
Phenotypic Investigation and RNA-seq of KN1 Involved in Leaf Angle Formation in Maize (Zea mays L.)

Abstract
1. Introduction
2. Results
2.1. Identification and Basic Bioinformatics Analysis of KN1
2.2. Identification of Phenotype and Grain Characteristics of Mutant kn1
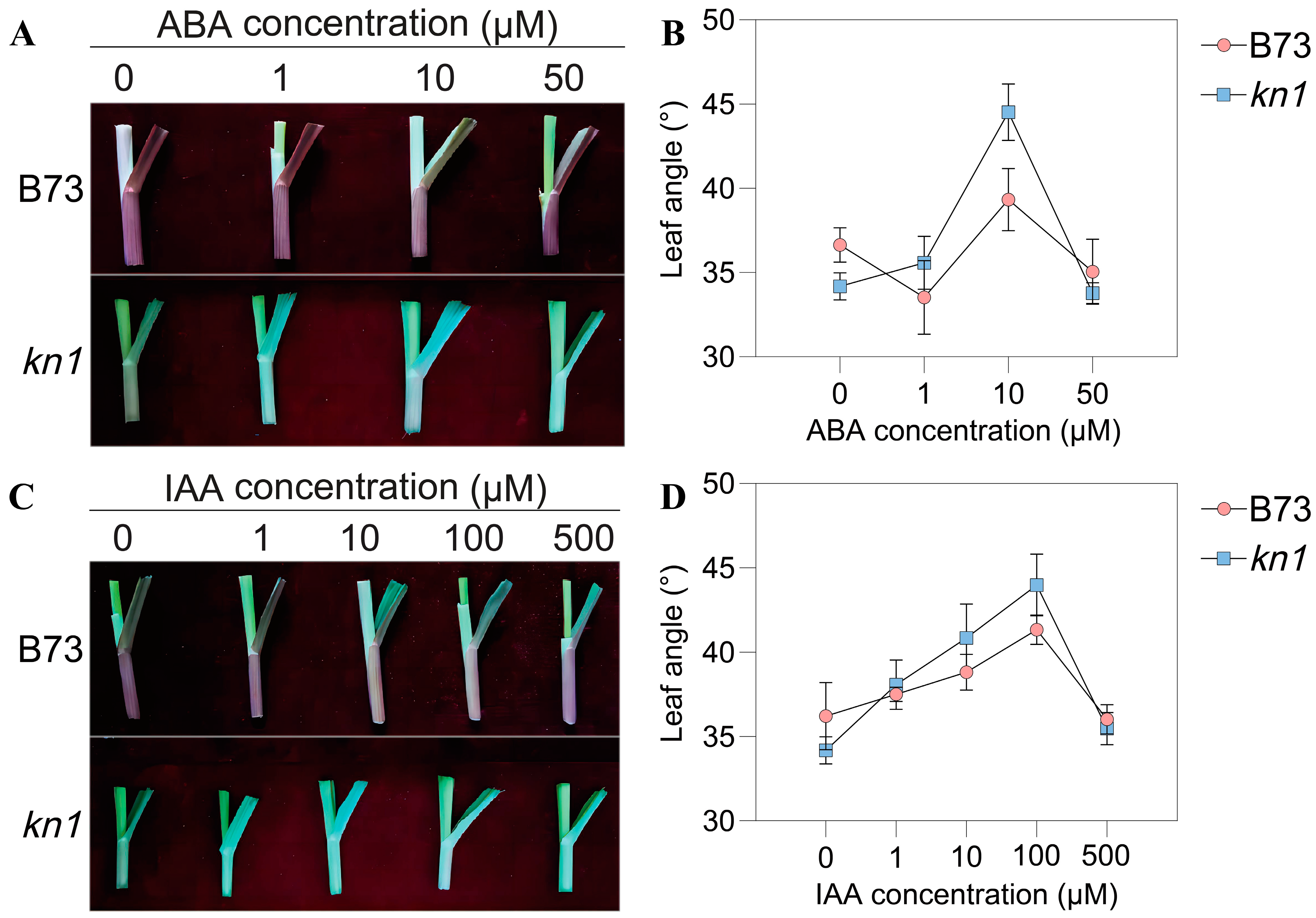
2.3. Spraying of Exogenous Phytohormones and Determination of Optimal Concentration
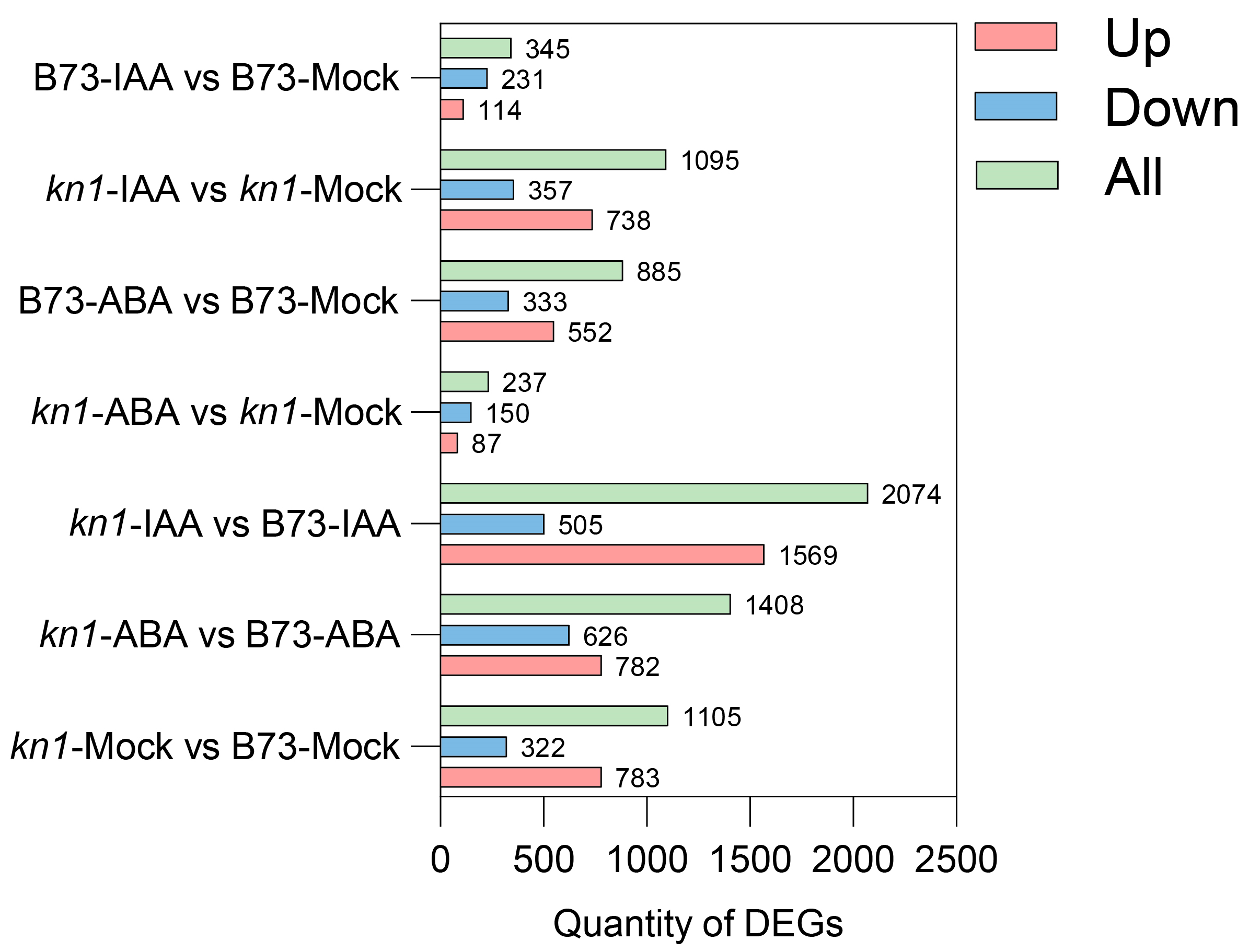
2.4. Transcriptome Sequencing (RNA-seq) Statistics and Transcriptomic Profile of B73 and kn1
2.5. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of LA Mutant kn1
2.6. GO and KEGG Enrichment Analysis of kn1 and B73 Treated with Exogenous ABA and IAA
2.7. qRT-PCR Verification of Differentially Expressed Genes
2.8. Identification of Potential Hub Genes Involved in Regulation of LA via Weighted Gene Co-Expression Network Analysis (WGCNA)
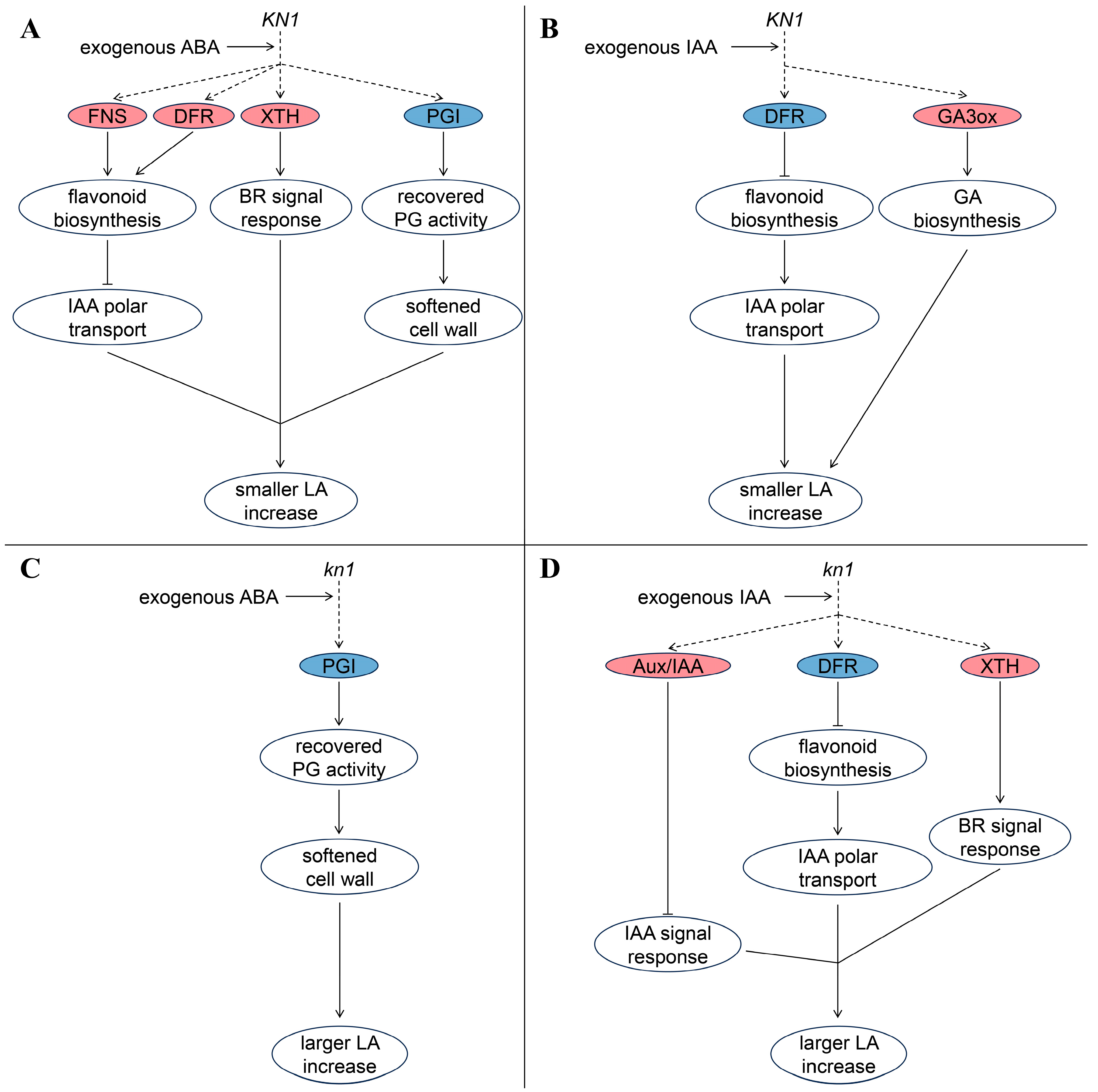
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Planting Circumstances
4.2. Analysis of kn1 Mutant Phenotype
4.3. Bioinformatics-Related Analysis of KN1
4.4. Identification of Homozygous Mutant kn1
4.5. Expression Pattern Analysis of KN1
4.6. Determination of the Optimal Concentration of Exogenous Phytohormones
4.7. Transcriptomic Profiles and Data Analysis
4.8. Reliability Verification of RNA-seq via qRT-PCR
4.9. Weighted Gene Co-Expression Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Warburton, M.L.; Setter, T.; Liu, H.; Xue, Y.; Yang, N.; Yan, J.; Xiao, Y. Genome-wide association studies of drought-related metabolic changes in maize using an enlarged SNP panel. Theor. Appl. Genet. 2016, 9, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.T.B.L.; Wu, T.Y. A review on solvent systems for furfural production from lignocellulosic biomass. Renew. Sustain. Energy Rev. 2021, 137, 110172. [Google Scholar] [CrossRef]
- He, Y.; Ding, Y.; Ma, C.; Di, J.; Jiang, C.; Li, A. One-pot conversion of biomass-derived xylose to furfuralcohol by a chemo-enzymatic sequential acid-catalyzed dehydration and bioreduction. Green Chem. 2017, 19, 3844–3850. [Google Scholar] [CrossRef]
- Peng, B.; Ma, C.L.; Zhang, P.Q.; Wu, C.Q.; Wang, Z.W.; Li, A.T.; He, Y.C.; Yang, B. An effective hybrid strategy for converting rice straw to furoic acid by tandem catalysis via Sn-sepiolite combined with recombinant E. coli whole cells harboring horse liver alcohol dehydrogenase. Green Chem. 2019, 21, 5914–5923. [Google Scholar] [CrossRef]
