
Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers


Abstract
1. Introduction
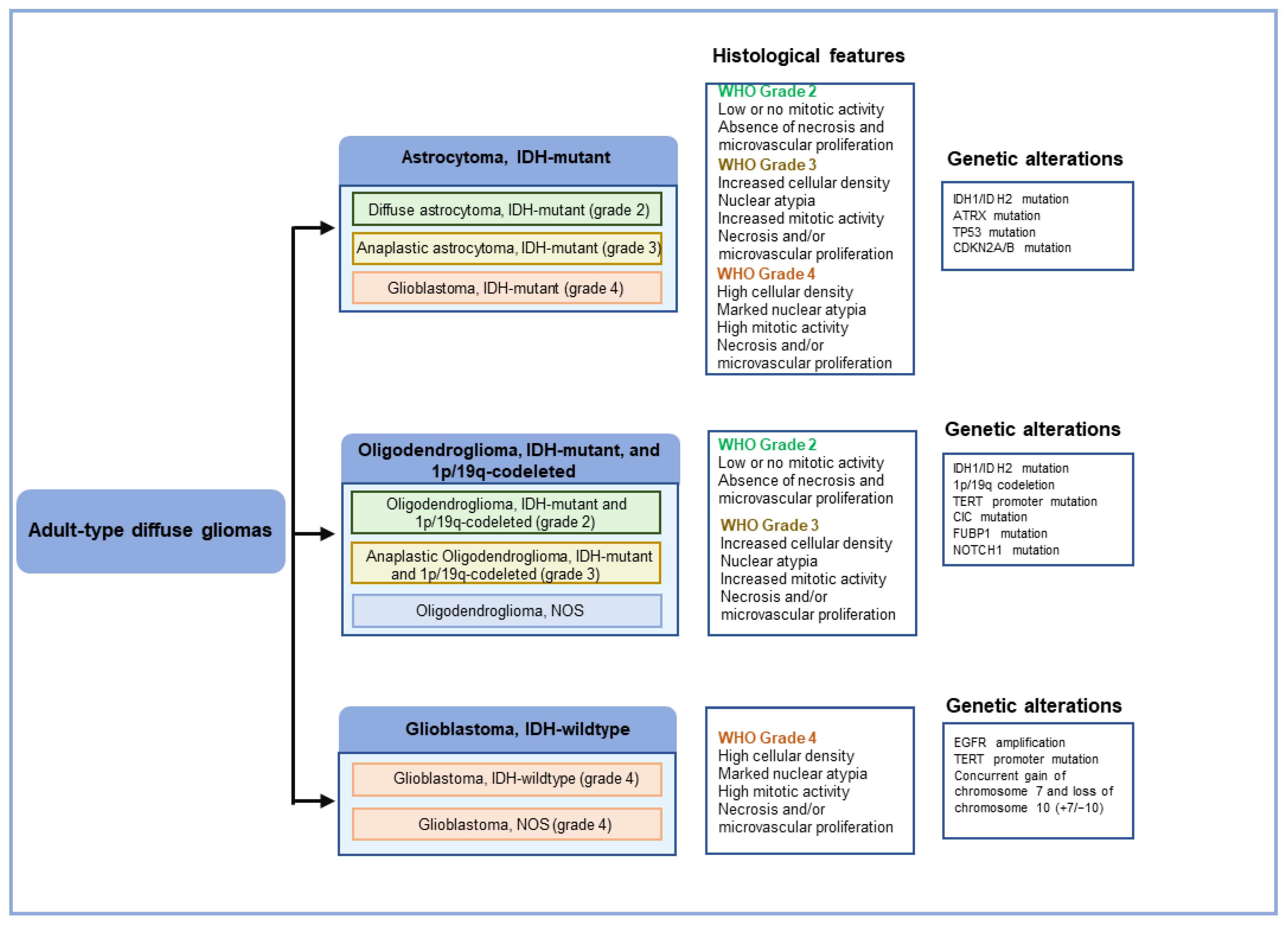
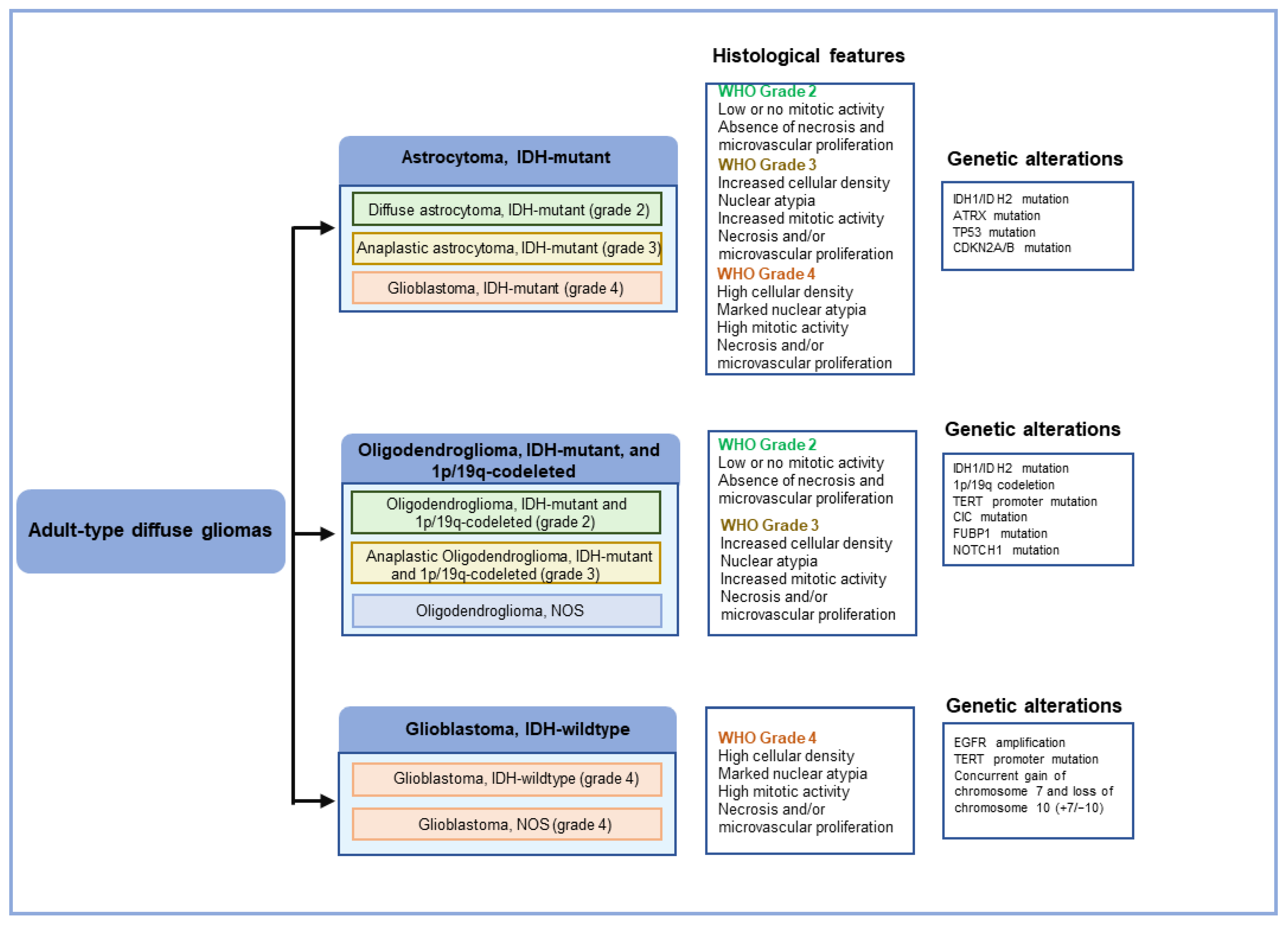
2. Histopathology of GBM
3. Molecular Pathology of GBM
3.1. Genetic Changes in GBM
3.2. Epigenetic Changes in GBM
3.3. Transcriptomic Changes in GBM
3.4. Correlation between Genetic, Epigenetic and Transcriptomic Alterations in GBM
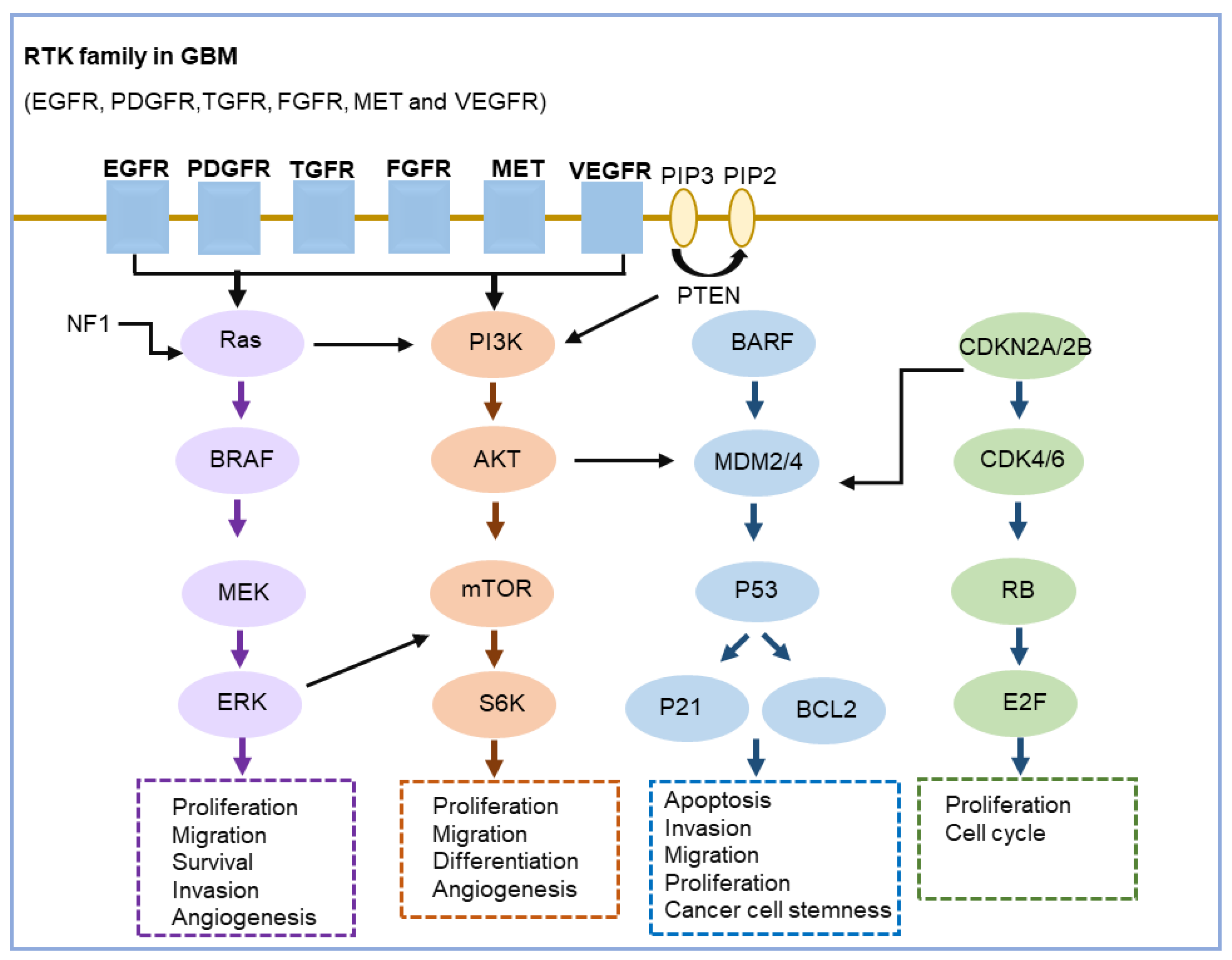
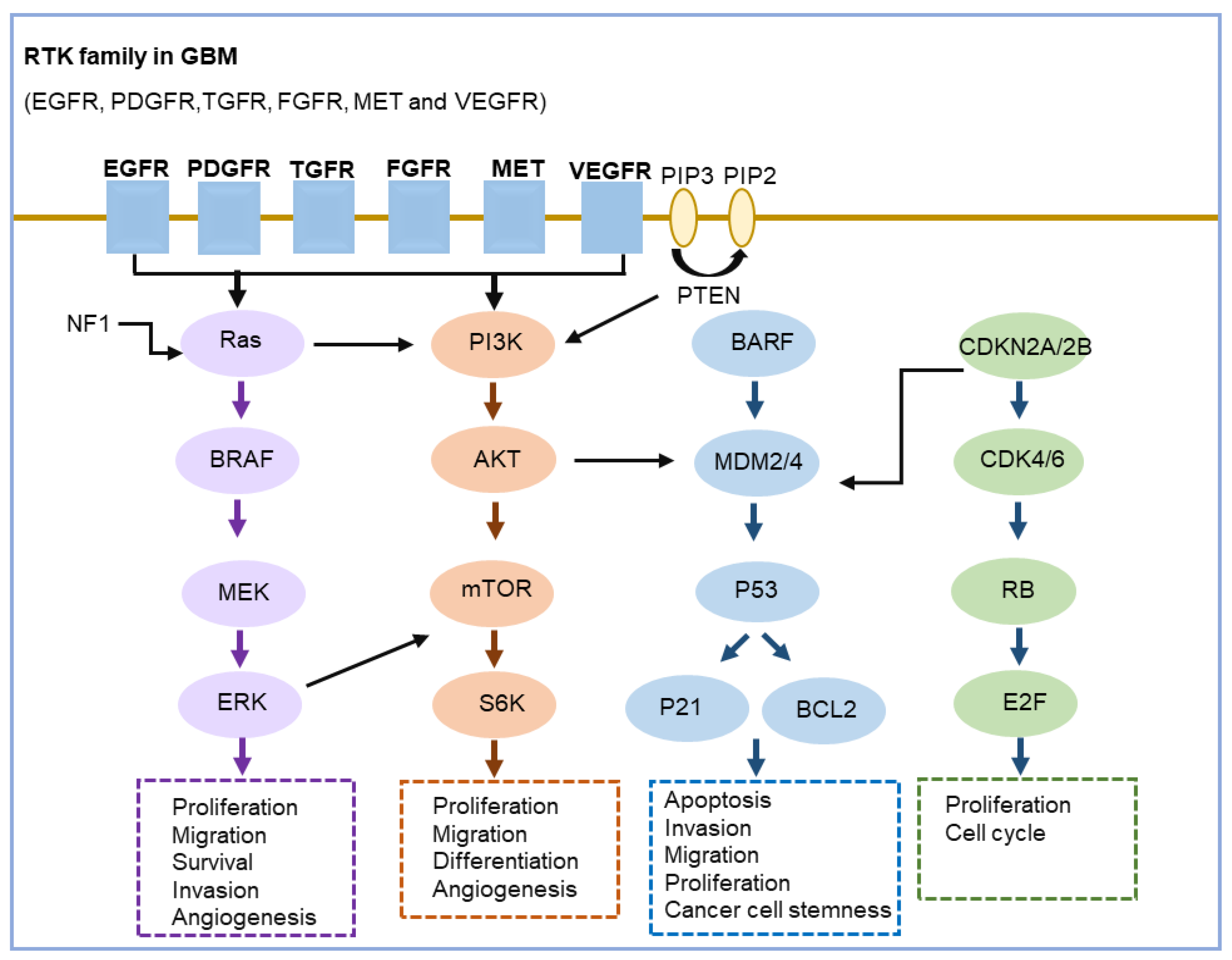
3.5. Key Signaling Pathways Altered in GBM
4. Clinically Relevant Molecular Biomarkers in GBM
5. Challenges and Future Directions
6. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef]
- Xiao, D.; Yan, C.; Li, D.; Xi, T.; Liu, X.; Zhu, D.; Huang, G.; Xu, J.; He, Z.; Wu, A.; et al. National Brain Tumour Registry of China (NBTRC) statistical report of primary brain tumours diagnosed in China in years 2019–2020. Lancet Reg. Health. West. Pac. 2023, 34, 100715. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016–2020. Neuro-Oncology 2023, 25, iv1–iv99. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2021, 10, 574012. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA-J. Am. Med. Assoc. 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Molinaro, A.M.; Taylor, J.W.; Wiencke, J.K.; Wrensch, M.R. Genetic and molecular epidemiology of adult diffuse glioma. Nat. Rev. Neurol. 2019, 15, 405–417. [Google Scholar] [CrossRef]
- Esemen, Y.; Awan, M.; Parwez, R.; Baig, A.; Rahman, S.; Masala, I.; Franchini, S.; Giakoumettis, D. Molecular Pathogenesis of Glioblastoma in Adults and Future Perspectives: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2607. [Google Scholar] [CrossRef]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Verdugo, E.; Puerto, I.; Medina, M.A. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun. 2022, 42, 1083–1111. [Google Scholar] [CrossRef]
- Liebermana, F. Glioblastoma update: Molecular biology, diagnosis, treatment, response assessment, and translational clinical trials. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Ludwig, K.; Muthukrishnan, S.D.; Alvarado, A.G.; Kornblum, H.I. Overview of glioblastoma biological hallmarks and molecular pathology. In Glioblastoma Resistance to Chemotherapy: Molecular Mechanisms and Innovative Reversal Strategies; Academic Press: Cambridge, MA, USA, 2021; Volume 15, pp. 1–15. [Google Scholar]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers 2019, 11, 469. [Google Scholar] [CrossRef]
