
Glut-3 Gene Knockdown as a Potential Strategy to Overcome Glioblastoma Radioresistance


 ,
,  ,
,  ,
,  ,
,  , and
, and 
Abstract
1. Introduction
2. Results
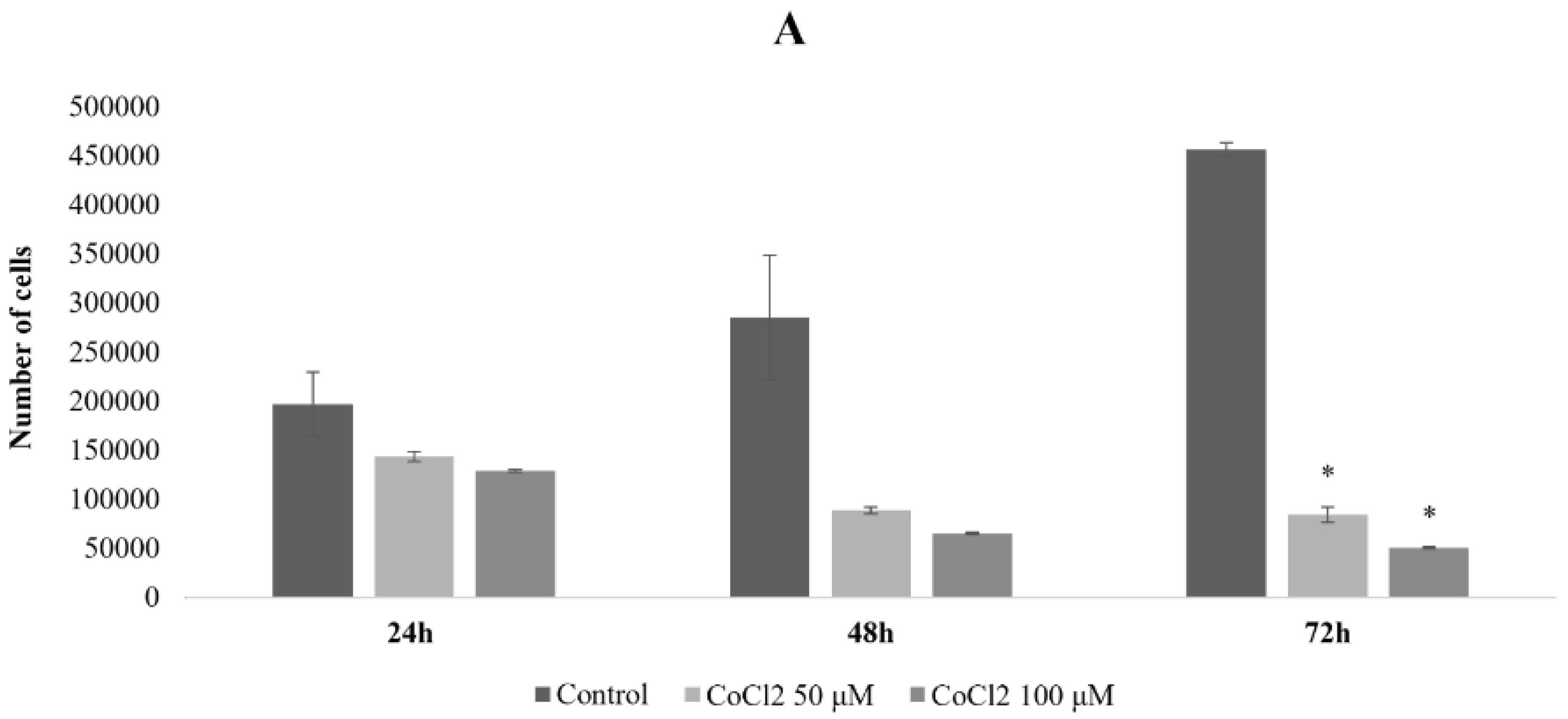
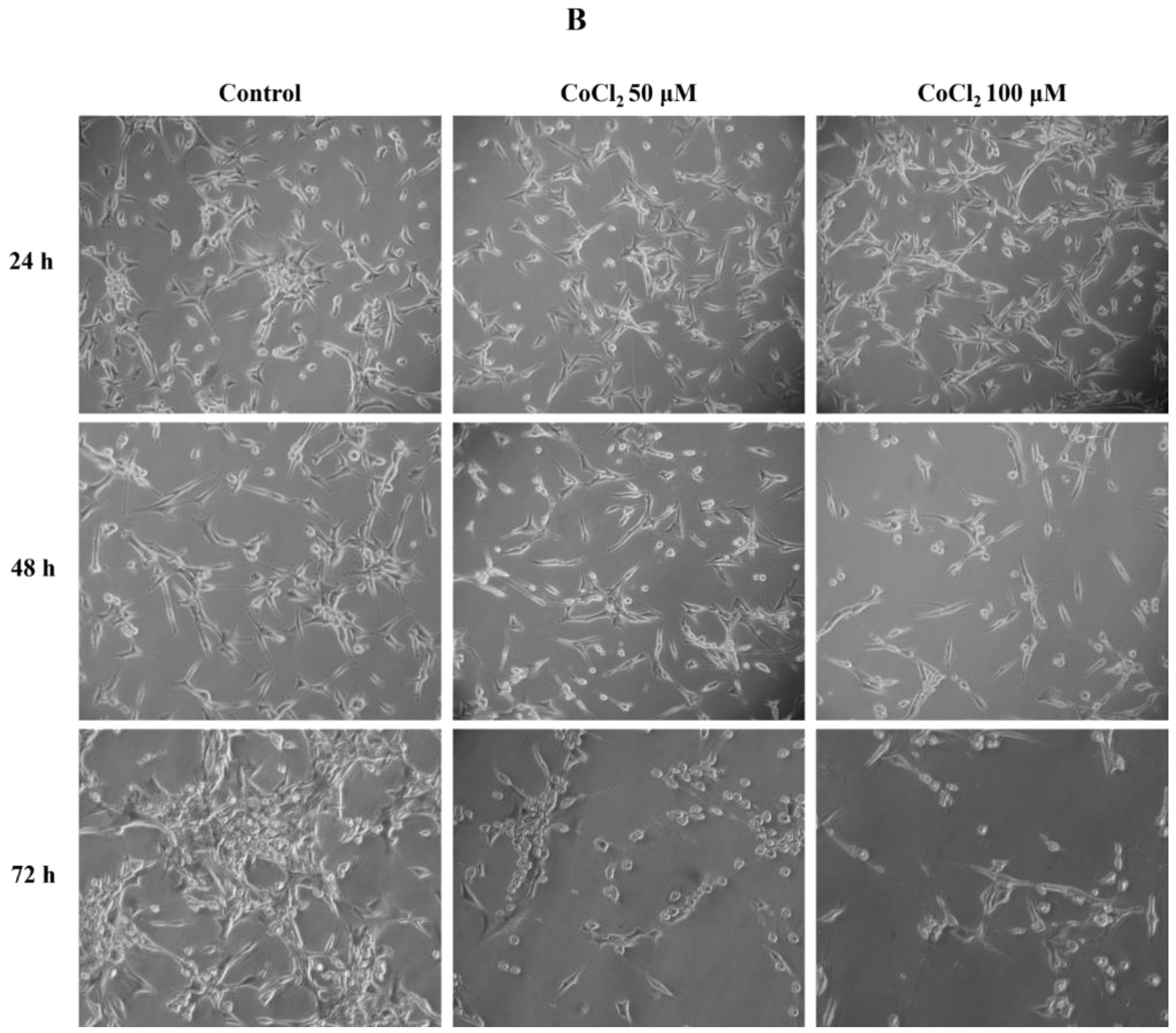
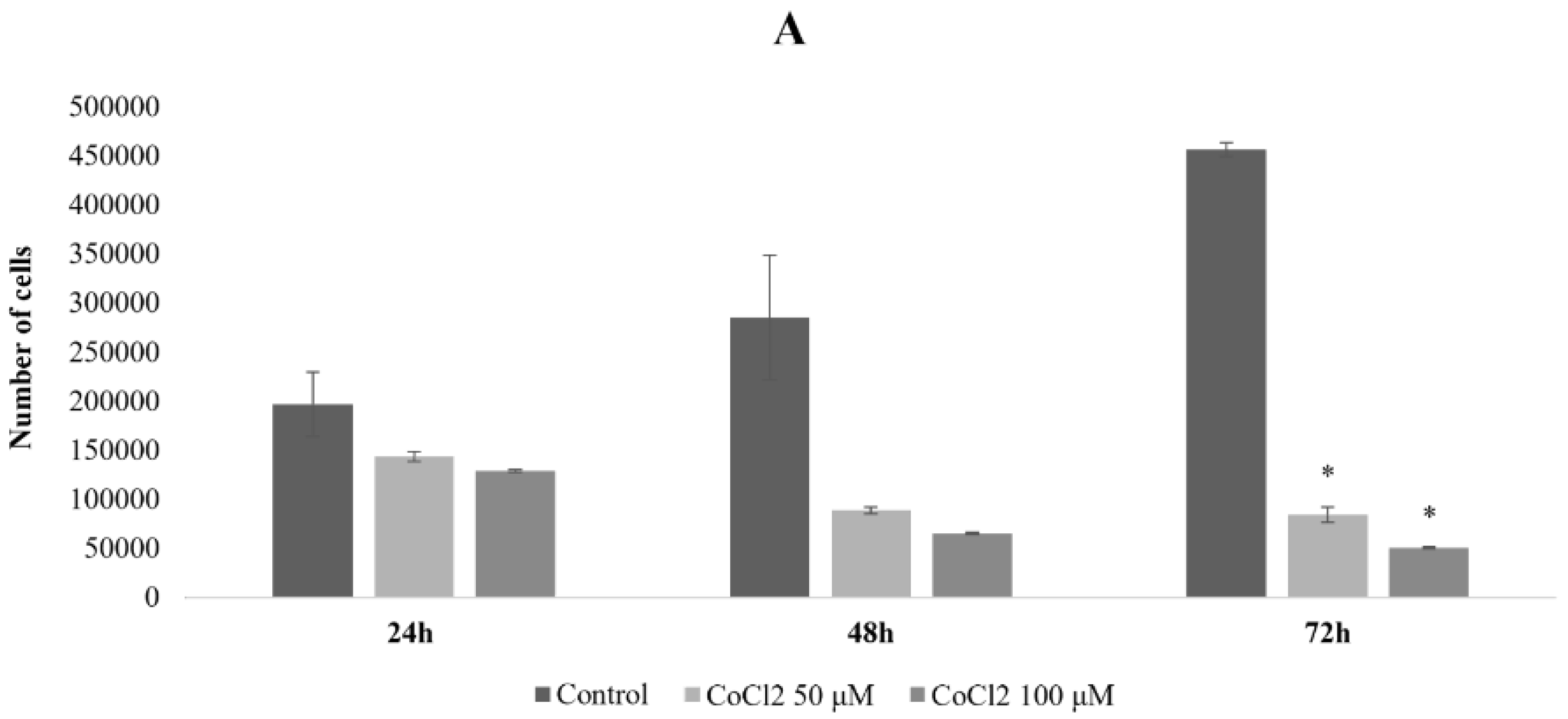
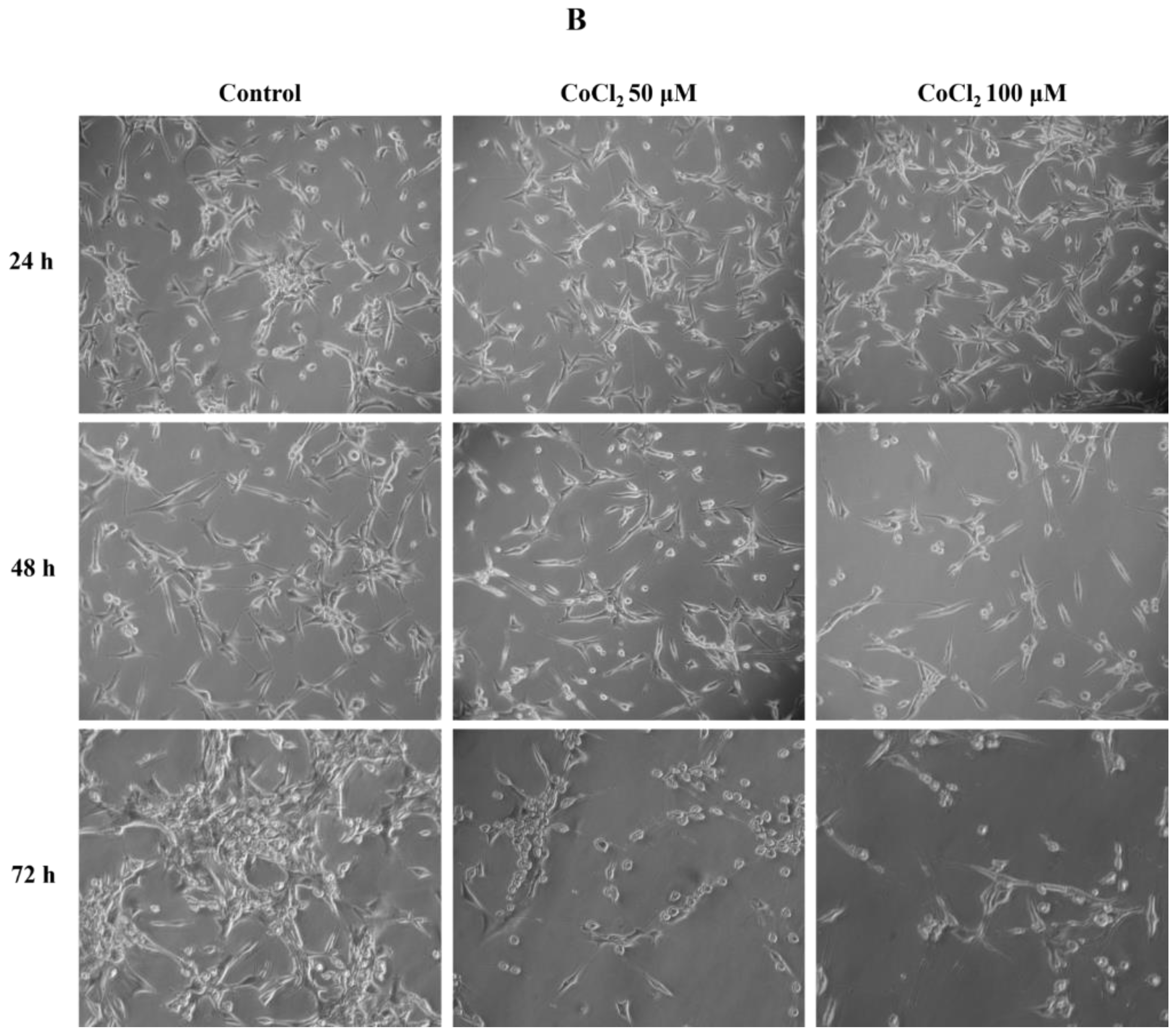
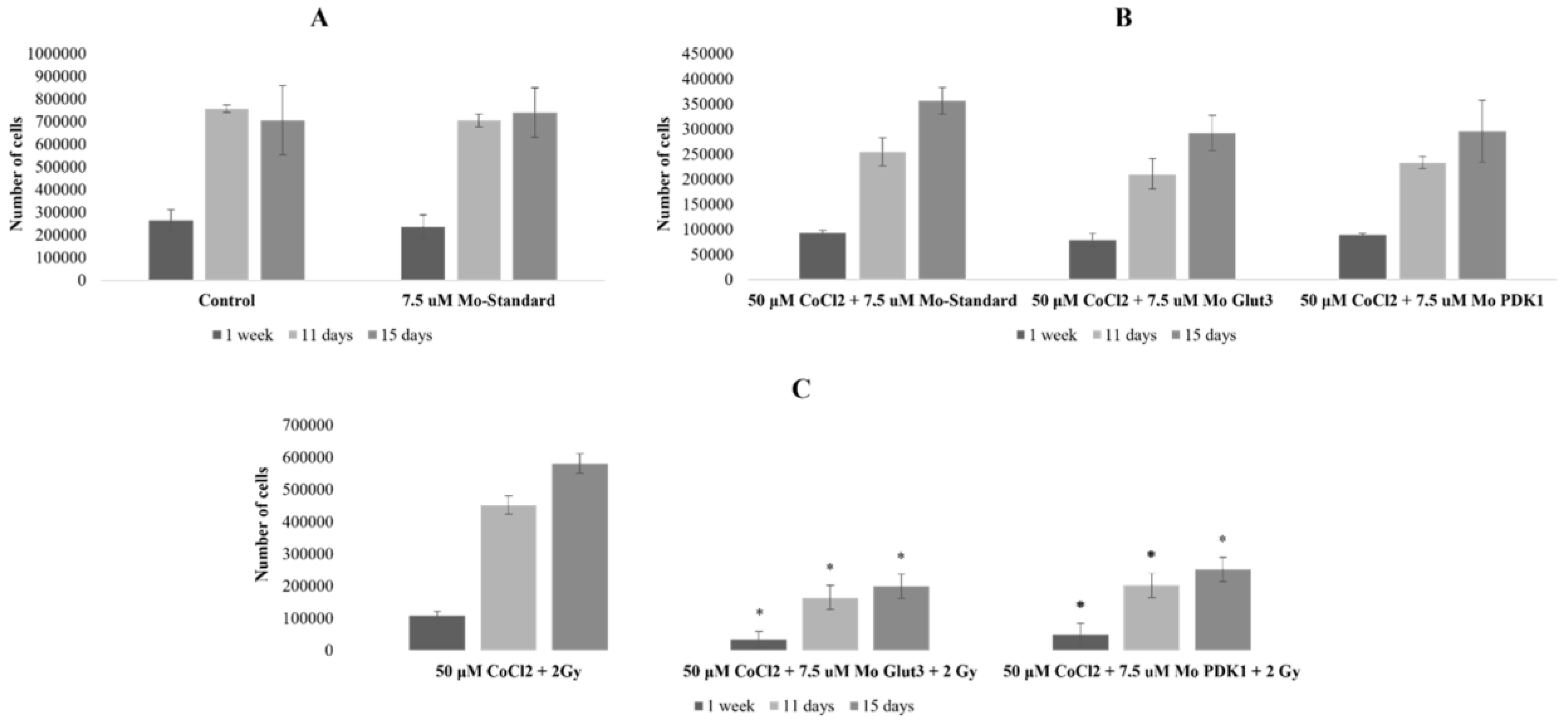
2.1. Effects of CoCl2 on U87 Cell Proliferation and Morphology
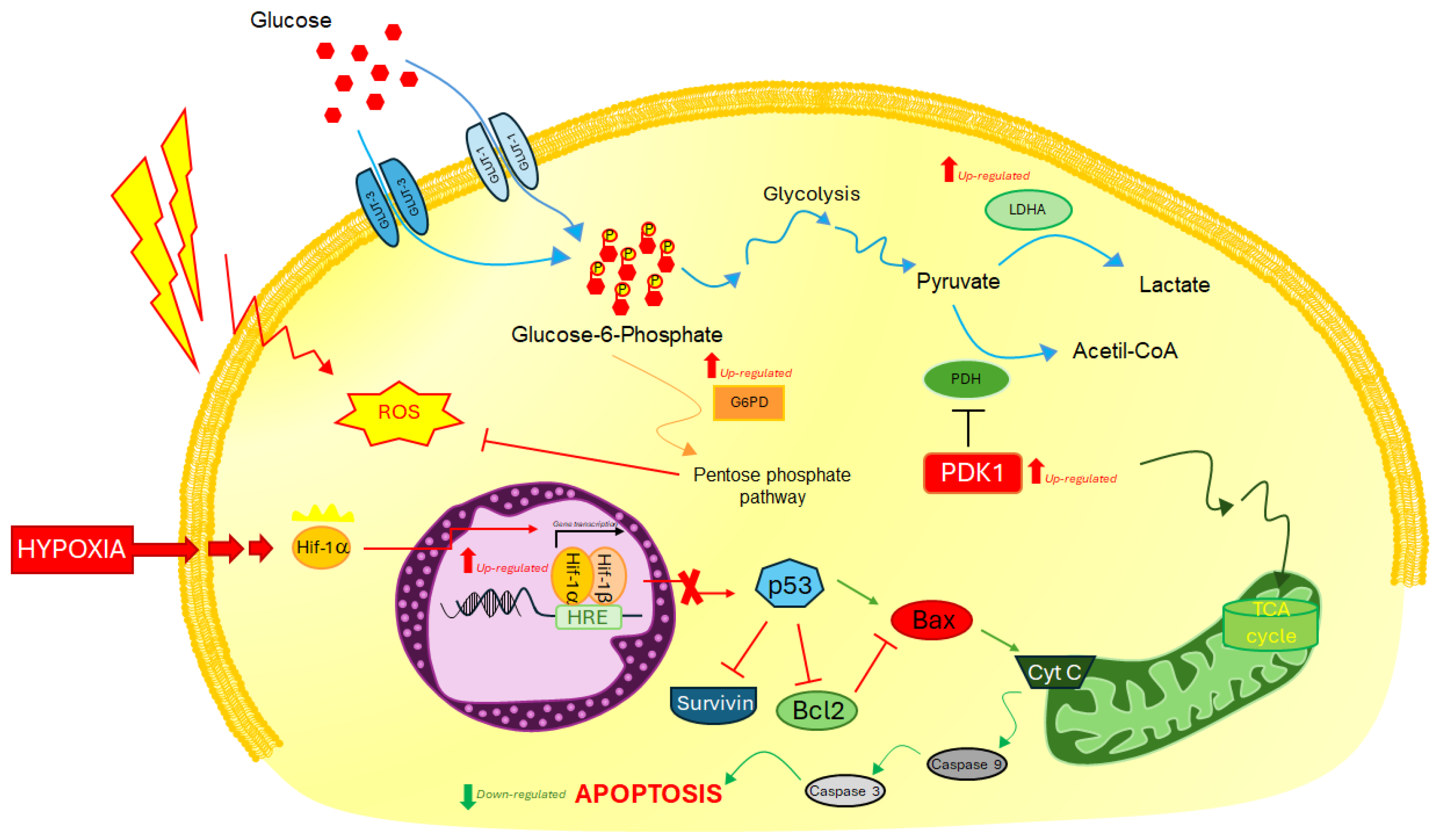
2.2. Analysis of Molecular Response to Induced Hypoxia through Specific HIF-1ɑ Downstream Biomarkers
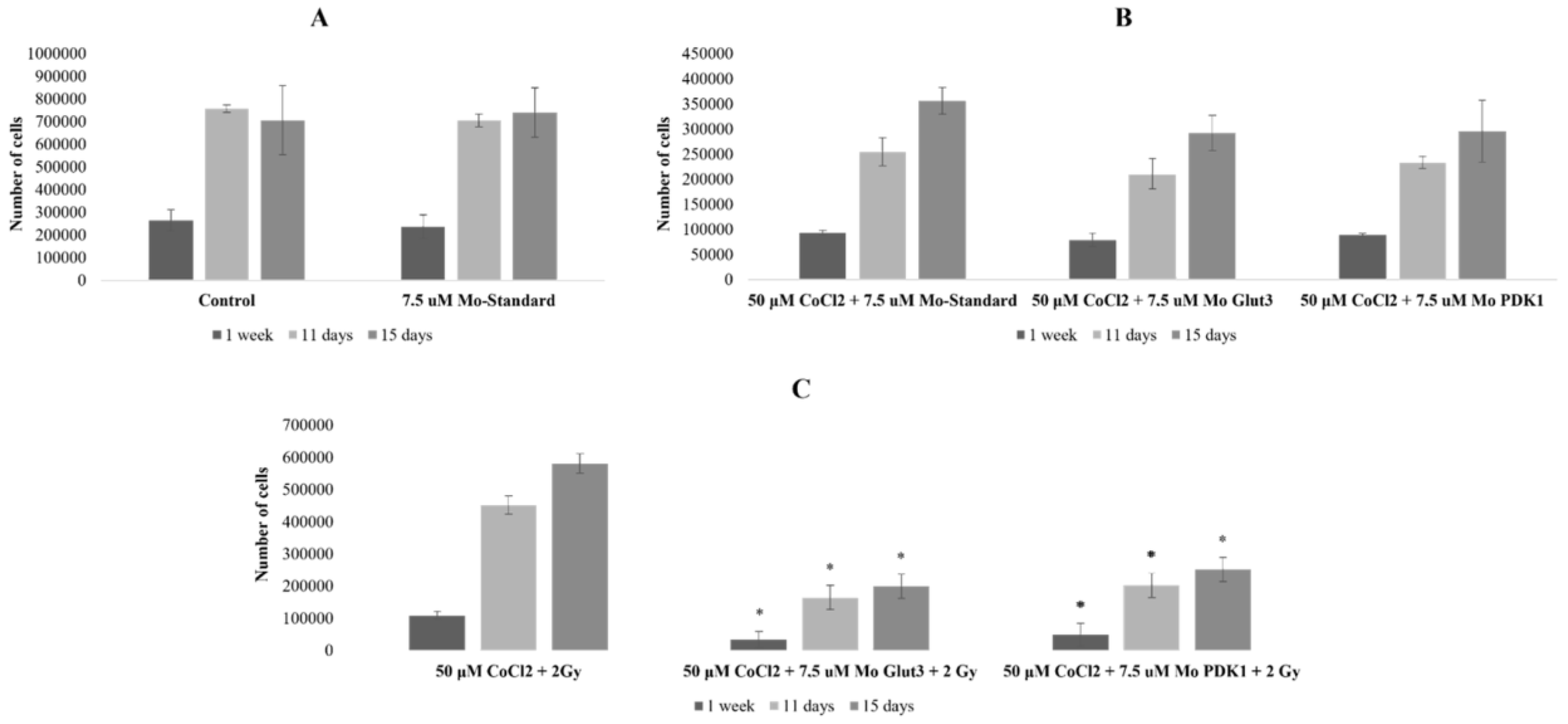
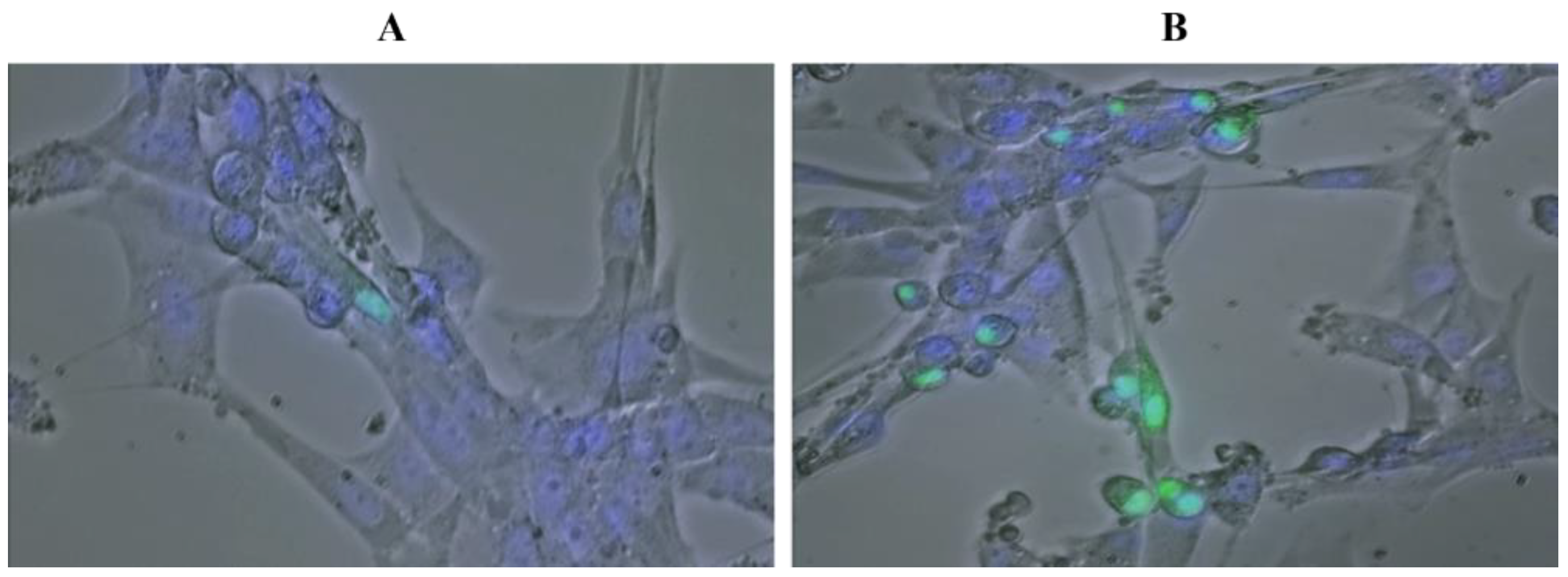
2.3. Radiosensitizing Effects of glut-3 and pdk-1 Morpholino-Induced Gene Knockdown
2.4. Molecular Response in U87 Cells under CoCl2-Induced Hypoxia Condition and glut-1/pdk-1 Gene Knockdown
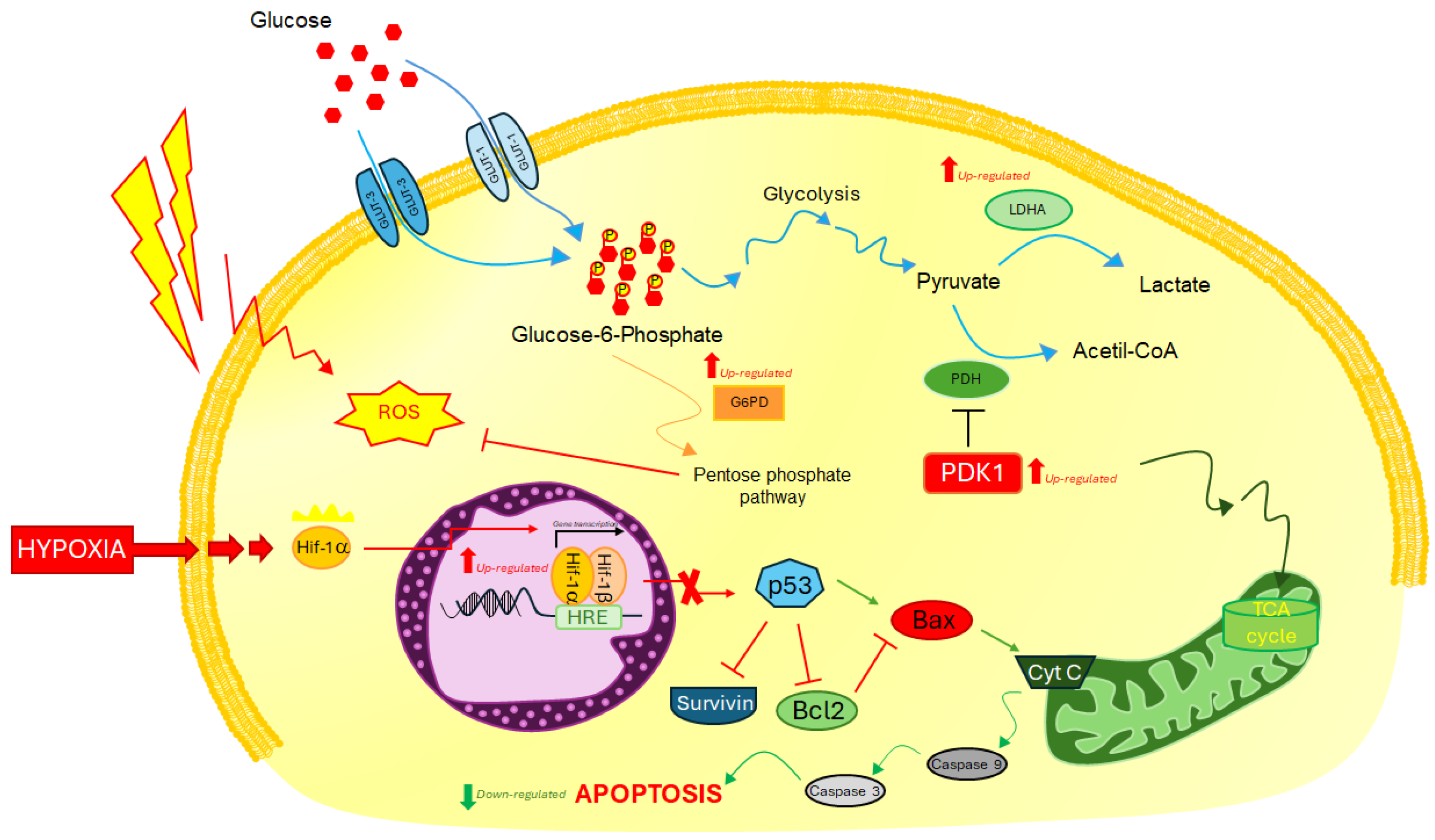
3. Discussion
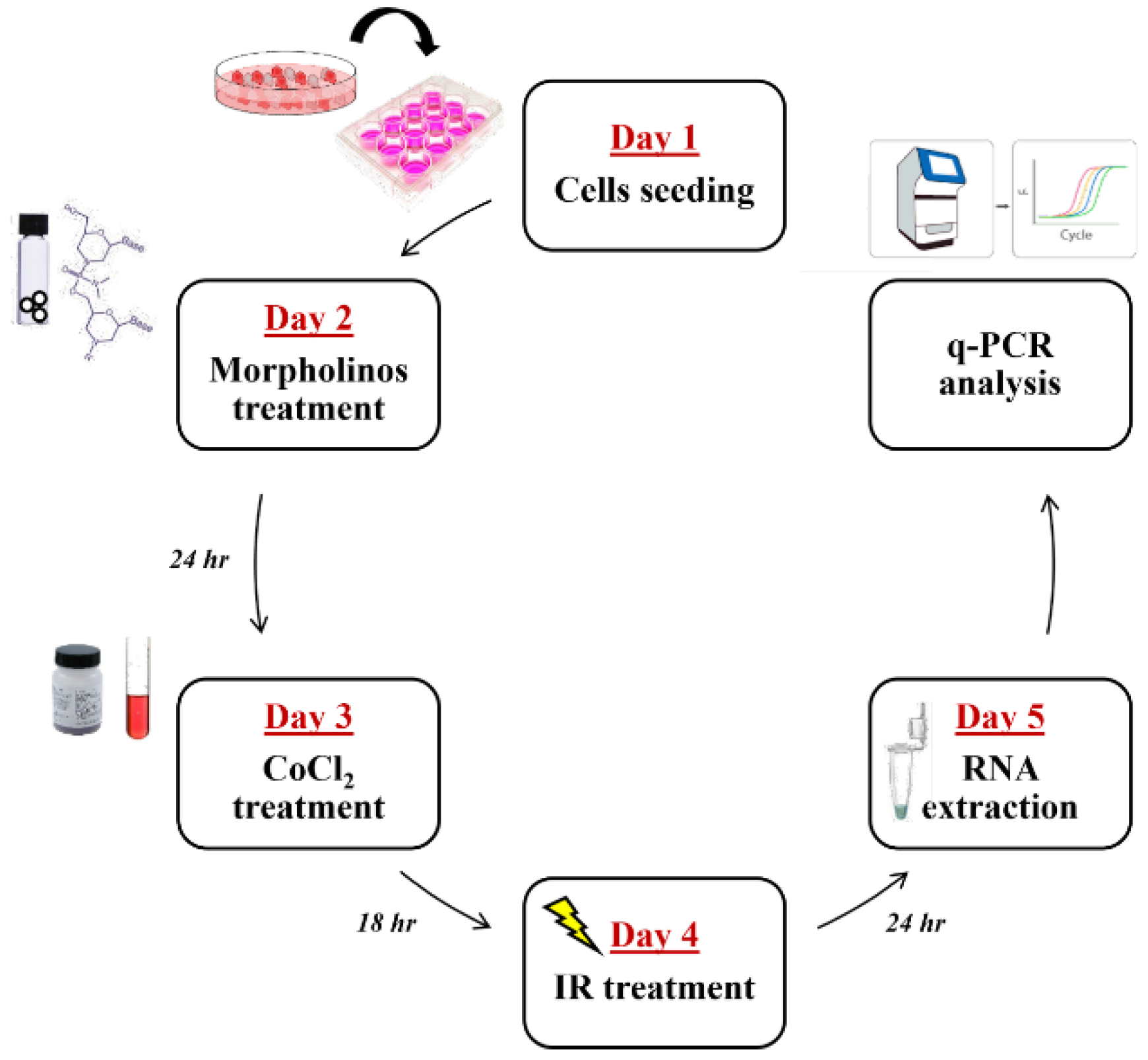
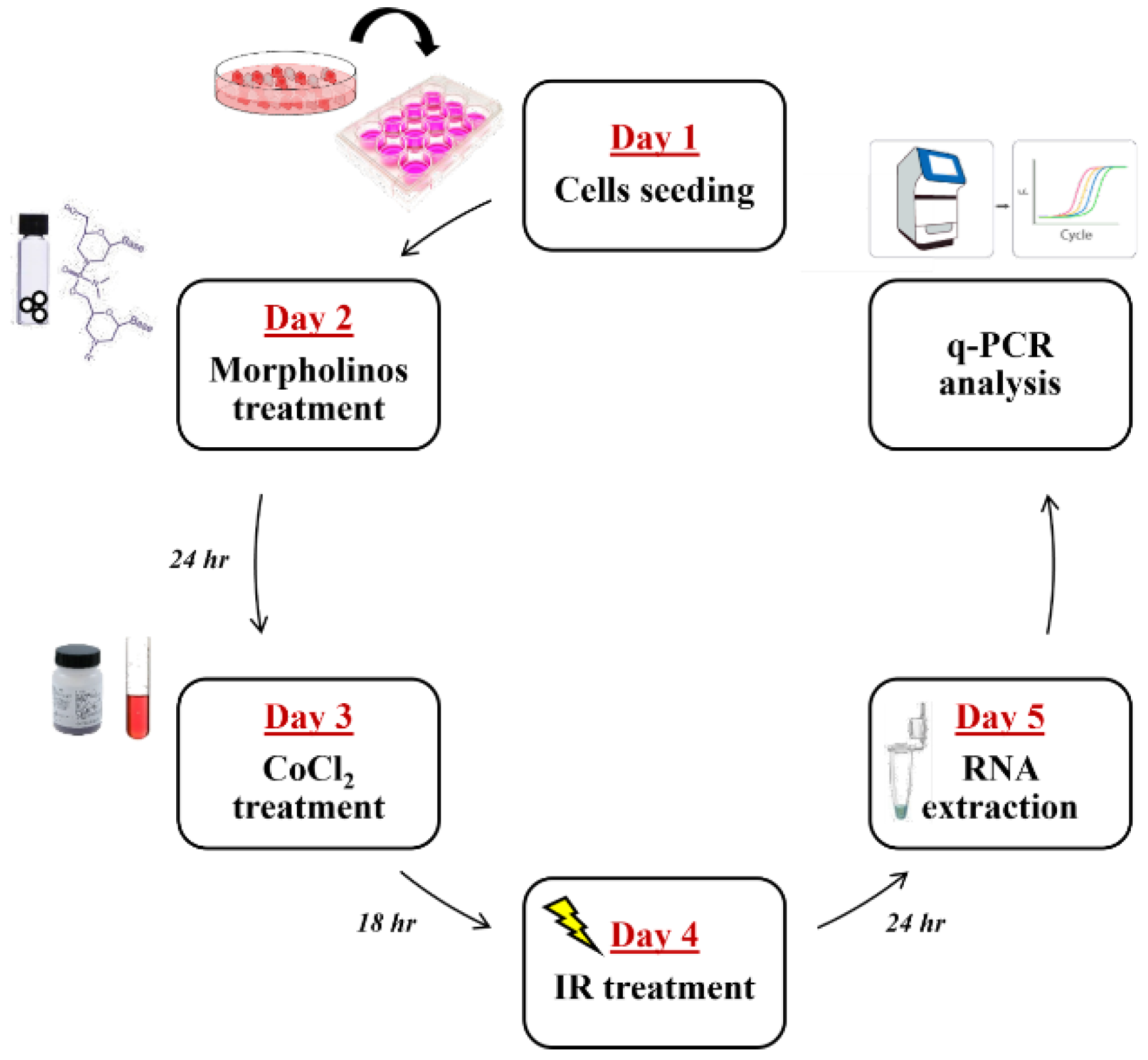
4. Material and Methods
4.1. Cell Culture, CoCl2 Treatments, and Cell Proliferation
4.2. Cytotoxicity Assays
4.3. Irradiation and Combined Treatments
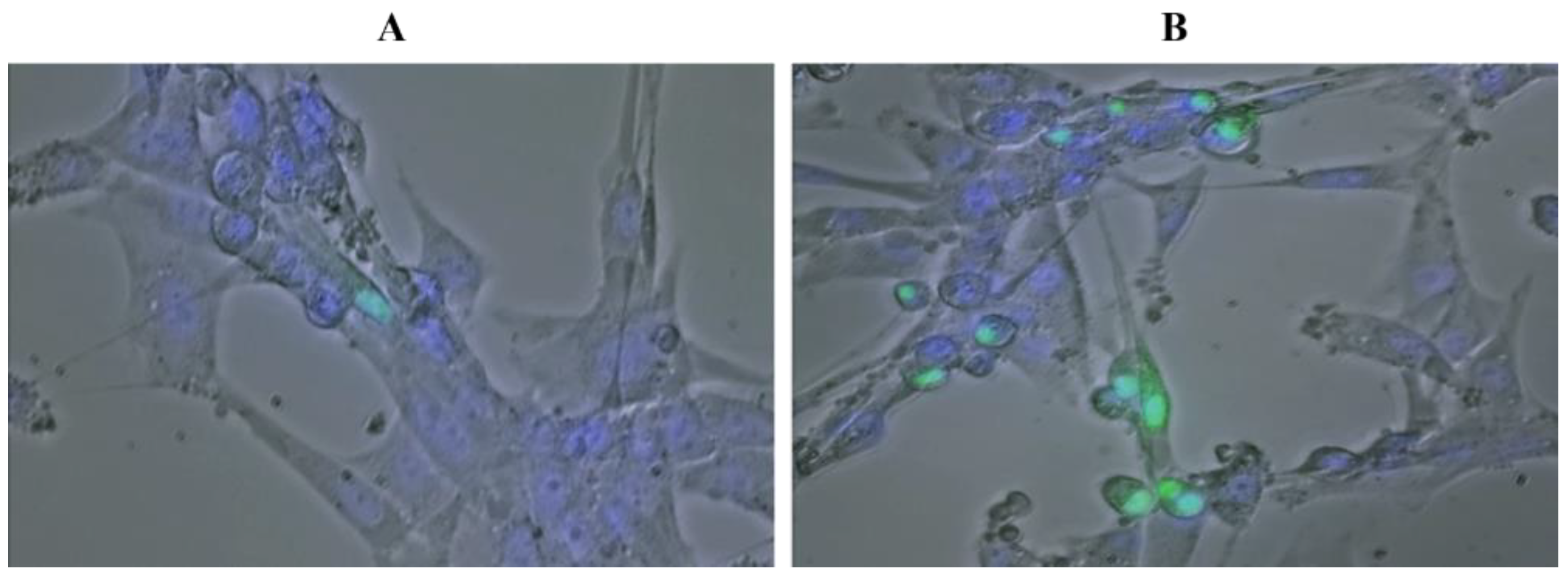
4.4. Optical and Fluorescence Microscopy
4.5. HIF1α-GFP Transgene Construction and Transfection
4.6. RNA Silencing by Using Vivo-Morpholino
4.7. Total RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR (qPCR)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, E.; Puerto, I.; Medina, M.Á. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun. 2022, 42, 1083–1111. [Google Scholar] [CrossRef]
- Habib, A.; Pease, M.; Kodavali, C.V.; Amankulor, N.; Zinn, P.O. A contemporary update on glioblastoma: Molecular biology, current management, and a vision towards bio-adaptable personalized care. J. Neuro-Oncol. 2021, 151, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Di Cintio, F.; Dal Bo, M.; Baboci, L.; De Mattia, E.; Polano, M.; Toffoli, G. The Molecular and Microenvironmental Landscape of Glioblastomas: Implications for the Novel Treatment Choices. Front. Neurosci. 2020, 14, 603647. [Google Scholar] [CrossRef]
- Glaviano, A.; Foo, A.S.C.; Lam, H.Y.; Yap, K.C.H.; Jacot, W.; Jones, R.H.; Eng, H.; Nair, M.G.; Makvandi, P.; Geoerger, B.; et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 2023, 22, 138. [Google Scholar] [CrossRef]
