
Study on the Mechanism of the Adrenaline-Evoked Procoagulant Response in Human Platelets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
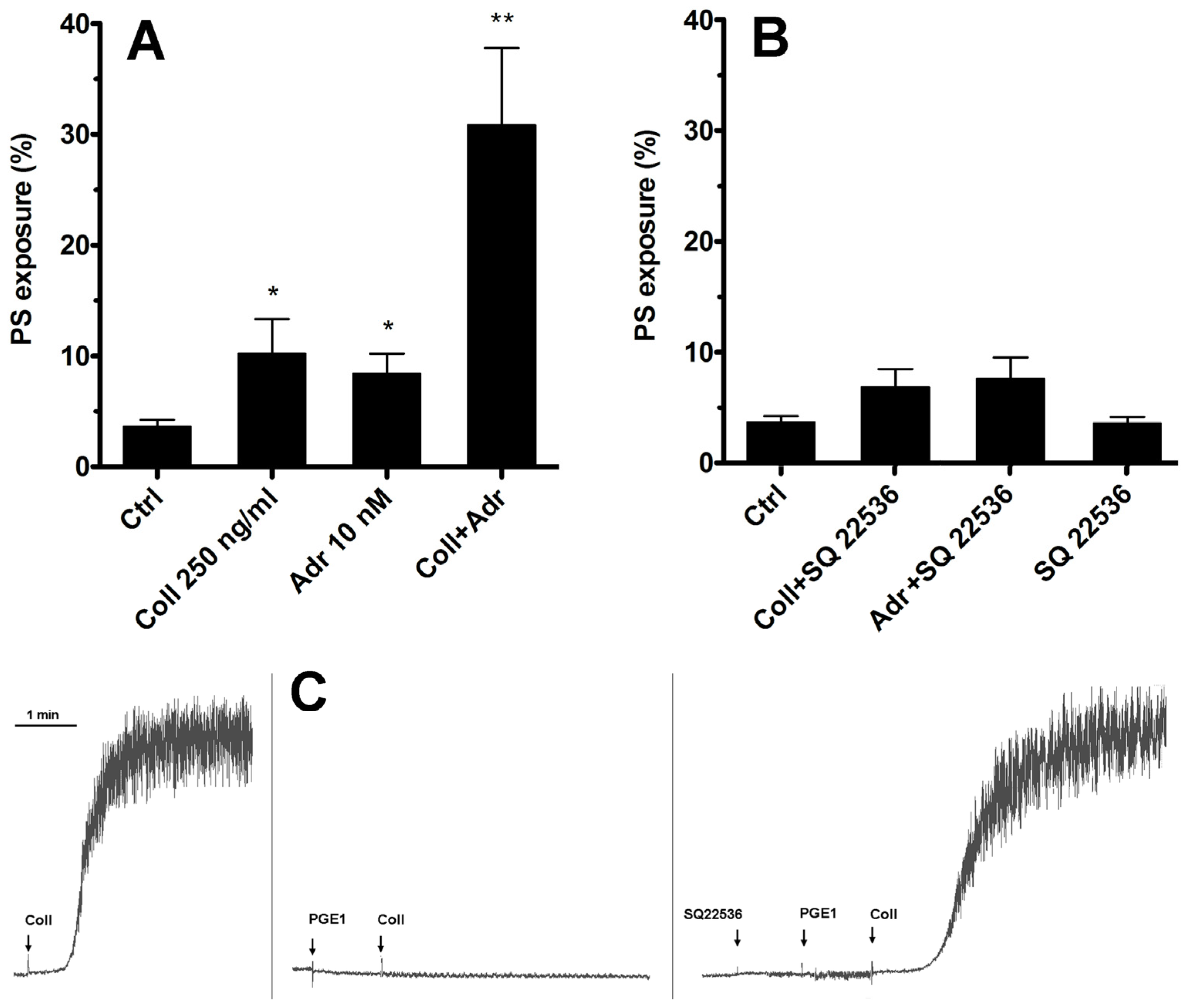
2.1. Inhibition of Only Adenylate Cyclase Is Not Sufficient to Produce Elevation of the PS-Exposing Platelets Number in the Presence of Subthreshold Collagen
