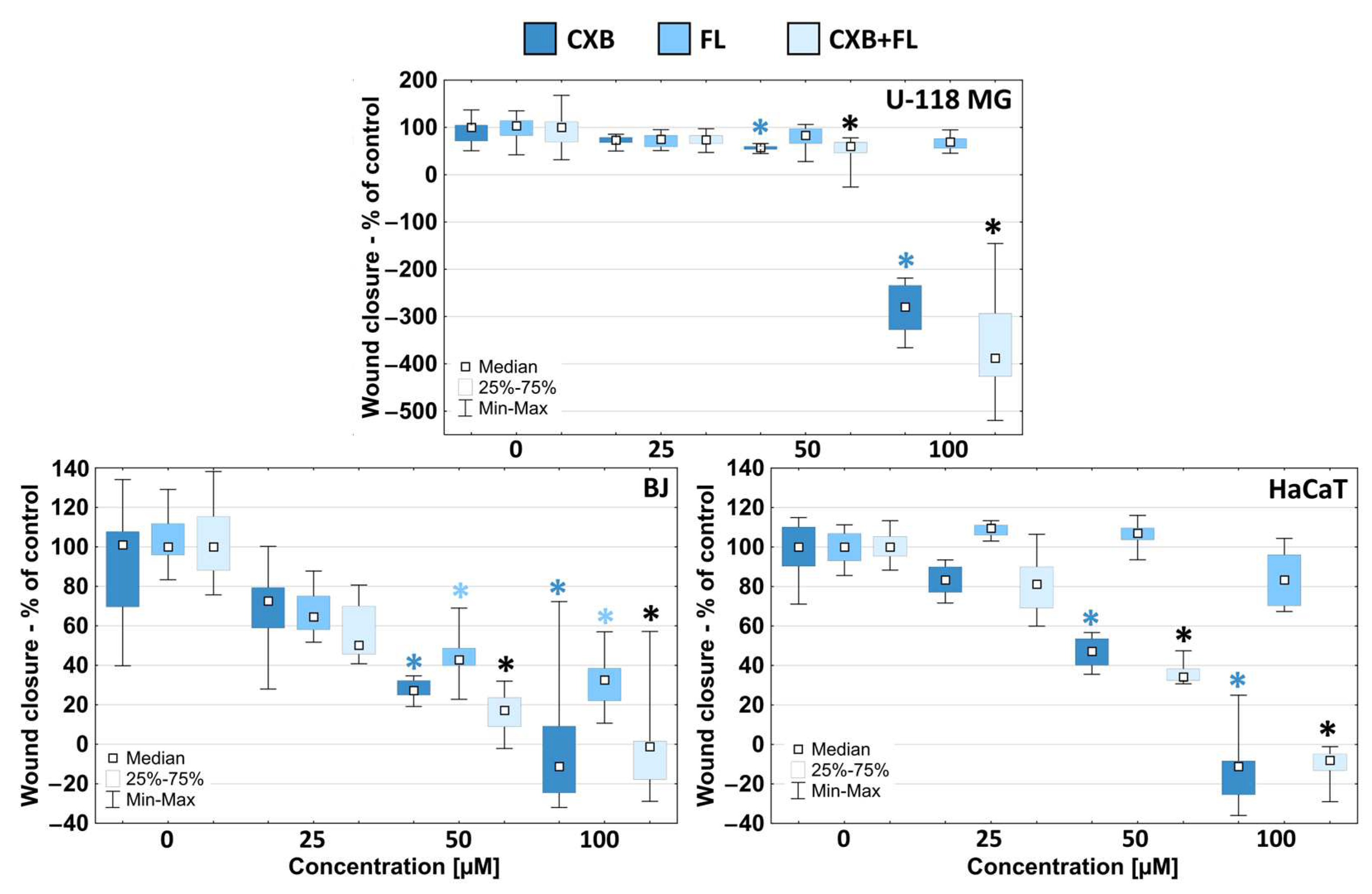
2.4. Migration
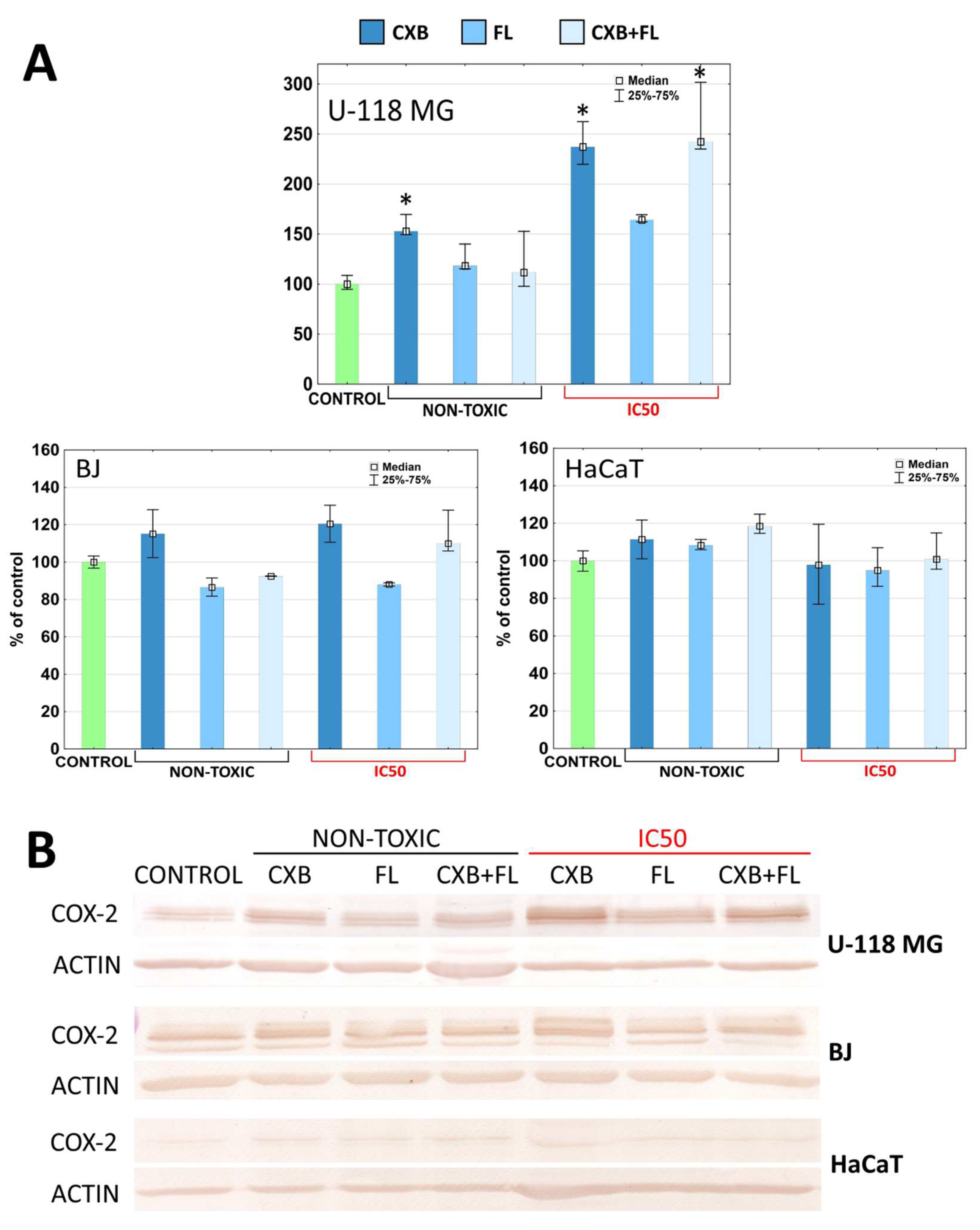
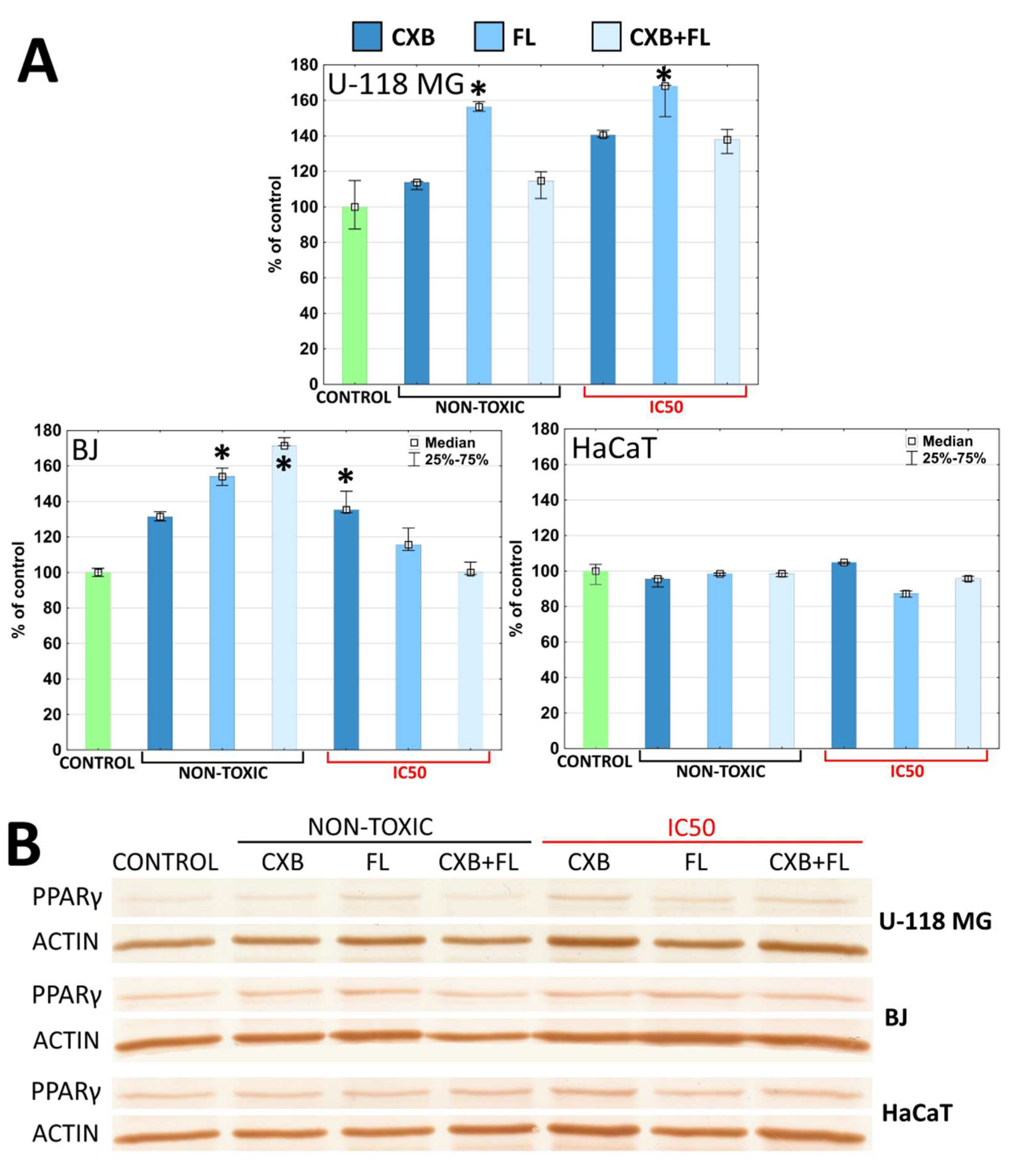
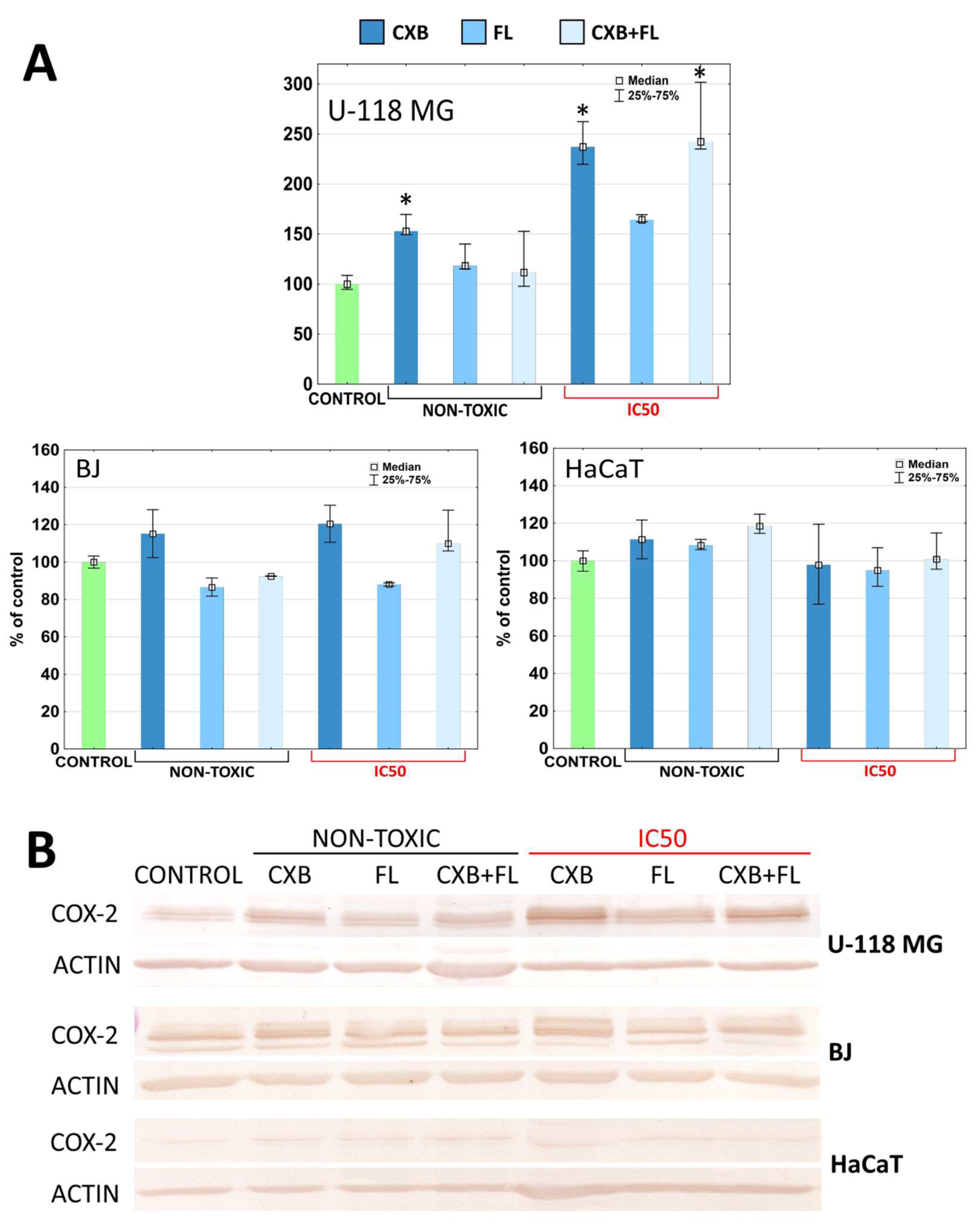
2.5. COX-2 and PPARγ Level
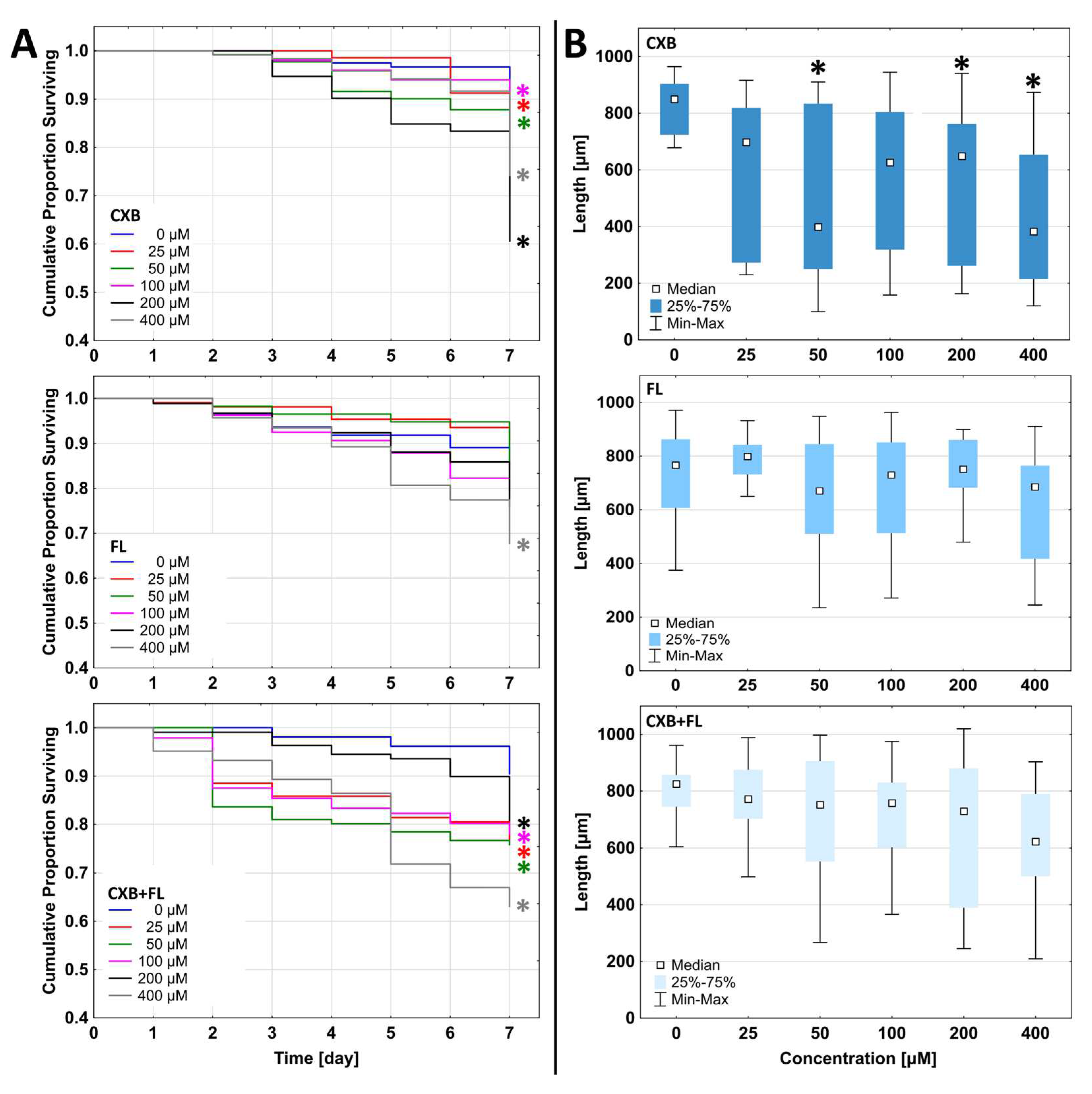
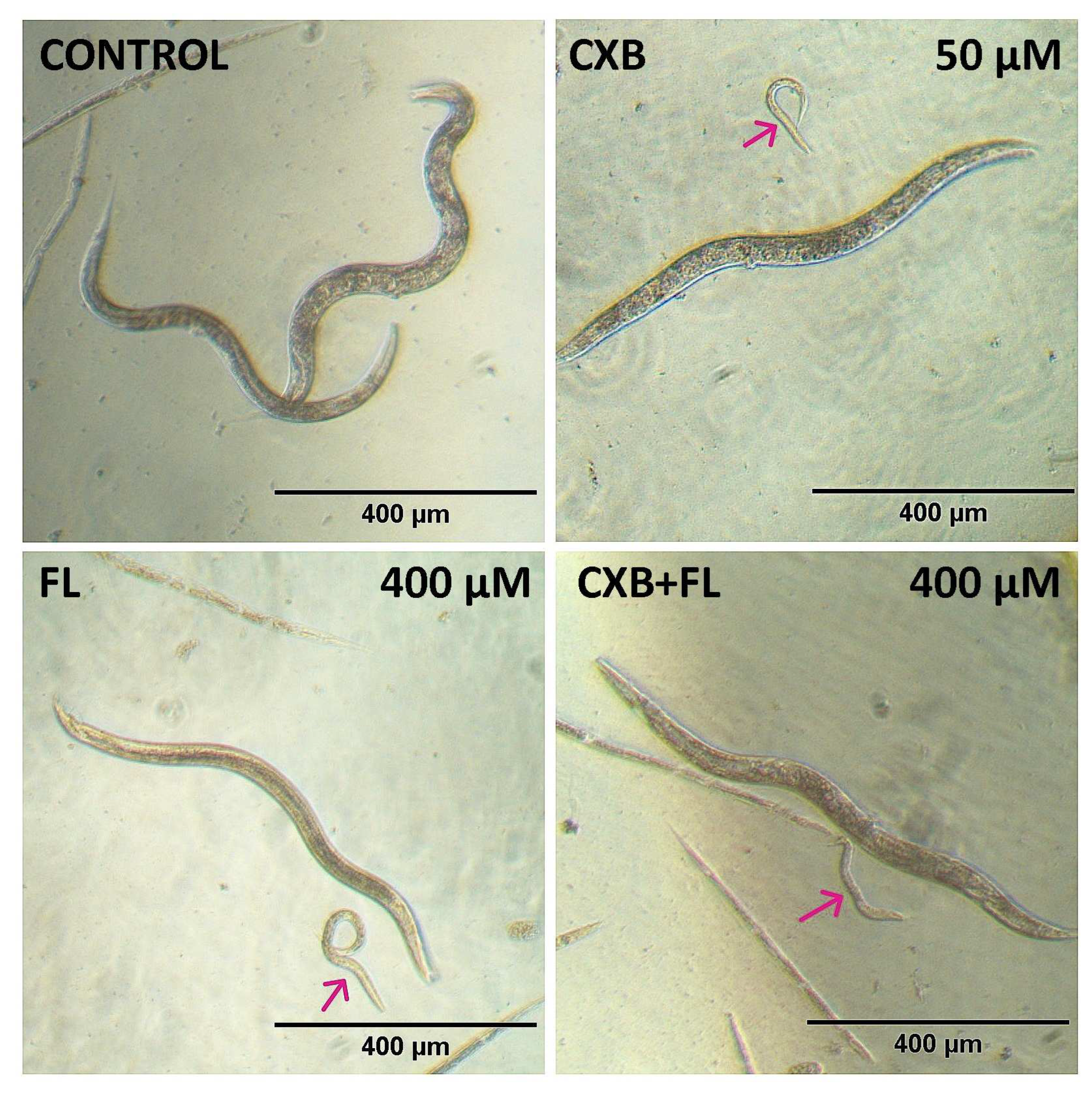
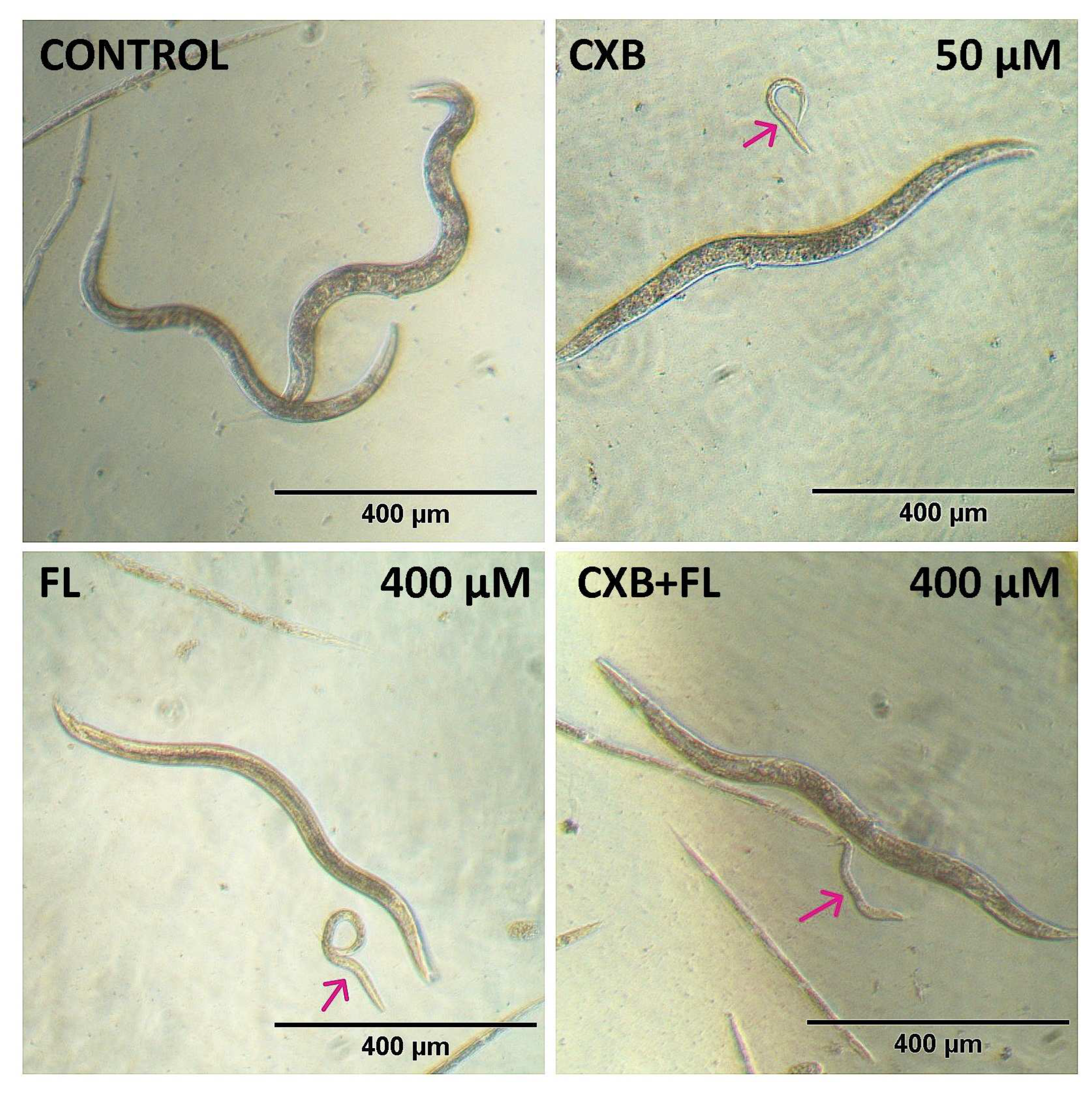
2.6. In Vivo Toxicity
3. Materials and Methods
3.1. Materials
3.2. Cell Cultures
3.3. Cytotoxicity
3.4. Proliferation
3.5. Intracellular ATP Level and Apoptosis
3.6. Migration Assay
3.7. Western Blot Analysis
3.8. In Vivo Toxicity
3.9. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro-Oncology 2022, 24, v1–v95. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Sonnberger, M.; Dunzinger, A.; Voglmayr, E.; Aichholzer, M.; Kleiser, R.; Strasser, P. Imaging Brain Diseases A Neuroradiology, Nuclear Medicine, Neurosurgery, Neuropathology and Molecular Biology-Based Approach; Springer Nature: Berlin, Germany, 2019. [Google Scholar]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Pineda, E.; Domenech, M.; Hernández, A.; Comas, S.; Balaña, C. Recurrent Glioblastoma: Ongoing Clinical Challenges and Future Prospects. OncoTargets Ther. 2023, 16, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Kort, E.; Jovinge, S. Drug Repurposing: Claiming the Full Benefit from Drug Development. Curr. Cardiol. Rep. 2021, 23, 62. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Preuss, C.V. Celecoxib. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Zhang, Z.; Chen, F.; Shang, L. Advances in Antitumor Effects of NSAIDs. Cancer Manag. Res. 2018, 10, 4631–4640. [Google Scholar] [CrossRef] [PubMed]
- Khafaga, A.F.; Shamma, R.N.; Abdeen, A.; Barakat, A.M.; Noreldin, A.E.; Elzoghby, A.O.; Sallam, M.A. Celecoxib Repurposing in Cancer Therapy: Molecular Mechanisms and Nanomedicine-Based Delivery Technologies. Nanomedicine 2021, 16, 1691–1712. [Google Scholar] [CrossRef]
- Yerokun, T.; Winfield, L.L. Celecoxib and LLW-3-6 Reduce Survival of Human Glioma Cells Independently and Synergistically with Sulfasalazine. Anticancer. Res. 2015, 35, 6419–6424. [Google Scholar]
- Kang, K.B.; Zhu, C.; Yong, S.K.; Gao, Q.; Wong, M.C. Enhanced Sensitivity of Celecoxib in Human Glioblastoma Cells: Induction of DNA Damage Leading to P53-Dependent G1 Cell Cycle Arrest and Autophagy. Mol. Cancer 2009, 8, 66. [Google Scholar] [CrossRef]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib Enhances Radiosensitivity of Hypoxic Glioblastoma Cells through Endoplasmic Reticulum Stress. Neuro-Oncology 2013, 15, 1186–1199. [Google Scholar] [CrossRef]