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes 2021, 12, 1596. [Google Scholar] [CrossRef]
- Uddin, M.S.; Mamun, A.A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef]
- Pottoo, F.H.; Javed, M.N.; Rahman, J.U.; Abu-Izneid, T.; Khan, F.A. Targeted delivery of miRNA based therapeuticals in the clinical management of Glioblastoma Multiforme. Semin. Cancer Biol. 2021, 69, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Zhu, J.; Zhang, X.; Mao, X. The Role of Hypoxia and Cancer Stem Cells in Development of Glioblastoma. Cancers 2023, 15, 2613. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1alpha. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef]
- Sebestyén, A.; Kopper, L.; Dankó, T.; Tímár, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 27, 1609802. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218, Erratum in Trends Biochem. Sci. 2016, 41, 287.. [Google Scholar] [CrossRef]
- Emami Nejad, A.; Najafgholian, S.; Rostami, A.; Sistani, A.; Shojaeifar, S.; Esparvarinha, M.; Nedaeinia, R.; Haghjooy Javanmard, S.; Taherian, M.; Ahmadlou, M.; et al. The role of hypoxia in the tumor microenvironment and development of cancer stem cell: A novel approach to developing treatment. Cancer Cell Int. 2021, 21, 62. [Google Scholar] [CrossRef]
- Bravatà, V.; Tinganelli, W.; Cammarata, F.P.; Minafra, L.; Calvaruso, M.; Sokol, O.; Petringa, G.; Cirrone, G.A.P.; Scifoni, E.; Forte, G.I.; et al. Hypoxia Transcriptomic Modifications Induced by Proton Irradiation in U87 Glioblastoma Multiforme Cell Line. J. Pers. Med. 2021, 11, 308. [Google Scholar] [CrossRef]
- Torrisi, F.; Vicario, N.; Spitale, F.M.; Cammarata, F.P.; Minafra, L.; Salvatorelli, L.; Russo, G.; Cuttone, G.; Valable, S.; Gulino, R.; et al. The Role of Hypoxia and SRC Tyrosine Kinase in Glioblastoma Invasiveness and Radioresistance. Cancers 2020, 12, 2860. [Google Scholar] [CrossRef]