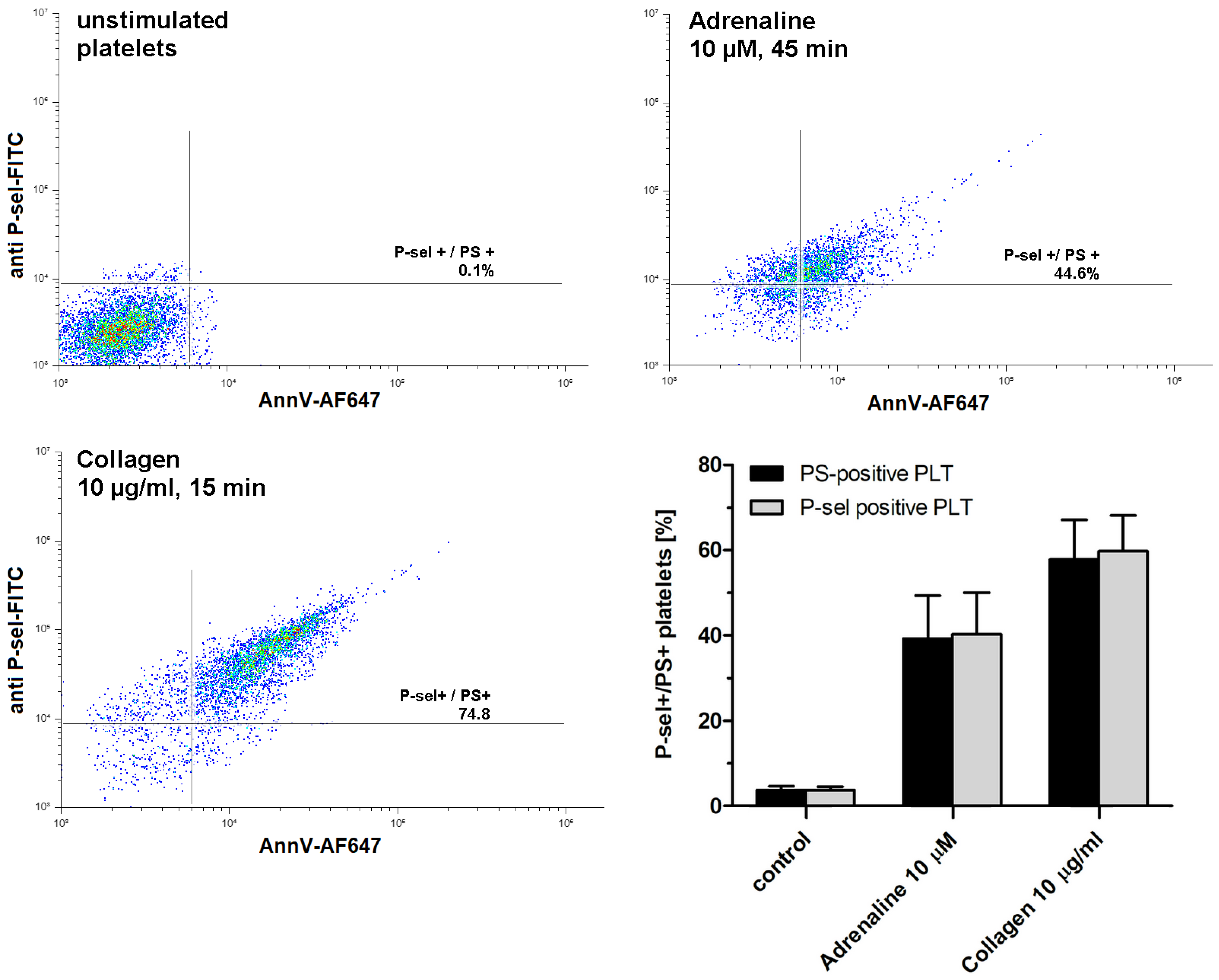
2.2. Adrenaline Produces Population of Procoagulant Human Platelets in a Concentration- and Time-Dependent Fashion
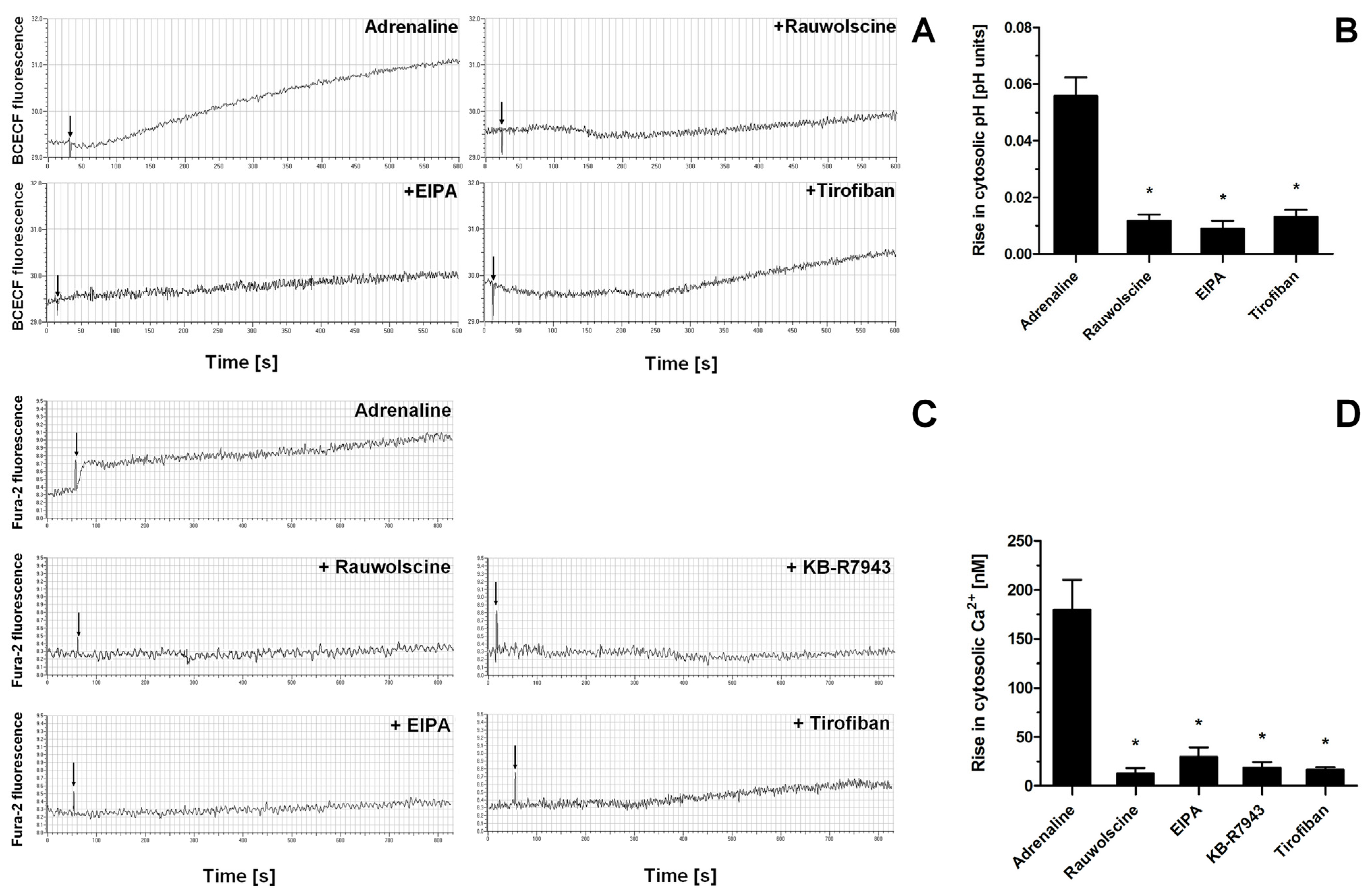
2.3. Effect of Selected Receptor Blockages on Adrenaline-Induced Platelet PS Exposure
2.4. Impact of a Modulation of Different Signaling Pathways on Adrenaline-Induced PS Exposure
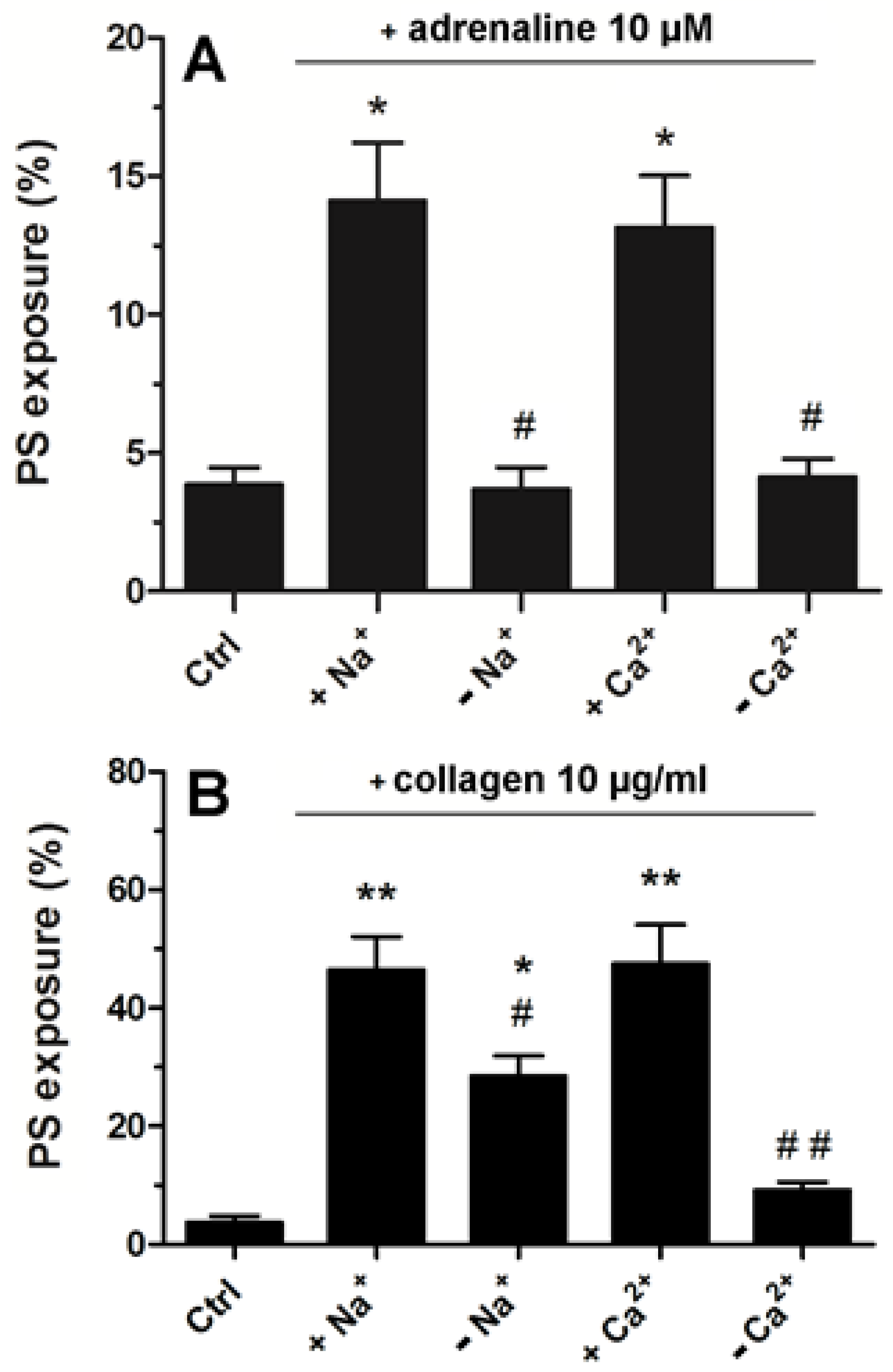
2.5. Strong Dependence of Adrenaline-Evoked PS Exposure on the Presence of Extracellular Calcium and Sodium Ions
3. Discussion
3.1. Role of AC and cAMP in Adrenaline-Evoked PS Exposure
3.2. Receptors’ Engagement in Adrenaline-Triggered PS Exposure
3.3. Role of Ca2+ in the Development of the Procoagulant Response of Platelets Exposed to Adrenaline
3.4. Significance of Na+ and Platelet Swelling in Adrenaline-Produced PS Exposure
3.5. Role of GPIIb/IIIa-Mediated Outside-in Signaling in Adrenaline-Triggered PS Exposure
3.6. Non-Redundant Role of PI3-K in Adrenaline-Evoked PS Exposure
3.7. How Does This Study Adhere to Clinical Reality?
4. Materials and Methods
4.1. Chemicals
4.2. Blood Collection and Preparation
4.3. PS and P-Selectin Exposure Quantification by Flow Cytometry
4.4. Fluorimetric Determination of Na+/H+ Exchange and Calcium Rise in Platelets Exposed to Adrenaline
4.5. Measurement of Platelet Aggregation
4.6. Electronic Measurements of Platelet Volume