- Delgado-Martin, B.; Medina, M.A. Advances in the Knowledge of the Molecular Biology of Glioblastoma and Its Impact in Patient Diagnosis, Stratification, and Treatment. Adv. Sci. 2020, 7, 1902971. [Google Scholar] [CrossRef]
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas—Implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, Q.; Liu, L.Q.; Li, S.W.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- del Pilar Guillermo Prieto, M.; de La Fuente, M.I. The Role of Molecular Genetics of Glioblastoma in the Clinical Setting. In Precision Molecular Pathology of Glioblastoma; Springer: Berlin/Heidelberg, Germany, 2021; pp. 21–33. [Google Scholar]
- Yang, K.Y.; Wu, Z.J.; Zhang, H.; Zhang, N.; Wu, W.T.; Wang, Z.Y.; Dai, Z.Y.; Zhang, X.; Zhang, L.Y.; Peng, Y.; et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Eisenbarth, D.; Wang, Y.A. Glioblastoma heterogeneity at single cell resolution. Oncogene 2023, 42, 2155–2165. [Google Scholar] [CrossRef]
- Johnson, K.C.; Anderson, K.J.; Courtois, E.T.; Barthel, F.P.; Varn, F.S.; Luo, D.N.; Seignon, M.; Yi, E.; Kim, H.; Estecio, M.R.H.; et al. Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response. Nat. Genet. 2021, 53, 1456–1468. [Google Scholar] [CrossRef]
- LeBlanc, V.G.; Trinh, D.L.; Aslanpour, S.; Hughes, M.; Livingstone, D.; Jin, D.; Ahn, B.Y.; Blough, M.D.; Cairncross, J.G.; Chan, J.A.; et al. Single-cell landscapes of primary glioblastomas and matched explants and cell lines show variable retention of inter- and intratumor heterogeneity. Cancer Cell 2022, 40, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jung, J.; Babikir, H.; Shamardani, K.; Jain, S.; Feng, X.; Gupta, N.; Rosi, S.; Chang, S.; Raleigh, D.; et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Cancer 2022, 3, 1534–1552. [Google Scholar] [CrossRef] [PubMed]
- Koh, L.; Novera, W.; Lim, S.W.; Chong, Y.K.; Pang, Q.Y.; Low, D.; Ang, B.T.; Tang, C. Integrative multi-omics approach to targeted therapy for glioblastoma. Pharmacol. Res. 2022, 182, 106308. [Google Scholar] [CrossRef]
- Heo, Y.J.; Hwa, C.; Lee, G.H.; Park, J.M.; An, J.Y. Integrative Multi-Omics Approaches in Cancer Research: From Biological Networks to Clinical Subtypes. Mol. Cells 2021, 44, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salié, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially resolved multi-omics deciphers bidirectional tumor-host interdependence in glioblastoma. Cancer Cell 2022, 40, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Blaavand, M.S.; Daubon, T.; Kruyt, F.A.; Thomsen, M.K. Three-dimensional culture models to study glioblastoma—Current trends and future perspectives. Curr. Opin. Pharmacol. 