- Liu, C.; Lin, Q.; Yun, Z. Cellular and molecular mechanisms underlying oxygen-dependent radiosensitivity. Radiat. Res. 2015, 183, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Cammarata, F.P.; Torrisi, F.; Forte, G.I.; Minafra, L.; Bravatà, V.; Pisciotta, P.; Savoca, G.; Calvaruso, M.; Petringa, G.; Cirrone, G.A.P.; et al. Proton Therapy and Src Family Kinase Inhibitor Combined Treatments on U87 Human Glioblastoma Multiforme Cell Line. Int. J. Mol. Sci. 2019, 20, 4745. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, F.; Minafra, L.; Cammarata, F.P.; Savoca, G.; Calvaruso, M.; Vicario, N.; Maccari, L.; Pérès, E.A.; Özçelik, H.; Bernaudin, M.; et al. SRC Tyrosine Kinase Inhibitor and X-rays Combined Effect on Glioblastoma Cell Lines. Int. J. Mol. Sci. 2020, 21, 3917. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Atas, E.; Oberhuber, M.; Kenner, L. The Implications of PDK-1-4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front. Oncol. 2020, 10, 583217. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Q.; Fu, Y.L.; Zhang, J.; Zhang, K.Y.; Ma, J.; Tang, J.Y.; Zhang, Z.W.; Zhou, Z.Y. Targeting glycolysis in non-small cell lung cancer: Promises and challenges. Front. Pharmacol. 2022, 13, 1037341. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, H.; Mei, Y.; Wu, M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Mol. Cell 2014, 53, 88–100. [Google Scholar] [CrossRef]
- Ivan, M.; Huang, X. miR-210: Fine-tuning the hypoxic response. Adv. Exp. Med. Biol. 2014, 772, 205–227. [Google Scholar]
- Jin, X.; Kuang, Y.; Li, L.; Li, H.; Zhao, T.; He, Y.; Di, C.; Kang, J.; Yuan, L.; Yu, B.; et al. A positive feedback circuit comprising p21 and HIF-1α aggravates hypoxia-induced radioresistance of glioblastoma by promoting Glut1/LDHA-mediated glycolysis. FASEB J. 2022, 36, e22229. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Lin, Y.J.; Wu, C.P.; Lee, H.T.; Shyu, W.C.; Wang, C.C. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clin. Cancer Res. 2015, 21, 460–470. [Google Scholar] [CrossRef]
- Bai, H.; Ge, S.; Lu, J.; Qian, G.; Xu, R. Hypoxia inducible factor-1α-mediated activation of survivin in cervical cancer cells. J. Obstet. Gynaecol. Res. 2013, 39, 555–563. [Google Scholar] [CrossRef]