- Zhang, P.; Liao, X.; Ma, C.; Li, Q.; Li, A.; He, Y. Chemoenzymatic conversion of corncob to furfurylamine via tandem catalysis with tin-based solid acid and transaminase biocatalyst. ACS Sustain. Chem. Eng. 2019, 7, 17636–17642. [Google Scholar] [CrossRef]
- Gong, F.; Wu, X.; Zhang, H.; Chen, Y.; Wang, W. Making better maize plants for sustainable grain production in a changing climate. Front. Plant Sci. 2015, 6, 835. [Google Scholar] [CrossRef]
- Savary, S.; Akter, S.; Almekinders, C.; Harris, J.; Korsten, L.; Rötter, R.; Waddington, S.; Watson, D. Mapping disruption and resilience mechanisms in food systems. Food Secur. 2020, 12, 695–717. [Google Scholar] [CrossRef]
- Mansfield, B.D.; Mumm, R.H. Survey of plant density tolerance in U.S. maize germplasm. Crop Sci. 2014, 54, 157–173. [Google Scholar] [CrossRef]
- Leivar, P.; Tepperman, J.M.; Cohn, M.M.; Monte, E.; Al-Sady, B.; Erickson, E.; Quail, P.H. Dynamic antagonism between phytochromes and PIF family basic helix-loop-helix factors induces selective reciprocal responses to light and shade in a rapidly responsive transcriptional network in Arabidopsis. Plant Cell 2012, 24, 1398–1419. [Google Scholar] [CrossRef]
- Hill, W.G. Understanding and using quantitative genetic variation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 73–85. [Google Scholar] [CrossRef]
- El-Soda, M.; Malosetti, M.; Zwaan, B.J.; Koornneef, M.; Aarts, M.G. Genotype × environment interaction QTL mapping in plants: Lessons from Arabidopsis. Trends Plant Sci. 2014, 19, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhong, Z.; Wang, H.; Shen, R. Leaf angle: A target of genetic improvement in cereal crops tailored for high-density planting. Plant Biotechnol. J. 2022, 20, 426–436. [Google Scholar] [CrossRef]
- Song, Y.; You, J.; Xiong, L. Characterization of OsIAA1 gene, a member of rice Aux/IAA family involved in auxin and brassinosteroid hormone responses and plant morphogenesis. Plant Mol. Biol. 2009, 70, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.Q.; Xiang, J.J.; Xue, H.W. Studies on the rice LEAF INCLINATION1 (LC1), an IAA–amido synthetase, reveal