
Comparative Analysis and Phylogenetic Study of Dawkinsia filamentosa and Pethia nigrofasciata Mitochondrial Genomes


Abstract
1. Introduction
2. Results
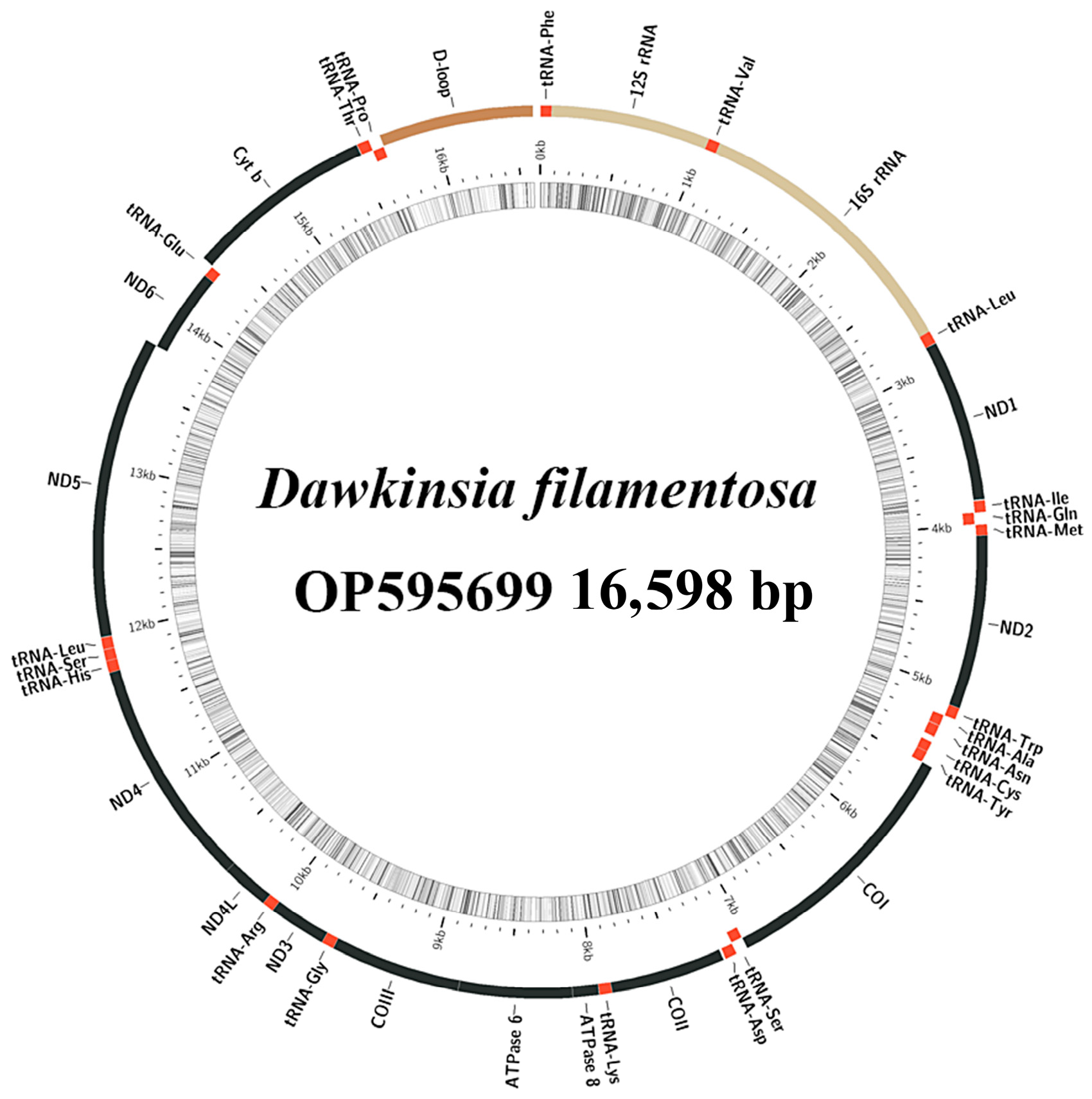
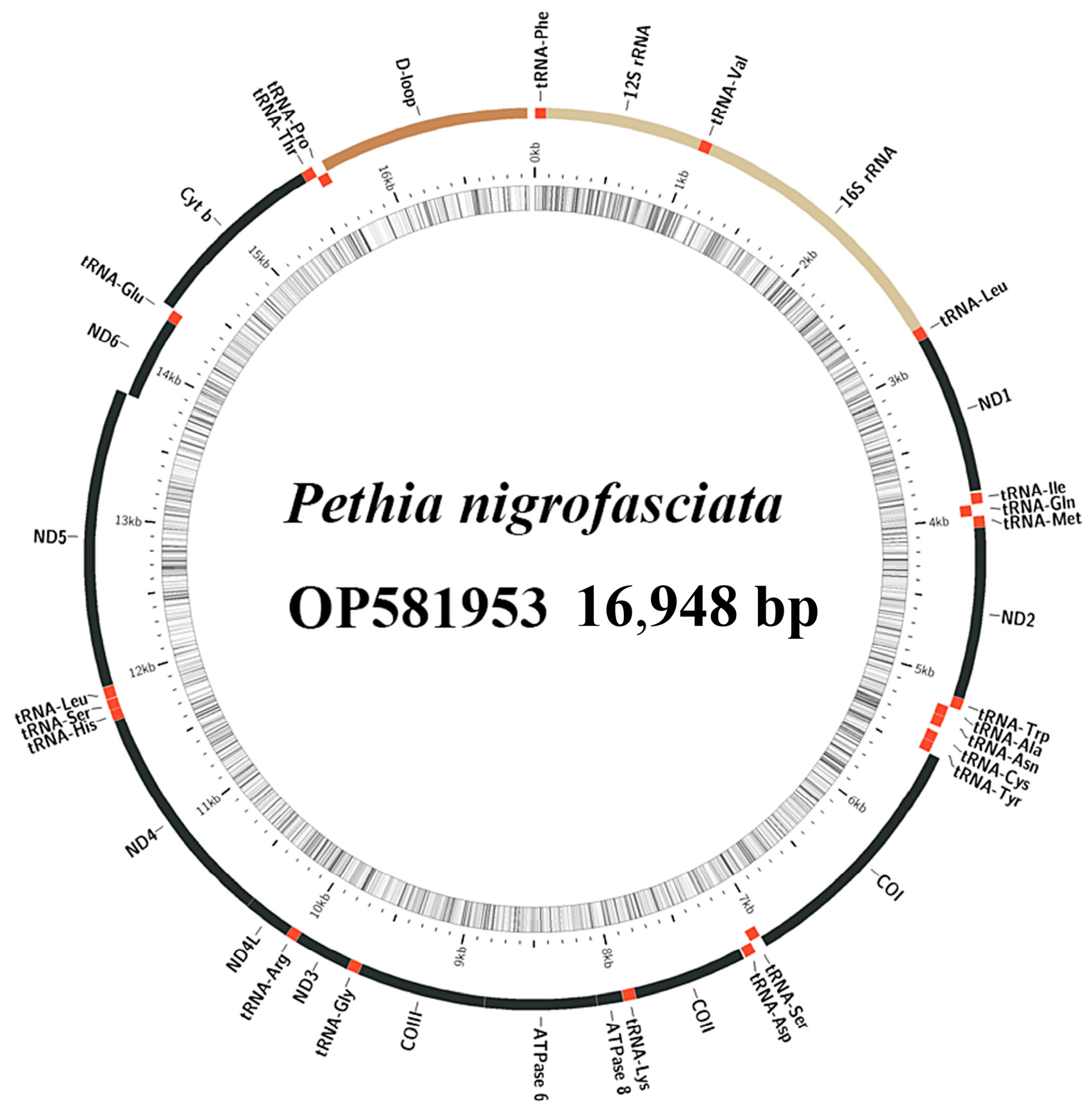
2.1. Mitochondrial Genome Analysis
2.2. Protein-Coding Gene Analysis
2.3. tRNAs and rRNAs Gene Analysis
2.4. Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Experimental Materials and DNA Extraction
4.2. High-Throughput Sequencing and Gene Annotation
4.3. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, M.; Armbruster, J.W. Phylogenetic classification of extant genera of fishes of the order Cypriniformes (Teleostei: Ostariophysi). Zootaxa 2018, 4476, 6–39. [Google Scholar] [CrossRef] [PubMed]
- Mian, S.; Ferdous, M.J.; Sarker, M.Y.; Islam, M.J.; Reza, A.M.; Iqbal, M.M.; Hossain, M.A.R. Status of biodiversity and conservation of freshwater barbs in Bangladesh. World J. Fish Mar. Sci. 2013, 5, 701–708. [Google Scholar]
- Mohsin, A.B.M.; Haque, S.M.M.; Chaki, N.; Fazad, F.H. Seasonal abundance of finfish in the Padma River in the Rajshahi District, Bangladesh. World J. Fish Mar. Sci. 2013, 5, 680–685. [Google Scholar]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. A mitogenomic perspective on basal teleostean phylogeny: Resolving higher-level relationships with longer DNA sequences. Mol. Phylogenet. Evol. 2001, 20, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K.; Severson, D.W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Biol. Rev. Camb. Philos. Soc. 2013, 88, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.H.; Sun, P.Y.; Lao, Y.L.; Wu, T.; Zhang, Y.N.; Huang, Q.; Zhang, Q. Mitogenome of a monotypic genus, Oliotius Kottelat, 2013 (Cypriniformes: Cyprinidae): Genomic characterization and phylogenetic position. Gene 2023, 851, 147035. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at four-fold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Mullens, N.; Sonet, G.; Decru, E.; Virgilio, M.; Snoeks, K.; Emmanuel, V. Mitogenomic characterization and systematic placement of the Congo blind barb Caecobarbus geertsii (Cypriniformes: Cyprinidae). Int. J. Biol. Macromol. 2020, 161, 292–298. [Google Scholar] [CrossRef]