2021, 61, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, A.; Goranci-Buzhala, G.; Ricci-Vitiani, L.; Pallini, R.; Gopalakrishnan, J. Trends and challenges in modeling glioma using 3D human brain organoids. Cell Death Differ. 2021, 28, 15–23. [Google Scholar] [CrossRef]
- Zhang, C.C.; Jin, M.Z.; Zhao, J.N.; Chen, J.X.; Jin, W.L. Organoid models of glioblastoma: Advances, applications and challenges. Am. J. Cancer Res. 2020, 10, 2242–2257. [Google Scholar] [PubMed]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Aaroe, A.; Liang, J.Y.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, N.; Nobusawa, S.; Nakata, S.; Nakada, M.; Yamazaki, T.; Matsumura, N.; Harada, K.; Matsuda, H.; Funata, N.; Nagai, S.; et al. BRAF V600E, TERT promoter mutations and CDKN2A/B homozygous deletions are frequent in epithelioid glioblastomas: A histological and molecular analysis focusing on intratumoral heterogeneity. Brain Pathol. 2018, 28, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.E.; Soshnev, A.A.; Allis, C.D. Epigenomic Reprogramming as a Driver of Malignant Glioma. Cancer Cell 2020, 38, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Montella, L.; Cuomo, M.; Del Gaudio, N.; Buonaiuto, M.; Costabile, D.; Visconti, R.; Di Risi, T.; Vinciguerra, R.; Trio, F.; Ferraro, S.; et al. Epigenetic alterations in glioblastomas: Diagnostic, prognostic and therapeutic relevance. Int. J. Cancer 2023, 153, 476–488. [Google Scholar] [CrossRef]
- Uddin, M.S.; Al Mamun, A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2022, 83, 100–120. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro-Oncology 2019, 21, 167–178. [Google Scholar] [CrossRef]
- Della Monica, R.; Cuomo, M.; Buonaiuto, M.; Costabile, D.; Franca, R.A.; De Caro, M.D.; Catapano, G.; Chiariotti, L.; Visconti, R. MGMT and Whole-Genome DNA Methylation Impacts on Diagnosis, Prognosis and Therapy of Glioblastoma Multiforme. Int. J. Mol. Sci. 2022, 23, 7148. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef]
- McClellan, B.L.; Haase, S.; Nunez, F.J.; Alghamri, M.S.; Dabaja, A.A.; Lowenstein, P.R.; Castro, M.G. Impact of epigenetic reprogramming on antitumor immune responses in glioma. J. Clin. Investig. 2023, 133, e163450. [Google Scholar] [CrossRef]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front. Oncol. 2018, 8, 448. [Google Scholar] [CrossRef]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E.; Mcsherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncologist 2018, 23, 157-e21. [Google Scholar] [CrossRef] [PubMed]