the effects of auxin in leaf inclination control. Mol. Plant 2013, 6, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Xie, Y.; Guo, F.; Han, N.; Ma, S.; Zeng, Z.; Wang, J.; Yang, Y.; Zhu, M. Distinctive expression patterns and roles of the miRNA393/TIR1 homolog module in regulating flag leaf inclination and primary and crown root growth in rice (Oryza sativa). New Phytol. 2012, 196, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wei, J.; Wang, H.; Fang, Y.; Yin, S.; Xu, Y.; Liu, J.; Yang, Z.; Xu, C. Natural variation and domestication selection of ZmPGP1 affects plant architecture and yield-related traits in maize. Genes 2019, 10, 664. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhang, X.; Zhang, Z.; Liu, H.; Lin, Z. A new allele of the Brachytic2 gene in maize can efficiently modify plant architecture. Heredity 2018, 121, 75–86. [Google Scholar] [CrossRef]
- Li, X.; Sun, S.; Li, C.; Qiao, S.; Wang, T.; Leng, L.; Shen, H.; Wang, X. The Strigolactone-related mutants have enhanced lamina joint inclination phenotype at the seedling stage. J. Genet. Genom. 2014, 41, 605–608. [Google Scholar] [CrossRef]
- Fang, Z.; Ji, Y.; Hu, J.; Guo, R.; Sun, S.; Wang, X. Strigolactones and brassinosteroids antagonistically regulate the stability of the D53-OsBZR1 complex to determine FC1 expression in rice tillering. Mol. Plant 2020, 13, 586–597. [Google Scholar] [CrossRef]
- Jang, S.; Cho, J.Y.; Do, G.R.; Kang, Y.; Li, H.Y.; Song, J.; Kim, H.Y.; Kim, B.G.; Hsing, Y.I. Modulation of rice leaf angle and grain size by expressing and under the control of promoter. Int. J. Mol. Sci. 2021, 22, 7792. [Google Scholar] [CrossRef]
- Tong, H.; Xiao, Y.; Liu, D.; Gao, S.; Liu, L.; Yin, Y.; Jin, Y.; Qian, Q.; Chu, C. Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. Plant Cell 2014, 26, 4376–4393. [Google Scholar] [CrossRef]
- Tian, J.; Wang, C.; Xia, J.; Wu, L.; Xu, G.; Wu, W.; Li, D.; Qin, W.; Han, X.; Chen, Q.; et al. Teosinte ligule allele narrows plant architecture and enhances high-density maize yields. Science 2019, 365, 658–664. [Google Scholar] [CrossRef]