- Frank, A.C.; Lobry, J.R. Asymmetric substitution patterns: A review of possible underlying mutational or selective mechanisms. Gene 1999, 238, 65–77. [Google Scholar] [CrossRef]
- Lazarou, M.; Thorburn, D.R.; Ryan, M.T.; McKenzie, M. Assembly of mitochondrial complex I and defects in disease. Biochim. Biophys. Acta 2009, 1793, 78–88. [Google Scholar] [CrossRef]
- Efremov, R.G.; Baradaran, R.; Sazanov, L.A. The architecture of respiratory complex I. Nature 2010, 465, 441–445. [Google Scholar] [CrossRef]
- Miya, M.; Sato, Y.; Fukunaga, T.; Sado, T.; Poulsen, J.Y.; Sato, K.; Minamoto, T.; Yamamoto, S.; Yamanaka, H.; Araki, H.; et al. MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fish: Detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2015, 2, 150088. [Google Scholar] [CrossRef]
- Lynch, M. Mutation accumulation in nuclear, organelle and prokaryotic transfer RNA genes. Mol. Biol. Evol. 1997, 14, 914–925. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Islam, N.N.; Akter, J.A.; Sanzida, N.J. DNA barcoding of Smiliogastrinae (Teleostei: Cypriniformes) of Bangladesh based on cytochrome c oxidase subunit I (coi) sequences. Zool. Ecol. 2021, 67, 78. [Google Scholar] [CrossRef]
- Sudasinghe, H.; Rüber, L.; Meegaskumbura, M. Molecular phylogeny and systematics of the South Asian freshwater-fish genus Puntius (Teleostei: Cyprinidae). Zool. Scr. 2023, 52, 571–587. [Google Scholar] [CrossRef]
- Mamiatis, T.; Fritsch, F.; Sambrook, J.; Engel, J. Molecular Cloning of a Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory. Acta Biotechnol. 1985, 5, 104. [Google Scholar]
- Li, R.Q.; Zhu, H.M.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genome using massive parallel short-read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef]
- Zhao, Q.Y.; Wang, Y.; Kong, Y.M.; Luo, D.; Li, X.; Hao, P. Optimizing de novo transcriptome assembly from short-read RNA-seq data: A comparative study. BMC Bioinform. 2011, 12, S2. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole-genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar]