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.Z.; Zhou, W.; Shi, G.Q.; Imitola, J.; Li, C.L.; Hsu, S.; Wang, J.; et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro-Oncology 2018, 20, 753–763. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.H.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef] [PubMed]
- Antunes, A.R.P.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Di Nunno, V.; Franceschi, E.; Tosoni, A.; Gatto, L.; Bartolini, S.; Brandes, A.A. Glioblastoma Microenvironment: From an Inviolable Defense to a Therapeutic Chance. Front. Oncol. 2022, 12, 852950. [Google Scholar] [CrossRef]
- Martínez, A.H.; Madurga, R.; García-Romero, N.; Ayuso-Sacido, A. Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer Lett. 2022, 527, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Meyer, M.; Reimand, J.; Lan, X.Y.; Head, R.; Zhu, X.M.; Kushida, M.; Bayani, J.; Pressey, J.C.; Lionel, A.C.; Clarke, I.D.; et al. Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. USA 2015, 112, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef]
- Meng, Q.K.; Zhang, Y.; Li, G.Q.; Li, Y.N.; Xie, H.B.; Chen, X.J. New insights for precision treatment of glioblastoma from analysis of single-cell lncRNA expression. J. Cancer Res. Clin. Oncol. 2021, 147, 1881–1895. [Google Scholar] [CrossRef]
- Wu, H.B.; Guo, C.C.; Wang, C.Y.; Xu, J.; Zheng, S.Y.; Duan, J.; Li, Y.Y.; Bai, H.M.; Xu, Q.Y.; Ning, F.L.; et al. Single-cell RNA sequencing reveals tumor heterogeneity, microenvironment, and drug-resistance mechanisms of recurrent glioblastoma. Cancer Sci. 2023, 114, 2609–2621. [Google Scholar] [CrossRef]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Korshunov, A.; Schrimpf, D.; Ryzhova, M.; Sturm, D.; Chavez, L.; Hovestadt, V.; Sharma, T.; Habel, A.; Burford, A.; Jones, C.; et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 2017, 134, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Khabibov, M.; Garifullin, A.; Boumber, Y.; Khaddour, K.; Fernandez, M.; Khamitov, F.; Khalikova, L.; Kuznetsova, N.; Kit, O.; Kharin, L. Signaling pathways and therapeutic approaches in glioblastoma multiforme (Review). Int. J. Oncol. 2022, 60, 69. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, O.; Horescu, C.; Sevastre, A.S.; Cioc, C.E.; Baloi, C.; Oprita, A.; Dricu, A. Receptor tyrosine kinase targeting in glioblastoma: Performance, limitations and future approaches. Wspolczesna Onkol. 2020, 24, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Wu, C.J.; Chen, N.C.; Gu, H.D.; Yen, A.; Cao, L.; Wang, E.H.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Dewdney, B.; Jenkins, M.R.; Best, S.A.; Freytag, S.; Prasad, K.; Holst, J.; Endersby, R.; Johns, T.G. From signalling pathways to targeted therapies: Unravelling glioblastoma’s secrets and harnessing two decades of progress. Signal Transduct. Target. Ther. 2023, 8, 400. [Google Scholar] [CrossRef] [PubMed]
- Oprita, A.; Baloi, S.C.; Staicu, G.A.; Alexandru, O.; Tache, D.E.; Danoiu, S.; Micu, E.S.; Sevastre, A.S. Updated Insights on EGFR Signaling Pathways in Glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef] [PubMed]
- Qin, A.; Musket, A.; Musich, P.R.; Schweitzer, J.B.; Xie, Q. Receptor tyrosine kinases as druggable targets in glioblastoma: Do signaling pathways matter? Neuro-Oncol. Adv. 2021, 3, vdab133. [Google Scholar] [CrossRef]
- Kim, G.; Ko, Y.T. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch. Pharm. Res. 2020, 43, 385–394. [Google Scholar] [CrossRef]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.M.; Coughlan, M.; Yang, Y.Z.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The p53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Chkheidze, R.; Raisanen, J.; Gagan, J.; Richardson, T.E.; Pinho, M.C.; Raj, K.; Achilleos, M.; Slepicka, C.; White, C.L.; Evers, B.M.; et al. Alterations in the RB Pathway With Inactivation of Characterize Glioblastomas With a Primitive Neuronal Component. J. Neuropathol. Exp. Neurol. 2021, 80, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Suwala, A.K.; Stichel, D.; Schrimpf, D.; Maas, S.L.N.; Sill, M.; Dohmen, H.; Banan, R.; Reinhardt, A.; Sievers, P.; Hinz, F.; et al. Glioblastomas with primitive neuronal component harbor a distinct methylation and copy-number profile with inactivation of TP53, PTEN, and RB1. Acta Neuropathol. 2021, 142, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Tien, A.C.; Li, J.; Bao, X.; Derogatis, A.; Kim, S.; Mehta, S.; Sanai, N. A Phase 0 Trial of Ribociclib in Recurrent Glioblastoma Patients Incorporating a Tumor Pharmacodynamic- and Pharmacokinetic-Guided Expansion Cohort. Clin. Cancer Res. 2019, 25, 5777–5786. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Arasaratnam, M.; Chan, D.L.H.; Khasraw, M.; Howell, V.M.; Wheeler, H. Anti-epidermal growth factor receptor therapy for glioblastoma in adults. Cochrane Syst. Rev. 2020, 5, CD013238. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Kamel, A.; Ciubotaru, G.V.; Onose, G.; Sevastre, A.S.; Sfredel, V.; Danoiu, S.; Dricu, A.; Tataranu, L.G. An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma. Int. J. Mol. Sci. 2023, 24, 11110. [Google Scholar] [CrossRef]
- Khan, I.; Waqas, M.; Shamim, M.S. Prognostic significance of IDH 1 mutation in patients with glioblastoma multiforme. J. Pak. Med. Assoc. 2017, 67, 816. [Google Scholar]
- Han, S.; Liu, Y.; Cai, S.R.J.; Qian, M.Y.; Ding, J.Y.; Larion, M.; Gilbert, M.R.; Yang, C.Z. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Butler, M.; Pongor, L.; Su, Y.T.; Xi, L.Q.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Brar, H.K.; Jose, J.; Wu, Z.M.; Sharma, M. Tyrosine Kinase Inhibitors for Glioblastoma Multiforme: Challenges and Opportunities for Drug Delivery. Pharmaceutics 2023, 15, 59. [Google Scholar] [CrossRef] [PubMed]
- Aldaz, P.; Arozarena, I. Tyrosine Kinase Inhib. Adult Glioblastoma: (Un)Closed Chapter? Cancers 2021, 13, 5799. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J. Neuro-Oncol. 2018, 140, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Q.; Trippa, L.; Fell, G.; Rahman, R.; Arrillaga-Romany, I.; Touat, M.; Drappatz, J.; Welch, M.R.; Galanis, E.; Ahluwalia, M.S.; et al. Preliminary results of the abemaciclib arm in the Individualized Screening Trial of Innovative Glioblastoma Therapy (INSIGhT): A phase II platform trial using Bayesian adaptive randomization. J. Clin. Oncol. 2021, 39. [Google Scholar] [CrossRef]
- Sepúlveda-Sánchez, J.M.; Gil-Gil, M.; Alonso-García, M.; Salgado, M.A.V.; Vicente, E.; Barroso, C.M.; Sánchez, A.R.; Durán, G.; De Las Peñas, R.; Muñoz-Langa, J.; et al. Phase II Trial of Palbociclib in Recurrent Retinoblastoma-Positive Anaplastic Oligodendroglioma: A Study from the Spanish Group for Research in Neuro-Oncology (GEINO). Target. Oncol. 2020, 15, 613–622. [Google Scholar] [CrossRef]
- van den Bent, M.J.; Klein, M.; Smits, M.; Reijneveld, J.C.; French, P.J.; Clement, P.; de Vos, F.Y.F.; Wick, A.; Mulholland, P.J.; Taphoorn, M.J.B.; et al. Bevacizumab and temozolomide in patients with first recurrence of WHO grade II and III glioma, without 1p/19q co-deletion (TAVAREC): A randomised controlled phase 2 EORTC trial. Lancet Oncol. 2018, 19, 1170–1179. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Weller, M.; Lim, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.; Ashby, L.; Ansstas, G.; Baehring, J.; Taylor, J.; et al. A Randomized Phase 3 Study of Nivolumab or Placebo Combined with Radiotherapy Plus Temozolomide in Patients with Newly Diagnosed Glioblastoma with Methylated Mgmt Promoter: Checkmate 548. Neuro-Oncology 2021, 23, 55–56. [Google Scholar] [CrossRef]
- Sampson, J.H.; Omuro, A.M.P.; Preusser, M.; Lim, M.; Butowski, N.A.; Cloughesy, T.F.; Strauss, L.C.; Latek, R.R.; Paliwal, P.; Weller, M.; et al. A randomized, phase 3, open-label study of nivolumab versus temozolomide (TMZ) in combination with radiotherapy (RT) in adult patients (pts) with newly diagnosed, O-6-methylguanine DNA methyltransferase (MGMT)-unmethylated glioblastoma (GBM): CheckMate-498. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- van den Bent, M.; Eoli, M.; Sepulveda, J.M.; Smits, M.; Walenkamp, A.; Frenel, J.S.; Franceschi, E.; Clement, P.M.; Chinot, O.; de Vos, F.Y.F.L.; et al. INTELLANCE 2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2020, 22, 684–693. [Google Scholar] [CrossRef]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro-Oncology 2018, 20, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.X.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.C.; Tang, L.; Li, X.Z.; Fan, F.; Liu, Z.X. Immunotherapy for glioma: Current management and future application. Cancer Lett. 2020, 476, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.B.; Ajina, R.; Aref, S.; Darwish, M.; Alsayb, M.; Taher, M.; AlSharif, S.A.; Hashem, A.M.; Alkayyal, A.A. Advances in immunotherapy for glioblastoma multiforme. Front. Immunol. 2022, 13, 944452. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Tumor Type | CNS WHO Grade |
|---|---|
| Adult-type diffuse gliomas | |
| Astrocytoma, IDH-mutant | 2, 3, 4 |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | 2, 3 |
| Glioblastoma, IDH-wildtype | 4 |
| Pediatric-type diffuse low-grade gliomas | |
| Diffuse astrocytoma, MYB- or MYBL1-altered # | 1 |
| Angiocentric glioma | 1 |
| Polymorphous low-grade neuroepithelial tumor of the young # | 1 |
| Diffuse low-grade glioma, MAPK pathway-altered *# | - |
| Pediatric-type diffuse high-grade gliomas | |
| Diffuse midline glioma, H3 K27-altered | 4 |
| Diffuse hemispheric glioma, H3 G34-mutant # | 4 |
| Diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype # | 4 |
| Infant-type hemispheric glioma *# | - |
| Circumscribed astrocytic gliomas | |