- Ma, H.I.; Chiou, S.H.; Hueng, D.Y.; Tai, L.K.; Huang, P.I.; Kao, C.L.; Chen, Y.W.; Sytwu, H.K. Celecoxib and Radioresistant Glioblastoma-Derived CD133+ Cells: Improvement in Radiotherapeutic Effects. Laboratory Investigation. J. Neurosurg. 2011, 114, 651–662. [Google Scholar] [CrossRef]
- Yin, D.; Jin, G.; He, H.; Zhou, W.; Fan, Z.; Gong, C.; Zhao, J.; Xiong, H. Celecoxib Reverses the Glioblastoma Chemo-Resistance to Temozolomide through Mitochondrial Metabolism. Aging 2021, 13, 21268–21282. [Google Scholar] [CrossRef]
- Tavakolian, F.; Khezrnia, S.S.; Dehghani, M.; Mansouri, A. The Role of Celecoxib in Glioblastoma Treatment—A Review of Literature. World Cancer Res. J. 2020, 7, e1617. [Google Scholar]
- Ahmadi, M.; Bekeschus, S.; Weltmann, K.D.; von Woedtke, T.; Wende, K. Non-Steroidal Anti-Inflammatory Drugs: Recent Advances in the Use of Synthetic COX-2 Inhibitors. RSC Med. Chem. 2022, 13, 471–496. [Google Scholar] [CrossRef]
- Yao, H.; Guo, Q.; Wang, M.; Wang, R.; Xu, Z. Discovery of Pyrazole N-Aryl Sulfonate: A Novel and Highly Potent Cyclooxygenase-2 (COX-2) Selective Inhibitors. Bioorganic Med. Chem. 2021, 46, 116344. [Google Scholar] [CrossRef]
- Sohrab, S.S.; Raj, R.; Nagar, A.; Hawthorne, S.; Paiva-Santos, A.C.; Kamal, M.A.; El-Daly, M.M.; Azhar, E.I.; Sharma, A. Chronic Inflammation’s Transformation to Cancer: A Nanotherapeutic Paradigm. Molecules 2023, 28, 4413. [Google Scholar] [CrossRef]
- Dean, P.T.; Hooks, S.B. Pleiotropic Effects of the COX-2/PGE2 Axis in the Glioblastoma Tumor Microenvironment. Front. Oncol. 2023, 12, 1116014. [Google Scholar] [CrossRef]
- Lombardi, F.; Augello, F.R.; Artone, S.; Gugu, M.K.; Cifone, M.G.; Cinque, B.; Palumbo, P. Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines. Int. J. Mol. Sci. 2022, 23, 1545. [Google Scholar] [CrossRef]
- Wu, M.; Guan, J.; Li, C.; Gunter, S.; Nusrat, L.; Ng, S.; Dhand, K.; Morshead, C.; Kim, A.; Das, S. Aberrantly Activated Cox-2 and Wnt Signaling Interact to Maintain Cancer Stem Cells in Glioblastoma. Oncotarget 2017, 8, 82217–82230. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak-Celińska, A.; Misiorek, J.O.; Kruhlenia, N.; Przybyl, L.; Kleszcz, R.; Rolle, K.; Krajka-Kuźniak, V. COXIBs and 2,5-Dimethylcelecoxib Counteract the Hyperactivated Wnt/β-Catenin Pathway and COX-2/PGE2/EP4 Signaling in Glioblastoma Cells. BMC Cancer 2021, 21, 493. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Quiles, M.; Broekema, M.F.; Kalkhoven, E. PPARgamma in Metabolism, Immunity, and Cancer: Unified and Diverse Mechanisms of Action. Front. Endocrinol. 2021, 12, 624112. [Google Scholar] [CrossRef]
- Basilotta, R.; Lanza, M.; Casili, G.; Chisari, G.; Munao, S.; Colarossi, L.; Cucinotta, L.; Campolo, M.; Esposito, E.; Paterniti, I. Potential Therapeutic Effects of PPAR Ligands in Glioblastoma. Cells 2022, 11, 621. [Google Scholar] [CrossRef]
- Heudobler, D.; Rechenmacher, M.; Lüke, F.; Vogelhuber, M.; Pukrop, T.; Herr, W.; Ghibelli, L.; Gerner, C.; Reichle, A. Peroxisome Proliferator-Activated Receptors (PPAR)γ Agonists as Master Modulators of Tumor Tissue. Int. J. Mol. Sci. 2018, 19, 3540. [Google Scholar] [CrossRef]
- Rocchi, S.; Picard, F.; Vamecq, J.; Gelman, L.; Potier, N.; Zeyer, D.; Dubuquoy, L.; Bac, P.; Champy, M.F.; Plunket, K.D.; et al. A Unique PPARγ Ligand with Potent Insulin-Sensitizing yet Weak Adipogenic Activity. Mol. Cell 2001, 8, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Ballav, S.; Biswas, B.; Sahu, V.K.; Ranjan, A.; Basu, S. PPAR-γ Partial Agonists in Disease-Fate Decision with Special Reference to Cancer. Cells 2022, 11, 3215. [Google Scholar] [CrossRef]
- Sheu, M.L.; Pan, L.Y.; Hu, H.Y.; Su, H.L.; Sheehan, J.; Tsou, H.K.; Pan, H.C. Potential Therapeutic Effects of Thiazolidinedione on Malignant Glioma. Int. J. Mol. Sci. 2022, 23, 13510. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Knopfová, L.; Smarda, J. The Use of Cox-2 and PPARγ Signaling in Anti-Cancer Therapies. Exp. Ther. Med. 2010, 1, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Hau, P.; Kunz-Schughart, L.; Bogdahn, U.; Baumgart, U.; Hirschmann, B.; Weimann, E.; Muhleisen, H.; Ruemmele, P.; Steinbrecher, A.; Reichle, A. Low-Dose Chemotherapy in Combination with COX-2 Inhibitors and PPAR-Gamma Agonists in Recurrent High-Grade Gliomas—A Phase II Study. Oncology 2007, 73, 21–25. [Google Scholar] [CrossRef]
- Carmo, A.; Carvalheiro, H.; Crespo, I.; Nunes, I.; Lopes, M.C. Effect of Temozolomide on the U-118 Glioma Cell Line. Oncol. Lett. 2011, 2, 1165–1170. [Google Scholar] [CrossRef]