- Lowe, T.M.; Chan, P.P. TRNAscan-SE online: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Chandra, S.; Abhilash, R.; Sidharthan, A.; Raghavan, R.; Dahanukar, N. Complete mitogenome of Lepidopygopsis typus, an evolutionarily distinct, endangered cyprinid fish from the Western Ghats Biodiversity Hotspot: Phylogenetic relationships and implications for conservation. Gene 2023, 898, 148098. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm for estimating large phylogenies using maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MBayes: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Vrieze, S.I. Model selection and psychological theory: A discussion of the differences between the Akaike information criterion (AIC) and Bayesian information criterion (BIC). Psychol. Methods 2012, 17, 228–243. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Mark, A.M.; Terri, S.; Wayne, P. Embedding CIPRES Science Gateway Capabilities in Phylogenetics Software Environments. In XSEDE ‘13: Proceedings of the Conference on Extreme Science and Engineering Discovery Environment: Gateway to Discovery; Association for Computing Machinery: New York, NY, USA, 2013; Volume 40, pp. 1–8. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Gene | Position | Size | Intergenic Nucleotides | Codon | Strand | ||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| tRNA-Phe | 1 | 69 | 69 | 0 | H | ||
| 12S rRNA | 70 | 1024 | 955 | 0 | H | ||
| tRNA-Val | 1025 | 1096 | 72 | 0 | H | ||
| 16S rRNA | 1097 | 2791 | 1695 | 0 | H | ||
| tRNA-Leu2 | 2792 | 2868 | 77 | 0 | H | ||
| ND1 | 2870 | 3844 | 975 | 1 | ATG | TAA | H |
| tRNA-Ile | 3849 | 3920 | 72 | 4 | H | ||
| tRNA-Gln | 3919 | 3989 | 71 | −2 | L | ||
| tRNA-Met | 3992 | 4061 | 70 | 2 | H | ||
| ND2 | 4062 | 5106 | 1045 | 0 | ATG | T | H |
| tRNA-Trp | 5107 | 5177 | 71 | 0 | H | ||
| tRNA-Ala | 5179 | 5247 | 69 | 1 | L | ||
| tRNA-Asn | 5249 | 5321 | 73 | 1 | L | ||
| tRNA-Cys | 5357 | 5423 | 67 | 35 | L | ||
| tRNA-Tyr | 5423 | 5493 | 71 | −1 | L | ||
| COI | 5495 | 7045 | 1551 | 1 | GTG | TAA | H |
| tRNA-Ser2 | 7046 | 7116 | 71 | 0 | L | ||
| tRNA-Asp | 7118 | 7189 | 72 | 1 | H | ||
| COII | 7202 | 7892 | 691 | 12 | ATG | T | H |
| tRNA-Lys | 7893 | 7968 | 76 | 0 | H | ||
| ATP8 | 7970 | 8134 | 165 | 1 | ATG | TAG | H |
| ATP6 | 8128 | 8810 | 683 | −7 | ATG | TA | H |