- Chen, L.; Wang, W.; Zhu, S.; Jin, X.; Wang, J.; Zhu, J.; Zhou, Y. MicroRNA-590-3p enhances the radioresistance in glioblastoma cells by targeting LRIG1. Exp. Ther. Med. 2017, 14, 1818–1824. [Google Scholar] [CrossRef]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef] [PubMed]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes. Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.V.; Zheng, X.; Pereira, T.; Gradin, K.; Jin, S.; Lundkvist, J.; Ruas, J.L.; Poellinger, L.; Lendahl, U.; Bondesson, M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 2005, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhou, C.; Xu, L.; Xiao, H. Hypoxia enhances stemness of cancer stem cells in glioblastoma: An in vitro study. Int. J. Med. Sci. 2013, 10, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef] [PubMed]
- Briere, T.M.; McAleer, M.F.; Levy, L.B.; Yang, J.N. Sparing of normal tissues with volumetric arc radiation therapy for glioblastoma: Single institution clinical experience. Radiat. Oncol. 2017, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, R.; Nichol, A.M.; Vollans, E.; Fong, M.; Nakano, S.; Moiseenko, V.; Schmuland, M.; Ma, R.; McKenzie, M.; Otto, K. A comparison of volumetric modulated arc therapy and conventional intensity-modulated radiotherapy for frontal and temporal high-grade gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 1177–1184. [Google Scholar] [CrossRef]
- Cao, Y.; Tseng, C.L.; Balter, J.M.; Teng, F.; Parmar, H.A.; Sahgal, A. MR-guided radiation therapy: Transformative technology and its role in the central nervous system. Neuro-Oncology 2017, 19 (Suppl. S2), ii16–ii29. [Google Scholar] [CrossRef]
- Tsien, C.; Moughan, J.; Michalski, J.M.; Gilbert, M.R.; Purdy, J.; Simpson, J.; Kresel, J.J.; Curran, W.J.; Diaz, A.; Mehta, M.P. Radiation Therapy Oncology Group Trial 98-03. Phase I three-dimensional conformal radiation dose escalation study in newly diagnosed glioblastoma: Radiation Therapy Oncology Group Trial 98-03. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 699–708. [Google Scholar] [CrossRef]
- Combs, S.E.; Thilmann, C.; Edler, L.; Debus, J.; Schulz-Ertner, D. Efficacy of fractionated stereotactic reirradiation in recurrent gliomas: Long-term results in 172 patients treated in a single institution. J. Clin. Oncol. 2005, 23, 8863–8869. [Google Scholar] [CrossRef]
- Combs, S.E.; Debus, J.; Schulz-Ertner, D. Radiotherapeutic alternatives for previously irradiated recurrent gliomas. BMC Cancer 2007, 7, 167. [Google Scholar] [CrossRef]