- Li, L.; Hu, Y.; Ylivinkka, I.; Li, H.; Chen, P.; Keski-Oja, J.; Hyytiäinen, M. NETRIN-4 Protects Glioblastoma Cells FROM Temozolomide Induced Senescence. PLoS ONE 2013, 8, e80363. [Google Scholar] [CrossRef]
- Ye, X.; Liu, X.; Gao, M.; Gong, L.; Tian, F.; Shen, Y.; Hu, H.; Sun, G.; Zou, Y.; Gong, Y. CUL4B Promotes Temozolomide Resistance in Gliomas by Epigenetically Repressing CDNK1A Transcription. Front. Oncol. 2021, 11, 638802. [Google Scholar] [CrossRef]
- Sebaugh, J.L. Guidelines for Accurate EC50/IC50 Estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Belousov, A.; Titov, S.; Shved, N.; Garbuz, M.; Malykin, G.; Gulaia, V.; Kagansky, A.; Kumeiko, V. The Extracellular Matrix and Biocompatible Materials in Glioblastoma Treatment. Front. Bioeng. Biotechnol. 2019, 7, 341. [Google Scholar] [CrossRef] [PubMed]
- Vera, M.; Barcia, E.; Negro, S.; Marcianes, P.; García-García, L.; Slowing, K.; Fernández-Carballido, A. New Celecoxib Multiparticulate Systems to Improve Glioblastoma Treatment. Int. J. Pharm. 2014, 473, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, G.E.S.; Akim, A.M.; Sung, Y.Y.; Muhammad, T.S.T. Cancer and Apoptosis. Methods Mol. Biol. 2022, 2543, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Shono, K.; Yamaguchi, I.; Mizobuchi, Y.; Kagusa, H.; Sumi, A.; Fujihara, T.; Nakajima, K.; Kitazato, K.T.; Matsuzaki, K.; Saya, H.; et al. Downregulation of the CCL2/CCR2 and CXCL10/CXCR3 Axes Contributes to Antitumor Effects in a Mouse Model of Malignant Glioma. Sci. Rep. 2020, 10, 15286. [Google Scholar] [CrossRef] [PubMed]
- Elrod, H.A.; Sun, S.Y. PPARγ and Apoptosis in Cancer. PPAR Res. 2008, 2008, 704165. [Google Scholar] [CrossRef] [PubMed]
- Zamaraeva, M.V.; Sabirov, R.Z.; Maeno, E.; Ando-Akatsuka, Y.; Bessonova, S.V.; Okada, Y. Cells Die with Increased Cytosolic ATP during Apoptosis: A Bioluminescence Study with Intracellular Luciferase. Cell Death Differ. 2005, 12, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Apoptosis and Necrosis: Intracellular ATP Level as a Determinant for Cell Death Modes. Cell Death Differ. 1997, 4, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Sakamoto, S.; Yoshida, T.; Matsui, Y.; Penuela, S.; Laird, D.W.; Mizukami, S.; Kikuchi, K.; Kakizuka, A. Single-Cell Dynamics of Pannexin-1-Facilitated Programmed ATP Loss during Apoptosis. eLife 2020, 9, e61960. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.S.; Jiang, J.; Chen, B.J.; Wang, K.; Tang, Y.L.; Liang, X. Plasticity of Cancer Cell Invasion: Patterns and Mechanisms. Transl. Oncol. 2021, 14, 100899. [Google Scholar] [CrossRef]