| COIII | 8811 | 9595 | 785 | 0 | ATG | TA | H |
| tRNA-Gly | 9596 | 9668 | 73 | 0 | H | ||
| ND3 | 9669 | 10017 | 349 | 0 | ATG | T | H |
| tRNA-Arg | 10018 | 10087 | 70 | 0 | H | ||
| ND4L | 10088 | 10384 | 297 | 0 | ATG | TAA | H |
| ND4 | 10378 | 11758 | 1381 | −7 | ATG | T | H |
| tRNA-His | 11759 | 11827 | 69 | 0 | H | ||
| tRNA-Ser1 | 11828 | 11896 | 69 | 0 | H | ||
| tRNA-Leu1 | 11898 | 11970 | 73 | 1 | H | ||
| ND5 | 11972 | 13792 | 1821 | 1 | ATG | TAA | H |
| ND6 | 13789 | 14310 | 522 | −4 | ATG | TAA | L |
| tRNA-Glu | 14311 | 14378 | 68 | 0 | L | ||
| Cytb | 14385 | 15521 | 1137 | 6 | ATG | TAA | H |
| tRNA-Thr | 15525 | 15597 | 73 | 3 | H | ||
| tRNA-Pro | 15597 | 15667 | 71 | −1 | L | ||
| D-loop | 15668 | 16598 | 931 | 0 | H | ||
| Gene | Position | Size | Intergenic Nucleotides | Codon | Strand | ||
|---|---|---|---|---|---|---|---|
| From | To | Start | Stop | ||||
| tRNA-Phe | 1 | 69 | 69 | 0 | H | ||
| 12S rRNA | 70 | 1025 | 956 | 0 | H | ||
| tRNA-Val | 1026 | 1097 | 72 | 0 | H | ||
| 16S rRNA | 1098 | 2781 | 1684 | 0 | H | ||
| tRNA-Leu2 | 2782 | 2856 | 75 | 0 | H | ||
| ND1 | 2858 | 3832 | 975 | 1 | ATG | TAA | H |
| tRNA-Ile | 3845 | 3916 | 72 | 12 | H | ||
| tRNA-Gln | 3916 | 3986 | 71 | −1 | L | ||
| tRNA-Met | 3988 | 4057 | 70 | 1 | H | ||
| ND2 | 4058 | 5102 | 1045 | 0 | ATG | T | H |
| tRNA-Trp | 5103 | 5173 | 71 | 0 | H | ||
| tRNA-Ala | 5175 | 5243 | 69 | 1 | L | ||
| tRNA-Asn | 5245 | 5317 | 73 | 1 | L | ||
| tRNA-Cys | 5350 | 5416 | 67 | 32 | L | ||
| tRNA-Tyr | 5416 | 5485 | 70 | −1 | L | ||
| COI | 5487 | 7037 | 1551 | 1 | GTG | TAA | H |
| tRNA-Ser2 | 7038 | 7108 | 71 | 0 | L | ||
| tRNA-Asp | 7110 | 7181 | 72 | 1 | H | ||
| COII | 7191 | 7881 | 691 | 9 | ATG | T | H |
| tRNA-Lys | 7882 | 7958 | 77 | 0 | H | ||
| ATP8 | 7960 | 8124 | 165 | 1 | ATG | TAG | H |
| ATP6 | 8118 | 8800 | 683 | −7 | ATG | TA | H |
| COIII | 8801 | 9584 | 784 | 0 | ATG | T | H |
| tRNA-Gly | 9585 | 9657 | 73 | 0 | H | ||
| ND3 | 9658 | 10006 | 349 | 0 | ATG | T | H |
| tRNA-Arg | 10007 | 10076 | 70 | 0 | H | ||
| ND4L | 10077 | 10373 | 297 | 0 | ATG | TAA | H |
| ND4 | 10367 | 11747 | 1381 | −7 | ATG | T | H |
| tRNA-His | 11748 | 11816 | 69 | 0 | H | ||