| Pilocytic astrocytoma | 1 |
| High-grade astrocytoma with piloid features *# | - |
| Pleomorphic xanthoastrocytoma | 2, 3 |
| Subependymal giant cell astrocytoma | 1 |
| Chordoid glioma | 2 |
| Astroblastoma, MN1 altered * | - |
| Ependymal tumors |
| Tumor Type | Genes/Molecular Profiles Characteristically Altered |
|---|---|
| Astrocytoma, IDH-mutant | IDH1, IDH2, ATRX, TP53, CDKN2A/B |
| Oligodendroglioma, IDH-mutant, and 1p/19q-codeleted | IDH1, IDH2, 1p/19q, TERT promoter, CIC, FUBP1, NOTCH1 |
| Glioblastoma, IDH-wildtype | IDH-wild type, TERT promoter, chromosomes 7/10, EGFR |
| Diffuse astrocytoma, MYB- or MYBL1-altered | MYB, MYBL1 |
| Angiocentric glioma | MYB |
| Polymorphous low-grade neuroepithelial tumor of the young | BRAF, FGFR family |
| Diffuse low-grade glioma, MAPK pathway-altered | FGFR1, BRAF |
| Diffuse midline glioma, H3 K27-altered | H3 K27, TP53, ACVR1, PDGFRA, EGFR, EZHIP |
| Diffuse hemispheric glioma, H3 G34-mutant | H3 G34, TP53, ARTX |
| Diffuse pediatric-type high-grade glioma, H3-wildtype, and IDH-wildtype | IDH-wildtype, H3-wildtype, PDGFRA, MYCN, EGFR (methylome) |
| Infant-type hemispheric glioma | NTRK family, ALK, ROS, MET |
| Pilocytic astrocytoma | KIAA1549-BRAF, BRAF, NF1 |
| High-grade astrocytoma with piloid features | BRAF, NF1, ATRX, CDKN2A/B (methylome) |
| Pleomorphic xanthoastrocytoma | BRAF, CDKN2A/B |
| Subependymal giant cell astrocytoma | TSC1, TSC2 |
| Chordoid glioma | PRKCA |
| Astroblastoma, MN1-altered | MN1 |
| Supratentorial ependymomas | ZFTA, RELA, YAP1, MAML2 |
| Posterior fossa ependymomas | H3 K27me3, EZHIP (methylome) |
| Spinal ependymomas | NF2, MYCN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, Z.; Li, X.; Zhang, X. Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers. Int. J. Mol. Sci. 2024, 25, 3040. https://doi.org/10.3390/ijms25053040
Lan Z, Li X, Zhang X. Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers. International Journal of Molecular Sciences. 2024; 25(5):3040. https://doi.org/10.3390/ijms25053040
Chicago/Turabian StyleLan, Zhong, Xin Li, and Xiaoqin Zhang. 2024. "Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers" International Journal of Molecular Sciences 25, no. 5: 3040. https://doi.org/10.3390/ijms25053040
APA StyleLan, Z., Li, X., & Zhang, X. (2024). Glioblastoma: An Update in Pathology, Molecular Mechanisms and Biomarkers. International Journal of Molecular Sciences, 25(5), 3040. https://doi.org/10.3390/ijms25053040