- Mendes, M.; Branco, F.; Vitorino, R.; Sousa, J.; Pais, A.; Vitorino, C. A Two-Pronged Approach against Glioblastoma: Drug Repurposing and Nanoformulation Design for in Situ-Controlled Release. Drug Deliv. Transl. Res. 2023, 13, 3169. [Google Scholar] [CrossRef] [PubMed]
- Lichtor, T.; Spagnolo, A.; Glick, R.P.; Feinstein, D.L. PPAR-Gamma Thiazolidinedione Agonists and Immunotherapy in the Treatment of Brain Tumors. PPAR Res. 2008, 2008, 547470. [Google Scholar] [CrossRef]
- Qiu, J.; Shi, Z.; Jiang, J. Cyclooxygenase-2 in Glioblastoma Multiforme. Drug Discov. Today 2017, 22, 148–156. [Google Scholar] [CrossRef]
- Zhang, F.; Chu, J.; Wang, F. Expression and Clinical Significance of Cyclooxygenase 2 and Survivin in Human Gliomas. Oncol. Lett. 2017, 14, 1303–1308. [Google Scholar] [CrossRef]
- Uram, Ł.; Misiorek, M.; Pichla, M.; Filipowicz-Rachwał, A.; Markowicz, J.; Wołowiec, S.; Wałajtys-Rode, E. The Effect of Biotinylated PAMAM G3 Dendrimers Conjugated with COX-2 Inhibitor (Celecoxib) and PPARγ Agonist (Fmoc-L-Leucine) on Human Normal Fibroblasts, Immortalized Keratinocytes and Glioma Cells in Vitro. Molecules 2019, 24, 3801. [Google Scholar] [CrossRef]
- Uram, Ł.; Filipowicz, A.; Misiorek, M.; Pieńkowska, N.; Markowicz, J.; Wałajtys-Rode, E.; Wołowiec, S. Biotinylated PAMAM G3 Dendrimer Conjugated with Celecoxib and/or Fmoc-l-Leucine and Its Cytotoxicity for Normal and Cancer Human Cell Lines. Eur. J. Pharm. Sci. 2018, 124, 1–9. [Google Scholar] [CrossRef]
- Levitt, R.J.; Buckley, J.; Blouin, M.-J.; Schaub, B.; Triche, T.J.; Pollak, M. Growth Inhibition of Breast Epithelial Cells by Celecoxib Is Associated with Upregulation of Insulin-like Growth Factor Binding Protein-3 Expression. Biochem. Biophys. Res. Commun. 2004, 316, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.-H.; Choi, G.J.; Lee, J.H.; Han, Y.A.; Lim, S.J.; Kim, S.H. Differential Effects of Selective Cyclooxygenase-2 Inhibitors in Inhibiting Proliferation and Induction of Apoptosis in Oral Squamous Cell Carcinoma. Oncol. Rep. 2008, 19, 425–433. [Google Scholar] [CrossRef]