| tRNA-Ser1 | 11817 | 11884 | 68 | 0 | H | ||
| tRNA-Leu1 | 11886 | 11958 | 73 | 1 | H | ||
| ND5 | 11962 | 13785 | 1824 | 3 | ATG | TAA | H |
| ND6 | 13782 | 14303 | 522 | −4 | ATG | TAA | L |
| tRNA-Glu | 14304 | 14372 | 69 | 0 | L | ||
| Cytb | 14379 | 15519 | 1141 | 6 | ATG | T | H |
| tRNA-Thr | 15520 | 15591 | 72 | 0 | H | ||
| tRNA-Pro | 15591 | 15660 | 70 | −1 | L | ||
| D-loop | 15661 | 16948 | 1288 | 0 | H | ||
| Regions | Strand | Size (bp) | AT (%) | AT Skewness | GC Skewness |
|---|---|---|---|---|---|
| PCGs | H | 10,872 | 58.1 | 0.139 | −0.371 |
| PCGs | L | 522 | 57.9 | −0.497 | 0.509 |
| tRNAs | H | 1006 | 56 | 0.130 | −0.070 |
| tRNAs | L | 561 | 54.9 | −0.136 | 0.233 |
| rRNAs | H | 2650 | 55.4 | 0.290 | −0.107 |
| 16S rRNA | H | 1695 | 57.4 | 0.289 | −0.091 |
| 12S rRNA | H | 955 | 51.8 | 0.293 | −0.130 |
| Full genome | H | 16,598 | 58.1 | 0.166 | −0.300 |
| Regions | Strand | Size (bp) | AT (%) | AT Skewness | GC Skewness |
|---|---|---|---|---|---|
| PCGs | H | 10,878 | 60.2 | 0.067 | −0.301 |
| PCGs | L | 522 | 60.2 | −0.452 | 0.442 |
| tRNAs | H | 1003 | 56.9 | 0.103 | −0.042 |
| tRNAs | L | 560 | 54.4 | −0.121 | 0.224 |
| rRNAs | H | 2640 | 56.3 | 0.250 | −0.082 |
| 16S rRNA | H | 1684 | 59.1 | 0.260 | −0.078 |
| 12S rRNA | H | 956 | 51.7 | 0.231 | −0.087 |
| Full genome | H | 16,948 | 59.9 | 0.108 | −0.245 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU (F) | 79 | 0.7 | UCU (S) | 27 | 0.69 | UAU (Y) | 49 | 0.84 | UGU (C) | 6 | 0.48 |
| UUC (F) | 146 | 1.3 | UCC (S) | 51 | 1.31 | UAC (Y) | 68 | 1.16 | UGC (C) | 19 | 1.52 |
| UUA (L) | 113 | 1.12 | UCA (S) | 98 | 2.51 | UAA (*) | 6 | 3.43 | UGA (W) | 114 | 1.92 |
| UUG (L) | 14 | 0.14 | UCG (S) | 5 | 0.13 | UAG (*) | 1 | 0.57 | UGG (W) | 5 | 0.08 |
| CUU (L) | 62 | 0.61 | CCU (P) | 18 | 0.34 | CAU (H) | 16 | 0.32 | CGU (R) | 10 | 0.54 |
| CUC (L) | 66 | 0.65 | CCC (P) | 34 | 0.64 | CAC (H) | 85 | 1.68 | CGC (R) | 8 | 0.43 |
| CUA (L) | 333 | 3.29 | CCA (P) | 159 | 2.97 | CAA (Q) | 97 | 1.96 | CGA (R) | 54 | 2.92 |
| CUG (L) | 20 | 0.2 | CCG (P) | 3 | 0.06 | CAG (Q) | 2 | 0.04 | CGG (R) | 2 | 0.11 |
| AUU (I) | 140 | 0.93 | ACU (T) | 33 | 0.39 | AAU (N) | 29 | 0.47 | AGU (S) | 11 | 0.28 |