- McGee, M.C.; Hamner, J.B.; Williams, R.F.; Rosati, S.F.; Sims, T.L.; Ng, C.Y.; Gaber, M.W.; Calabrese, C.; Wu, J.; Nathwani, A.C.; et al. Improved intratumoral oxygenation through vascular normalization increases glioma sensitivity to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 1537–1545. [Google Scholar] [CrossRef]
- Graham, K.; Unger, E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int. J. Nanomed. 2018, 13, 6049–6058. [Google Scholar] [CrossRef]
- Gérard, M.; Corroyer-Dulmont, A.; Lesueur, P.; Collet, S.; Chérel, M.; Bourgeois, M.; Stefan, D.; Limkin, E.J.; Perrio, C.; Guillamo, J.S.; et al. Hypoxia Imaging and Adaptive Radiotherapy: A State-of-the-Art Approach in the Management of Glioma. Front. Med. 2019, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Gainer, J.L.; Sheehan, J.P.; Larner, J.M.; Jones, D.R. Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme. J. Neurosurg. 2017, 126, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Dwyer, D.; Malide, D.; Moley, K.H.; Travis, A.; Vannucci, S.J. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E242–E253. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Guda, M.R.; Asuthkar, S.; Labak, C.M.; Tsung, A.J.; Alexandrov, I.; Mackenzie, M.J.; Prasad, D.V.; Velpula, K.K. Targeting PDK4 inhibits breast cancer metabolism. Am. J. Cancer Res. 2018, 8, 1725–1738. [Google Scholar] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Chelakkot, V.S.; Shin, Y.; Song, K. Modulating Glycolysis to Improve Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 2606. [Google Scholar] [CrossRef]
- Bao, Y.Y.; Zhong, J.T.; Shen, L.F.; Dai, L.B.; Zhou, S.H.; Fan, J.; Yao, H.T.; Lu, Z.J. Effect of Glut-1 and HIF-1α double knockout by CRISPR/CAS9 on radiosensitivity in laryngeal carcinoma via the PI3K/Akt/mTOR pathway. J. Cell Mol. Med. 2022, 26, 2881–2894. [Google Scholar] [CrossRef]
- Su, J.; Gao, T.; Jiang, M.; Wu, L.; Zeng, W.; Zhao, S.; Peng, C.; Chen, X. CD147 silencing inhibits tumor growth by suppressing glucose transport in melanoma. Oncotarget 2016, 7, 64778–64784. [Google Scholar] [CrossRef]
- Oh, S.; Kim, H.; Nam, K.; Shin, I. Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells. BMB Rep. 2017, 50, 132–137. [Google Scholar] [CrossRef]