- Tong, H.; Liu, L.; Jin, Y.; Du, L.; Yin, Y.; Qian, Q.; Zhu, L.; Chu, C. Dwarf and Low-Tillering acts as a direct downstream target of a GSK3/SHAGGY-like kinase to mediate brassinosteroid responses in rice. Plant Cell 2012, 24, 2562–2577. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, S.; Xu, Y.; Yu, C.; Shen, C.; Qian, Q.; Geisler, M.; Jiang, D.A.; Qi, Y. The auxin response factor, OsARF19, controls rice leaf angles through positively regulating OsGH3-5 and OsBRI1. Plant Cell Environ. 2015, 38, 638–654. [Google Scholar] [CrossRef]
- Li, H.; Wang, L.; Liu, M.; Dong, Z.; Li, Q.; Fei, S.; Xiang, H.; Liu, B.; Jin, W. Maize plant architecture is regulated by the ethylene biosynthetic gene ZmACS7. Plant Physiol. 2020, 183, 1184–1199. [Google Scholar] [CrossRef]
- Li, Q.F.; Lu, J.; Zhou, Y.; Wu, F.; Tong, H.N.; Wang, J.D.; Yu, J.W.; Zhang, C.Q.; Fan, X.L.; Liu, Q.Q. Abscisic acid represses rice lamina joint inclination by antagonizing brassinosteroid biosynthesis and signaling. Int. J. Mol. Sci. 2019, 20, 4908. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lin, H.; Joo, S.; Goodenough, U. Early sexual origins of homeoprotein heterodimerization and evolution of the plant KNOX/BELL family. Cell 2008, 133, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Bürglin, T.R. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1997, 25, 4173–4180. [Google Scholar] [CrossRef] [PubMed]
- Hofer, J.; Gourlay, C.; Michael, A.; Ellis, T.H. Expression of a class 1 knotted1-like homeobox gene is down-regulated in pea compound leaf primordia. Plant Mol. Biol. 2001, 45, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Vollbrecht, E.; Veit, B.; Sinha, N.; Hake, S. The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature 1991, 350, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Bermudez-Lozano, C.L.; Clancy, M.A.; Davis, T.M.; Folta, K.M. A strawberry KNOX gene regulates leaf, flower and meristem architecture. PLoS ONE 2011, 6, e24752. [Google Scholar] [CrossRef]
- Bolduc, N.; Hake, S. The maize transcription factor KNOTTED1 directly regulates the gibberellin catabolism gene ga2ox1. Plant Cell 2009, 21, 1647–1658. [Google Scholar] [CrossRef]
- Bolduc, N.; Yilmaz, A.; Mejia-Guerra, M.K.; Morohashi, K.; O’Connor, D.; Grotewold, E.; Hake, S. Unraveling the KNOTTED1 regulatory network in maize meristems. Genes Dev. 2012, 26, 1685–1690. [Google Scholar] [CrossRef]