| AUC (I) | 161 | 1.07 | ACC (T) | 124 | 1.48 | AAC (N) | 94 | 1.53 | AGC (S) | 42 | 1.08 |
| AUA (M) | 168 | 1.73 | ACA (T) | 172 | 2.05 | AAA (K) | 76 | 1.9 | AGA (*) | 0 | 0 |
| AUG (M) | 26 | 0.27 | ACG (T) | 6 | 0.07 | AAG (K) | 4 | 0.1 | AGG (*) | 0 | 0 |
| GUU (V) | 42 | 0.81 | GCU (A) | 46 | 0.59 | GAU (D) | 14 | 0.37 | GGU (G) | 29 | 0.48 |
| GUC (V) | 28 | 0.54 | GCC (A) | 133 | 1.71 | GAC (D) | 62 | 1.63 | GGC (G) | 38 | 0.63 |
| GUA (V) | 126 | 2.42 | GCA (A) | 128 | 1.64 | GAA (E) | 98 | 1.9 | GGA (G) | 145 | 2.39 |
| GUG (V) | 12 | 0.23 | GCG (A) | 5 | 0.06 | GAG (E) | 5 | 0.1 | GGG (G) | 31 | 0.51 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU (F) | 124 | 1.09 | UCU(S) | 33 | 0.85 | UAU (Y) | 60 | 1.01 | UGU (C) | 11 | 0.88 |
| UUC (F) | 103 | 0.91 | UCC(S) | 42 | 1.08 | UAC (Y) | 59 | 0.99 | UGC (C) | 14 | 1.12 |
| UUA (L) | 176 | 1.73 | UCA(S) | 107 | 2.74 | UAA (*) | 5 | 3.33 | UGA (W) | 109 | 1.85 |
| UUG (L) | 9 | 0.09 | UCG(S) | 3 | 0.08 | UAG (*) | 1 | 0.67 | UGG (W) | 9 | 0.15 |
| CUU (L) | 92 | 0.9 | CCU(P) | 30 | 0.58 | CAU (H) | 38 | 0.72 | CGU (R) | 7 | 0.38 |
| CUC (L) | 65 | 0.64 | CCC(P) | 28 | 0.54 | CAC (H) | 67 | 1.28 | CGC (R) | 5 | 0.27 |
| CUA (L) | 243 | 2.38 | CCA(P) | 144 | 2.77 | CAA (Q) | 98 | 1.92 | CGA (R) | 58 | 3.14 |
| CUG (L) | 27 | 0.26 | CCG(P) | 6 | 0.12 | CAG (Q) | 4 | 0.08 | CGG (R) | 4 | 0.22 |
| AUU (I) | 204 | 1.43 | ACU(T) | 63 | 0.81 | AAU (N) | 46 | 0.8 | AGU (S) | 10 | 0.26 |
| AUC (I) | 81 | 0.57 | ACC(T) | 90 | 1.15 | AAC (N) | 69 | 1.2 | AGC (S) | 39 | 1 |
| AUA (M) | 165 | 1.66 | ACA(T) | 153 | 1.96 | AAA (K) | 81 | 1.91 | AGA (*) | 0 | 0 |
| AUG (M) | 34 | 0.34 | ACG(T) | 6 | 0.08 | AAG (K) | 4 | 0.09 | AGG (*) | 0 | 0 |
| GUU (V) | 55 | 1 | GCU(A) | 55 | 0.66 | GAU (D) | 18 | 0.47 | GGU (G) | 28 | 0.46 |
| GUC (V) | 30 | 0.55 | GCC(A) | 133 | 1.59 | GAC (D) | 58 | 1.53 | GGC (G) | 39 | 0.64 |
| GUA (V) | 120 | 2.19 | GCA(A) | 139 | 1.66 | GAA (E) | 91 | 1.77 | GGA (G) | 132 | 2.18 |
| GUG (V) | 14 | 0.26 | GCG(A) | 7 | 0.08 | GAG (E) | 12 | 0.23 | GGG (G) | 43 | 0.71 |
| Subfamily | Species | Length (bp) | A + T (%) | GenBank Accession No. |
|---|---|---|---|---|
| Smiliogastrinae | Barbodes binotatus | 16,573 | 57.0 | KY305681 |