- Zhong, J.T.; Yu, Q.; Zhou, S.H.; Yu, E.; Bao, Y.Y.; Lu, Z.J.; Fan, J. GLUT-1 siRNA Enhances Radiosensitization of Laryngeal Cancer Stem Cells via Enhanced DNA Damage, Cell Cycle Redistribution, and Promotion Of Apoptosis In Vitro and In Vivo. Onco Targets Ther. 2019, 12, 9129–9142. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, Y.; Zhu, L.; Li, J.; Liu, Y.; Luo, J.; Xie, T.; Chen, D. Tumor cell membrane-coated continuous electrochemical sensor for GLUT1 inhibitor screening. J. Pharm. Anal. 2023, 13, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Guda, M.R.; Labak, C.M.; Omar, S.I.; Asuthkar, S.; Airala, S.; Tuszynski, J.; Tsung, A.J.; Velpula, K.K. GLUT1 and TUBB4 in Glioblastoma Could be Efficacious Targets. Cancers 2019, 11, 1308. [Google Scholar] [CrossRef]
- Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined targeting of PDK-1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013, 73, 7277–7289. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.F.; Zhou, S.H.; Guo, Y. Role of GLUT-1 in the Upregulation of PD-L1 Expression After Radiotherapy and Association of PD-L1 with Favourable Overall Survival in Hypopharyngeal Cancer. Onco Targets Ther. 2020, 13, 11221–11235. [Google Scholar] [CrossRef]
- Yan, S.X.; Luo, X.M.; Zhou, S.H.; Bao, Y.Y.; Fan, J.; Lu, Z.J.; Liao, X.B.; Huang, Y.P.; Wu, T.T.; Wang, Q.Y. Effect of antisense oligodeoxynucleotides glucose transporter-1 on enhancement of radiosensitivity of laryngeal carcinoma. Int. J. Med. Sci. 2013, 10, 1375–1386. [Google Scholar] [CrossRef]
- Vartanian, A.; Agnihotri, S.; Wilson, M.R.; Burrell, K.E.; Tonge, P.D.; Alamsahebpour, A.; Jalali, S.; Taccone, M.S.; Mansouri, S.; Golbourn, B.; et al. Targeting hexokinase 2 enhances response to radio-chemotherapy in glioblastoma. Oncotarget 2016, 7, 69518–69535. [Google Scholar] [CrossRef]
- Minafra, L.; Porcino, N.; Bravatà, V.; Gaglio, D.; Bonanomi, M.; Amore, E.; Cammarata, F.P.; Russo, G.; Militello, C.; Savoca, G.; et al. Radiosensitizing effect of curcumin-loaded lipid nanoparticles in breast cancer cells. Sci. Rep. 2019, 9, 11134. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Expression Variations by qRT-PCR | ||||