- Lanza-Jacoby, S.; Dicker, A.P.; Miller, S.; Rosato, F.E.; Flynn, J.T.; Lavorgna, S.N.; Burd, R. Cyclooxygenase (COX)-2-Dependent Effects of the Inhibitor SC236 When Combined with Ionizing Radiation in Mammary Tumor Cells Derived from HER-2/Neu Mice. Mol. Cancer Ther. 2004, 3, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, F.; Augello, F.R.; Artone, S.; Ayroldi, E.; Giusti, I.; Dolo, V.; Cifone, M.G.; Cinque, B.; Palumbo, P. Cyclooxygenase-2 Upregulated by Temozolomide in Glioblastoma Cells Is Shuttled in Extracellular Vesicles Modifying Recipient Cell Phenotype. Front. Oncol. 2022, 12, 933746. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Chen, Y.; Su, Y.H.; Tseng, S.H. Celecoxib Increased Expression of 14-3-3sigma and Induced Apoptosis of Glioma Cells. Anticancer. Res. 2007, 27, 2547–2554. [Google Scholar]
- Kato, M.; Nagaya, T.; Fujieda, M.; Saito, K.; Yoshida, J.; Seo, H. Expression of PPARγ and Its Ligand-dependent Growth Inhibition in Human Brain Tumor Cell Lines. Jpn. J. Cancer Res. 2002, 93, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Ching, J.; Amiridis, S.; Stylli, S.S.; Bjorksten, A.R.; Kountouri, N.; Zheng, T.; Paradiso, L.; Luwor, R.B.; Morokoff, A.P.; O’Brien, T.J.; et al. The Peroxisome Proliferator Activated Receptor Gamma Agonist Pioglitazone Increases Functional Expression of the Glutamate Transporter Excitatory Amino Acid Transporter 2 (EAAT2) in Human Glioblastoma Cells. Oncotarget 2015, 6, 21301–21314. [Google Scholar] [CrossRef]
- Chu, T.H.; Chan, H.H.; Kuo, H.M.; Liu, L.F.; Hu, T.H.; Sun, C.K.; Kung, M.L.; Lin, S.W.; Wang, E.M.; Ma, Y.L.; et al. Celecoxib Suppresses Hepatoma Stemness and Progression by Up-Regulating PTEN. Oncotarget 2014, 5, 1475–1490. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.R. The C. elegans Model in Toxicity Testing. J. Appl. Toxicol. 2017, 37, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Kobet, R.A.; Pan, X.; Zhang, B.; Pak, S.C.; Asch, A.S.; Lee, M.-H. Caenorhabditis elegans: A Model System for Anti-Cancer Drug Discovery and Therapeutic Target Identification. Biomol. Ther. 2014, 22, 371–383. [Google Scholar] [CrossRef]
- Wittkowski, P.; Marx-Stoelting, P.; Violet, N.; Fetz, V.; Schwarz, F.; Oelgeschläger, M.; Schönfelder, G.; Vogl, S. Caenorhabditis elegans as a Promising Alternative Model for Environmental Chemical Mixture Effect Assessment—A Comparative Study. Environ. Sci. Technol. 2019, 53, 12725–12733. [Google Scholar] [CrossRef]
- Hunt, P.R.; Olejnik, N.; Sprando, R.L. Toxicity Ranking of Heavy Metals with Screening Method Using Adult Caenorhabditis elegans and Propidium Iodide Replicates Toxicity Ranking in Rat. Food Chem. Toxicol. 2012, 50, 3280–3290. [Google Scholar] [CrossRef]