4.7. Data Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bentur, O.S.; Sarig, G.; Brenner, B.; Jacob, G. Effects of Acute Stress on Thrombosis. Semin. Thromb. Hemost. 2018, 44, 662–668. [Google Scholar] [CrossRef]
- Goldstein, D.S. Catecholamines 101. Clin. Auton. Res. 2010, 20, 331–352. [Google Scholar] [CrossRef]
- Dimsdale, J.E.; Moss, J. Plasma catecholamines in stress and exercise. JAMA 1980, 243, 340–342. [Google Scholar] [CrossRef]
- von Känel, R. Acute mental stress and hemostasis: When physiology becomes vascular harm. Thromb. Res. 2015, 135, S52–S55. [Google Scholar] [CrossRef]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. Handb. Exp. Pharmacol. 2012, 210, 59–85. [Google Scholar] [CrossRef]
- Mills, D.C.; Roberts, G.C. Effects of adrenaline on human blood platelets. J. Physiol. 1967, 193, 443–453. [Google Scholar] [CrossRef]
- Cameron, H.A.; Ardlie, N.G. The facilitating effects of adrenaline on platelet aggregation. Prostaglandins Leukot. Med. 1982, 9, 117–128. [Google Scholar] [CrossRef]
- Wagner, C.T.; Kroll, M.H.; Chow, T.W.; Hellums, J.D.; Schafer, A.I. Epinephrine and shear stress synergistically induce platelet aggregation via a mechanism that partially bypasses VWF-GP IB interactions. Biorheology 1996, 33, 209–229. [Google Scholar] [CrossRef]