|---|---|---|---|---|
| CoCl2 | 2 Gy | CoCl2 + 2 Gy | ||
| HRE regulated genes | hif-1α | 1.05 | 1.22 | 1.38 |
| glut-3 | 4.05 | 0.70 | 1.62 | |
| glut-1 | 2.10 | 0.94 | 1.92 | |
| eno-1 | 1.78 | 0.79 | 1.37 | |
| ldha | 1.43 | 1.08 | 1.41 | |
| pdk1 | 2.06 | 0.96 | 2.59 | |
| p21 | 2.16 | 1.29 | 2.38 | |
| lon | 1.30 | 1.07 | 1.16 | |
| lincRNA-p21 | 1.5 | 0.73 | 0.60 | |
| mir-210 | 1.36 | 0.99 | 1.01 | |
| mir-590 | 3.51 | 1.04 | 2.03 | |
| survivin | 0.38 | 0.71 | 0.52 | |
| livin | 0.76 | 0.49 | 0.48 | |
| Genes indirectly regulated by HIF-1α | bax | 1.43 | 1.38 | 0.95 |
| casp-9 | 0.7 | 0.95 | 0.68 | |
| bcl2 | 1.01 | 0.16 | 1.20 | |
| Gene Expression Variations by qRT-PCR | ||||||||
|---|---|---|---|---|---|---|---|---|
| MoPDK1 | MoPDK1 + CoCl2 | MoPDK1 + 2 Gy | MoPDK1 + CoCl2 + 2 Gy | MoGLUT-3 | MoGLUT-3 + CoCl2 | MoGLUT-3 + 2 Gy | MoGLUT-3 + CoCl2 + 2 Gy | |
| glut-3 | 0.68 | 0.83 | 1.32 | 1.32 | - | - | - | - |
| glut-1 | 2.00 | 4.92 | 4.19 | 8.82 | 0.74 | 2.91 | 1.66 | 4.53 |
| pdk1 | - | - | - | - | 0.67 | 1.84 | 1.02 | 0.92 |
| Ldha | 1.21 | 2.31 | 1.83 | 2.34 | 0.69 | 1.50 | 1.51 | 1.32 |
| Bax | 0.90 | 1 | 1.28 | 1.38 | 0.74 | 0.95 | 0.90 | 1.60 |
| casp-9 | 0.72 | 1.07 | 1.72 | 1.05 | 0.76 | 1.65 | 1.5 | 1.63 |
| bcl2 | 1.64 | 2.05 | 1.64 | 2.30 | 1.65 | 1.68 | 1.93 | 3.15 |
| survivin | 1.51 | 1.1 | 1.33 | 1.75 | 1.32 | 1.41 | 1.52 | 2.34 |
| Gene Symbol | Gene Name | Forward Primer 5′>3′ | Reverse Primer 5′>3′ |
|---|---|---|---|
| hif-1α | Hypoxia Inducible Factor 1, Subunit α | GGGGGGCTAGCATGAACGACAAGAAAAAGATAAGT | GGGGGGGATCCTTAACTTGATCCAAAGCTCTG |
| hif-1α | Hypoxia Inducible Factor 1, Subunit α | GCTTGCTCATCAGTTGCCAC | ATCCAGAAGTTTCCTCACACG |
| glut-1 | Glucose transporter, Type 1 | CATCTTCACTGTGCTCCTGG | CCTCGGGTGTCTTGTCACTT |
| glut-3 | Glucose transporter, Type 3 | CACTTTGCTCTGGGTGGAAG | TCACTGACAAGGGTTTGGCTA |
| eno-1 | Enolase 1, (Alpha) | TTCGCCCGCACCACTACAG | AGAGCCGTCACTCATTCCCT |
| Ldha | Lactate Dehydrogenase A | CAGCCCGATTCCGTTACCTA | TCTTCAGAGAGACACCAGCAA |
| pdk1 | Pyruvate Dehydrogenase Kinase 1 | AACCAAAGCATCAGAGCCATC | TTGAGCCCAGAAGATTGAAGC |
| Livin | Baculoviral IAP repeat containing 7 | CTGGGCATATTCTGAGATTGG | AGGCACTTGGCACTGTCTTTA |