- Cole, R.D.; Anderson, G.L.; Williams, P.L. The Nematode Caenorhabditis elegans as a Model of Organophosphate-Induced Mammalian Neurotoxicity. Toxicol. Appl. Pharmacol. 2004, 194, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Melstrom, P.C.; Williams, P.L. Reversible AChE Inhibitors in C. elegans vs. Rats, Mice. Biochem. Biophys. Res. Commun. 2007, 357, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Gruber, J.; Ng, L.F.; Poovathingal, S.K.; Halliwell, B. Deceptively Simple but Simply Deceptive—Caenorhabditis elegans Lifespan Studies: Considerations for Aging and Antioxidant Effects. FEBS Lett. 2009, 583, 3377–3387. [Google Scholar] [CrossRef]
- So, S.; Miyahara, K.; Ohshima, Y. Control of Body Size in C. elegans Dependent on Food and Insulin/IGF-1 Signal. Genes Cells 2011, 16, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.T.; Hu, P.J. Insulin/Insulin-like Growth Factor Signaling in C. elegans; WormBook: Pasadena, CA, USA, 2013; pp. 1–43. [Google Scholar] [CrossRef]
- McCulloch, D.; Gems, D. Body Size, Insulin/IGF Signaling and Aging in the Nematode Caenorhabditis elegans. Exp. Gerontol. 2003, 38, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Liao, S.; Zou, Y.; Qu, Z.; Liu, F. Ins-7 Gene Expression Is Partially Regulated by the DAF-16/IIS Signaling Pathway in Caenorhabditis elegans under Celecoxib Intervention. PLoS ONE 2014, 9, e100320. [Google Scholar] [CrossRef]
- Ching, T.T.; Chiang, W.C.; Chen, C.S.; Hsu, A.L. Celecoxib Extends C. elegans Lifespan via Inhibition of Insulin-like Signaling but Not Cyclooxygenase-2 Activity. Aging Cell 2011, 10, 506–519. [Google Scholar] [CrossRef]
- Jia, W.; Wang, C.; Zheng, J.; Li, Y.; Yang, C.; Wan, Q.L.; Shen, J. Pioglitazone Hydrochloride Extends the Lifespan of Caenorhabditis elegans by Activating DAF-16/FOXO- and SKN-1/NRF2-Related Signaling Pathways. Oxid. Med. Cell Longev. 2022, 2022, e8496063. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral Red Uptake Assay for the Estimation of Cell Viability/Cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Stompor, M.; Uram, Ł.; Podgórski, R. In Vitro Effect of 8-Prenylnaringenin and Naringenin on Fibroblasts and Glioblastoma Cells-Cellular Accumulation and Cytotoxicity. Molecules 2017, 22, 1092. [Google Scholar] [CrossRef] [PubMed]