- Pozgajová, M.; Sachs, U.J.; Hein, L.; Nieswandt, B. Reduced thrombus stability in mice lacking the alpha2A-adrenergic receptor. Blood 2006, 108, 510–514. [Google Scholar] [CrossRef]
- Shen, J.; Sampietro, S.; Wu, J.; Tang, J.; Gupta, S.; Matzko, C.N.; Tang, C.; Yu, Y.; Brass, L.F.; Zhu, L.; et al. Coordination of platelet agonist signaling during the hemostatic response in vivo. Blood Adv. 2017, 1, 2767–2775. [Google Scholar] [CrossRef]
- Swieringa, F.; Kuijpers, M.J.; Lamers, M.M.; van der Meijden, P.E.; Heemskerk, J.W. Rate-limiting roles of the tenase complex of factors VIII and IX in platelet procoagulant activity and formation of platelet-fibrin thrombi under flow. Haematologica 2015, 100, 748–756. [Google Scholar] [CrossRef]
- Heemskerk, J.W.; Mattheij, N.J.; Cosemans, J.M. Platelet-based coagulation: Different populations, different functions. J. Thromb. Haemost. 2013, 11, 2–16. [Google Scholar] [CrossRef]
- Mazepa, M.; Hoffman, M.; Monroe, D. Superactivated platelets: Thrombus regulators, thrombin generators, and potential clinical targets. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1747–1752. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell. Biochem. 2017, 82, 405–456. [Google Scholar] [CrossRef]
- Golaszewska, A.; Misztal, T.; Marcinczyk, N.; Chabielska, E.; Rusak, T. Adrenaline May Contribute to Prothrombotic Condition via Augmentation of Platelet Procoagulant Response, Enhancement of Fibrin Formation, and Attenuation of Fibrinolysis. Front. Physiol. 2021, 12, 657881. [Google Scholar] [CrossRef]