| survivin | Baculoviral IAP repeat containing 5 | TCTAAGTTGGAGTGGAGTCTG | CAGTTTGGCTTGCTGGTCTC |
| p21 | Cyclin dependent kinase inhibitor 1A | CGGAACAAGGAGTCAGACATT | CGTTAGTGCCAGGAAAGACAA |
| Lon | lon peptidase 1, mitochondrial | CTGGAGAAGGACGACAAGGA | GGTAGTTGCGGGTGACATTG |
| lincRNA-p21 | P53 pathway corepressor 1 protein tumor | GGTGGGGCTGAAGTTTATGC | CACACACAGGTGGGTTGATG |
| miR-210 | microRNA 210 | CAGCCCCTGCCCACCGC | TGCCCAGGCACAGATCAGC |
| miR-590 | microRNA 590 | AAATGAGCTTATTCATAAAAGTGC | GCATGTTTCAATCAGAGACTAG |
| Morpholino Symbol | Morpholino Sequences 5′>3′ | Forward Primer 5′>3′ | Reverse Primer 5′>3′ |
|---|---|---|---|
| Mo-GLUT3 | GCCCAGTTTCTAGTCAATACCTGCC | GGTCATCAATGCTCCTGAG | TTCCAACAACGATGCCCAG |
| Mo-PDK1 | ACACAAGATGAGAATCTTACCAGCT | ATGAAGCAGTTCCTGGACTT | GCTGATTGAGTAACATTCTAA |
| Mo-ST | CCTCTTACCTCAGTTACAATTTATA | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pucci, G.; Minafra, L.; Bravatà, V.; Calvaruso, M.; Turturici, G.; Cammarata, F.P.; Savoca, G.; Abbate, B.; Russo, G.; Cavalieri, V.; et al. Glut-3 Gene Knockdown as a Potential Strategy to Overcome Glioblastoma Radioresistance. Int. J. Mol. Sci. 2024, 25, 2079. https://doi.org/10.3390/ijms25042079
Pucci G, Minafra L, Bravatà V, Calvaruso M, Turturici G, Cammarata FP, Savoca G, Abbate B, Russo G, Cavalieri V, et al. Glut-3 Gene Knockdown as a Potential Strategy to Overcome Glioblastoma Radioresistance. International Journal of Molecular Sciences. 2024; 25(4):2079. https://doi.org/10.3390/ijms25042079
Chicago/Turabian StylePucci, Gaia, Luigi Minafra, Valentina Bravatà, Marco Calvaruso, Giuseppina Turturici, Francesco P. Cammarata, Gaetano Savoca, Boris Abbate, Giorgio Russo, Vincenzo Cavalieri, and et al. 2024. "Glut-3 Gene Knockdown as a Potential Strategy to Overcome Glioblastoma Radioresistance" International Journal of Molecular Sciences 25, no. 4: 2079. https://doi.org/10.3390/ijms25042079
APA StylePucci, G., Minafra, L., Bravatà, V., Calvaruso, M., Turturici, G., Cammarata, F. P., Savoca, G., Abbate, B., Russo, G., Cavalieri, V., & Forte, G. I. (2024). Glut-3 Gene Knockdown as a Potential Strategy to Overcome Glioblastoma Radioresistance. International Journal of Molecular Sciences, 25(4), 2079. https://doi.org/10.3390/ijms25042079