- Muszynski, M.G.; Moss-Taylor, L.; Chudalayandi, S.; Cahill, J.; Del Valle-Echevarria, A.R.; Alvarez-Castro, I.; Petefish, A.; Sakakibara, H.; Krivosheev, D.M.; Lomin, S.N.; et al. The Maize Hairy Sheath Frayed1 (Hsf1) mutation alters leaf patterning through increased cytokinin signaling. Plant Cell 2020, 32, 1501–1518. [Google Scholar] [CrossRef]
- Sheng, M.; Ma, X.; Wang, J.; Xue, T.; Li, Z.; Cao, Y.; Yu, X.; Zhang, X.; Wang, Y.; Xu, W.; et al. KNOX II transcription factor HOS59 functions in regulating rice grain size. Plant J. 2022, 110, 863–880. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Manzoor, M.A.; Wang, G.; Chen, C.; Song, C. Comparative analysis of KNOX genes and their expression patterns under various treatments in Dendrobium huoshanense. Front. Plant Sci. 2023, 14, 1258533. [Google Scholar] [CrossRef] [PubMed]
- Laosatit, K.; Amkul, K.; Yimram, T.; Chen, J.; Lin, Y.; Yuan, X.; Wang, L.; Chen, X.; Somta, P. A Class II KNOX gene, KNAT7-1, regulates physical seed dormancy in mungbean [Vigna radiata (L.) Wilczek]. Front. Plant Sci. 2022, 13, 852373. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Zhang, L.; Zhou, W.; Vogelstein, J.; Basrai, M.A.; Bassett, D.E., Jr.; Hieter, P.; Vogelstein, B.; Kinzler, K.W. Characterization of the yeast transcriptome. Cell 1997, 88, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, D.J.; Winzeler, E.A. Genomics, gene expression and DNA arrays. Nature 2000, 405, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.; Wang, M.; Sun, Q.; Sylvester, A.W.; Hake, S.; Scanlon, M.J. Transcriptomic analyses indicate that maize ligule development recapitulates gene expression patterns that occur during lateral organ initiation. Plant Cell 2014, 26, 4718–4732. [Google Scholar] [CrossRef] [PubMed]
- Sentoku, N.; Sato, Y.; Matsuoka, M. Overexpression of rice OSH genes induces ectopic shoots on leaf sheaths of transgenic rice plants. Dev. Biol. 2000, 220, 358–364. [Google Scholar] [CrossRef][Green Version]
- Moon, J.; Candela, H.; Hake, S. The Liguleless narrow mutation affects proximal-distal signaling and leaf growth. Development 2013, 140, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Muehlbauer, G.J.; Fowler, J.E.; Freeling, M. Sectors expressing the homeobox gene liguleless3 implicate a time-dependent mechanism for cell fate acquisition along the proximal-distal axis of the maize leaf. Development 1997, 124, 5097–5106. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.A.; Harper, L.C.; Krueger, R.W.; Dellaporta, S.L.; Freeling, M. liguleless1 encodes a nuclear-localized protein required for induction of ligules and auricles during maize leaf organogenesis. Genes. Dev. 1997, 11, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.; Waters, C.A.; Freeling, M. The maize gene liguleless2 encodes a basic leucine zipper protein involved in the establishment of the leaf blade-sheath boundary. Genes. Dev. 1998, 12, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Wilkinson, E.G.; Sageman-Furnas, K.; Strader, L.C. Auxin and abiotic stress responses. J. Exp. Bot. 2023, 74, 7000–7014. [Google Scholar] [CrossRef]
- Yoshida, T.; Christmann, A.; Yamaguchi-Shinozaki, K.; Grill, E.; Fernie, A.R. Revisiting the basal role of ABA-roles outside of stress. Trends Plant Sci. 2019, 24, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, C.Y.; Miao, R.; Zhou, C.L.; Cao, P.H.; Lan, J.; Zhu, X.J.; Mou, C.L.; Huang, Y.S.; Liu, S.J.; et al. DS1/OsEMF1 interacts with OsARF11 to control rice architecture by regulation of brassinosteroid signaling. Rice 2018, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Deyholos, M.K. Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 2009, 69, 91–105. [Google Scholar] [CrossRef]
- Tian, X.; Li, X.; Zhou, W.; Ren, Y.; Wang, Z.; Liu, Z.; Tang, J.; Tong, H.; Fang, J.; Bu, Q. Transcription factor OsWRKY53 positively regulates brassinosteroid signaling and plant architecture. Plant Physiol. 2017, 175, 1337–1349. [Google Scholar] [CrossRef]
- Zhang, J.; Ku, L.X.; Han, Z.P.; Guo, S.L.; Liu, H.J.; Zhang, Z.Z.; Cao, L.R.; Cui, X.J.; Chen, Y.H. The ZmCLA4 gene in the qLA4-1 QTL controls leaf angle in maize (Zea mays L.). J. Exp. Bot. 2014, 65, 5063–5076. [Google Scholar] [CrossRef]
- Dou, D.; Han, S.; Cao, L.; Ku, L.; Liu, H.; Su, H.; Ren, Z.; Zhang, D.; Zeng, H.; Dong, Y.; et al. CLA4 regulates leaf angle through multiple hormone signaling pathways in maize. J. Exp. Bot. 2021, 72, 1782–1794. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Yu, Z.; Wang, J.; Huang, R.; Zhan, Q.; Li, S.; Lai, J.; Zhang, S.; Yang, C. SUMO E3 ligase AtMMS21-dependent SUMOylation of AUXIN/INDOLE-3-ACETIC ACID 17 regulates auxin signaling. Plant Physiol. 2023, 191, 1871–1883. [Google Scholar] [CrossRef]