| Barbodes semifasciolatus | 16,594 | 58.2 | KC113209 | |
| Dawkinsia denisonii | 16,899 | 58.6 | KF019637 | |
| Dawkinsia filamentosa | 16,598 | 58.1 | OP595699 | |
| Enteromius thysi | 16,688 | 60.5 | OP819561 | |
| Enteromius trimaculatus | 16,417 | 60.8 | NC_008666 | |
| Hampala macrolepidota | 16,766 | 58.2 | KF670818 | |
| Hampala salweenensis | 16,913 | 58.9 | MW548258 | |
| Oliotius oligolepis | 16,636 | 58.4 | ON864407 | |
| Oreichthys crenuchoides | 16,596 | 60.2 | MK456608 | |
| Osteobrama belangeri | 16,602 | 60.7 | KY887473 | |
| Pethia nigrofasciata | 16,948 | 59.9 | OP581953 | |
| Pethia padamya | 16,792 | 58.6 | ON864408 | |
| Pethia stoliczkana | 16,996 | 59.7 | OP785085 | |
| Pethia ticto | 17,302 | 60.0 | AB238969 | |
| Prolabeops melanhypopterus | 13,571 | 62.8 | ON323520 | |
| Puntigrus tetrazona | 16,550 | 59.8 | EU287909 | |
| Puntius sachsii | 16,587 | 58.2 | MZ364158 | |
| Puntius snyderi | 16,578 | 59.3 | KC113210 | |
| Sahyadria chalakkudiensis | 16,989 | 59.9 | JX311437 | |
| Systomus sarana | 16,590 | 58.6 | KU886061 | |
| Torinae | Lepidopygopsis typus | 16,729 | 56.7 | OR685011 |
| Cyprininae | Carassius auratus | 16,580 | 57.7 | OR950752 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, C.-H.; Lu, C.-H. Comparative Analysis and Phylogenetic Study of Dawkinsia filamentosa and Pethia nigrofasciata Mitochondrial Genomes. Int. J. Mol. Sci. 2024, 25, 3004. https://doi.org/10.3390/ijms25053004
Sun C-H, Lu C-H. Comparative Analysis and Phylogenetic Study of Dawkinsia filamentosa and Pethia nigrofasciata Mitochondrial Genomes. International Journal of Molecular Sciences. 2024; 25(5):3004. https://doi.org/10.3390/ijms25053004
Chicago/Turabian StyleSun, Cheng-He, and Chang-Hu Lu. 2024. "Comparative Analysis and Phylogenetic Study of Dawkinsia filamentosa and Pethia nigrofasciata Mitochondrial Genomes" International Journal of Molecular Sciences 25, no. 5: 3004. https://doi.org/10.3390/ijms25053004
APA StyleSun, C.-H., & Lu, C.-H. (2024). Comparative Analysis and Phylogenetic Study of Dawkinsia filamentosa and Pethia nigrofasciata Mitochondrial Genomes. International Journal of Molecular Sciences, 25(5), 3004. https://doi.org/10.3390/ijms25053004