- Uram, Ł.; Szuster, M.; Misiorek, M.; Filipowicz, A.; Wołowiec, S.; Wałajtys-Rode, E. The Effect of G3 PAMAM Dendrimer Conjugated with B-Group Vitamins on Cell Morphology, Motility and ATP Level in Normal and Cancer Cells. Eur. J. Pharm. Sci. 2017, 102, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, L.D.; Lund, S.P.; Coskun, S.H.; Hanna, S.K.; Johnson, M.E.; Sims, C.M.; Brignoni, K.; Lapasset, P.; Petersen, E.J.; Elliott, J.T.; et al. Counting Caenorhabditis elegans: Protocol Optimization and Applications for Population Growth and Toxicity Studies in Liquid Medium. Sci. Rep. 2018, 8, 904. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 [µM] | |||
|---|---|---|---|
| U-118 MG | BJ | HaCaT | |
| CXB | 78.56 | 77.61 | 59.52 |
| FL | 781 | 400 | ≫ |
| CXB+FL | 73.49 | 77.82 | 63.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uram, Ł.; Pieńkowska, N.; Misiorek, M.; Szymaszek, Ż.; Twardowska, M.; Siorek, M.; Wołowiec, S. Repurposed Drugs Celecoxib and Fmoc-L-Leucine Alone and in Combination as Temozolomide-Resistant Antiglioma Agents—Comparative Studies on Normal and Immortalized Cell Lines, and on C. elegans. Int. J. Mol. Sci. 2024, 25, 3226. https://doi.org/10.3390/ijms25063226
Uram Ł, Pieńkowska N, Misiorek M, Szymaszek Ż, Twardowska M, Siorek M, Wołowiec S. Repurposed Drugs Celecoxib and Fmoc-L-Leucine Alone and in Combination as Temozolomide-Resistant Antiglioma Agents—Comparative Studies on Normal and Immortalized Cell Lines, and on C. elegans. International Journal of Molecular Sciences. 2024; 25(6):3226. https://doi.org/10.3390/ijms25063226
Chicago/Turabian StyleUram, Łukasz, Natalia Pieńkowska, Maria Misiorek, Żaneta Szymaszek, Magdalena Twardowska, Michał Siorek, and Stanisław Wołowiec. 2024. "Repurposed Drugs Celecoxib and Fmoc-L-Leucine Alone and in Combination as Temozolomide-Resistant Antiglioma Agents—Comparative Studies on Normal and Immortalized Cell Lines, and on C. elegans" International Journal of Molecular Sciences 25, no. 6: 3226. https://doi.org/10.3390/ijms25063226
APA StyleUram, Ł., Pieńkowska, N., Misiorek, M., Szymaszek, Ż., Twardowska, M., Siorek, M., & Wołowiec, S. (2024). Repurposed Drugs Celecoxib and Fmoc-L-Leucine Alone and in Combination as Temozolomide-Resistant Antiglioma Agents—Comparative Studies on Normal and Immortalized Cell Lines, and on C. elegans. International Journal of Molecular Sciences, 25(6), 3226. https://doi.org/10.3390/ijms25063226