- Hernández-García, J.; Briones-Moreno, A.; Blázquez, M.A. Origin and evolution of gibberellin signaling and metabolism in plants. Semin. Cell Dev. Biol. 2021, 109, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Mierziak, J.; Kostyn, K.; Kulma, A. Flavonoids as important molecules of plant interactions with the environment. Molecules 2014, 19, 16240–16265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; He, M.; Jiang, Z.; Liu, L.; Pu, J.; Zhang, W.; Wang, S.; Xu, F. The xyloglucan endotransglucosylase/hydrolase gene XTH22/TCH4 regulates plant growth by disrupting the cell wall homeostasis in Arabidopsis under boron deficiency. Int. J. Mol. Sci. 2022, 23, 1250. [Google Scholar] [CrossRef] [PubMed]
- Safran, J.; Tabi, W.; Ung, V.; Lemaire, A.; Habrylo, O.; Bouckaert, J.; Rouffle, M.; Voxeur, A.; Pongrac, P.; Bassard, S.; et al. Plant polygalacturonase structures specify enzyme dynamics and processivities to fine-tune cell wall pectins. Plant Cell 2023, 35, 3073–3091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jin, Y.; Ma, Y.; Zhang, Q.; Wang, Q.; Jiang, N.; Zhao, H.; Qu, J.; Guan, S.; Wang, P. Identification of QTLs and candidate genes associated with leaf angle and leaf orientation value in maize (Zea mays L.) based on GBS. Trop. Plant Biol. 2021, 14, 34–49. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Name of Cis-Acting Element | Nucleotide Sequence | Function Annotation |
|---|---|---|---|
| KN1 | TCA-element | TCAGAAGAGG | Salicylic acid-responsive |
| CGTCA-motif | CGTCA | Methyl jasmonate-responsive | |
| TGACG-motif | TGACG | Methyl jasmonate-responsive | |
| CAAT-box | TGCCAAC/CAAAT | Cis-acting element shared by promoters and enhancers | |
| MBS | CAACTG | Drought response-related MYB binding sites | |
| ABRE | AACCCGG | Abscisic acid-responsive | |
| TATA-box | TATA | Core promoter element | |
| CCAAT-box | CAACGG | MYBHv1 binding site | |
| O2-site | GATGATGTGG | Zein metabolism-related cis-acting element | |
| A-box | CCGTCC | Cis-acting regulatory element | |
| CAT-box | GCCACT | Meristem-related cis-acting element | |
| TGA-element | AACGAC | Auxin-responsive | |
| GARE-motif | TCTGTTG | Gibberellins-responsive | |
| P-box | CCTTTTG | Gibberellins-responsive | |