- Pastori, D.; Cormaci, V.M.; Marucci, S.; Franchino, G.; Del Sole, F.; Capozza, A.; Fallarino, A.; Corso, C.; Valeriani, E.; Menichelli, D.; et al. A Comprehensive Review of Risk Factors for Venous Thromboembolism: From Epidemiology to Pathophysiology. Int. J. Mol. Sci. 2023, 24, 3169. [Google Scholar] [CrossRef]
- Carcaillon, L.; Alhenc-Gelas, M.; Bejot, Y.; Spaft, C.; Ducimetière, P.; Ritchie, K.; Dartigues, J.F.; Scarabin, P.Y. Increased thrombin generation is associated with acute ischemic stroke but not with coronary heart disease in the elderly: The Three-City cohort study. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1445–1451. [Google Scholar] [CrossRef]
- Singh, S.; Damén, T.; Nygren, A.; Shams Hakimi, C.; Ramström, S.; Dellborg, M.; Lindahl, T.L.; Hesse, C.; Jeppsson, A. Adrenaline Improves Platelet Reactivity in Ticagrelor-Treated Healthy Volunteers. Thromb. Haemost. 2019, 119, 735–743. [Google Scholar] [CrossRef]
- Singh, S.; Malm, C.J.; Ramström, S.; Hesse, C.; Jeppsson, A. Adrenaline enhances in vitro platelet activation and aggregation in blood samples from ticagrelor-treated patients. Res. Pract. Thromb. Haemost. 2018, 2, 718–725. [Google Scholar] [CrossRef]