| TCCC-motif | TCTCCCT | Light-responsive | |
| GATA-motif | AAGGATAAGG | Light-responsive | |
| Sp1 | GGGCGG | Light-responsive |
| Field Phenotype and Grain Traits | B73 | kn1 |
|---|---|---|
| Plant height (cm) | 250.39 ± 0.87 a | 203.00 ± 1.15 b |
| Ear height (cm) | 108.78 ± 1.95 a | 93.67 ± 0.73 b |
| Stem diameter (mm) | 22.29 ± 0.38 a | 21.3 ± 0.66 a |
| Chlorophyll content | 50.14 ± 1.64 a | 52.5 ± 0.45 a |
| Leaf length (cm) | 78.54 ± 2.08 a | 79.06 ± 0.97 a |
| Leaf width (cm) | 9.23 ± 0.18 a | 8.30 ± 0.17 b |
| Leaf area size (cm2) | 545.56 ± 23.84 a | 492.74 ± 14.17 a |
| Leaf direction value | 5.35 ± 0.28 a | 6.10 ± 0.38 a |
| Ear weight (g) | 81.52 ± 6.98 a | 64.82 ± 0.95 b |
| Ear length (cm) | 10.87 ± 0.49 a | 10.73 ± 0.23 a |
| Ear diameter (cm) | 3.97 ± 0.32 a | 4.13 ± 0.09 a |
| 100-kernel weight (g) | 16.61 ± 1.42 b | 21.65 ± 0.37 a |
| Quantity of rows | 26.67 ± 1.15 a | 16.67 ± 0.67 b |
| Ear rows | 16.67 ± 1.15 a | 15.33 ± 0.67 a |
| Cob weight (g) | 14.86 ± 1.80 a | 17.20 ± 0.49 a |
| Cob diameter (cm) | 2.77 ± 0.21 a | 3.10 ± 0.03 a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Zhang, Y.; Zhuang, Z.; Ji, X.; Bian, J.; Xian, J.; Wang, Y.; Peng, Y. Phenotypic Investigation and RNA-seq of KN1 Involved in Leaf Angle Formation in Maize (Zea mays L.). Int. J. Mol. Sci. 2024, 25, 3180. https://doi.org/10.3390/ijms25063180
Wu Y, Zhang Y, Zhuang Z, Ji X, Bian J, Xian J, Wang Y, Peng Y. Phenotypic Investigation and RNA-seq of KN1 Involved in Leaf Angle Formation in Maize (Zea mays L.). International Journal of Molecular Sciences. 2024; 25(6):3180. https://doi.org/10.3390/ijms25063180
Chicago/Turabian StyleWu, Yuanming, Yunfang Zhang, Zelong Zhuang, Xiangzhuo Ji, Jianwen Bian, Jinhong Xian, Yinxia Wang, and Yunling Peng. 2024. "Phenotypic Investigation and RNA-seq of KN1 Involved in Leaf Angle Formation in Maize (Zea mays L.)" International Journal of Molecular Sciences 25, no. 6: 3180. https://doi.org/10.3390/ijms25063180
APA StyleWu, Y., Zhang, Y., Zhuang, Z., Ji, X., Bian, J., Xian, J., Wang, Y., & Peng, Y. (2024). Phenotypic Investigation and RNA-seq of KN1 Involved in Leaf Angle Formation in Maize (Zea mays L.). International Journal of Molecular Sciences, 25(6), 3180. https://doi.org/10.3390/ijms25063180