- Mukherjee, A. Characterization of alpha 2-adrenergic receptors in human platelets by binding of a radioactive ligand [3H]yohimbine. Biochim. Biophys. Acta 1981, 676, 148–154. [Google Scholar] [CrossRef]
- Woulfe, D.; Jiang, H.; Mortensen, R.; Yang, J.; Brass, L.F. Activation of Rap1B by G(i) family members in platelets. J. Biol. Chem. 2002, 277, 23382–23390. [Google Scholar] [CrossRef]
- Zwaginga, J.J.; Nash, G.; King, M.R.; Heemskerk, J.W.; Frojmovic, M.; Hoylaerts, M.F.; Sakariassen, K.S. Biorheology Subcommittee of the SSC of the ISTH. Flow-based assays for global assessment of hemostasis. Part 1: Biorheologic considerations. J. Thromb. Haemost. 2006, 4, 2486–2487. [Google Scholar] [CrossRef]
- Rao, A.K.; Willis, J.; Kowalska, M.A.; Wachtfogel, Y.T.; Colman, R.W. Differential requirements for platelet aggregation and inhibition of adenylate cyclase by epinephrine. Studies of a familial platelet alpha 2-adrenergic receptor defect. Blood 1988, 71, 494–501. [Google Scholar] [CrossRef]
- Selheim, F.; Frøyset, A.K.; Strand, I.; Vassbotn, F.S.; Holmsen, H. Adrenaline potentiates PI 3-kinase in platelets stimulated with thrombin and SFRLLN: Role of secreted ADP. FEBS Lett. 2000, 485, 62–66. [Google Scholar] [CrossRef]
- Steen, V.M.; Cook, C.A.; Tysnes, O.B.; Holmsen, H. Potentiation by adrenaline of thrombin-induced elevation of pHi is not essential for synergistic activation of human platelets. FEBS Lett. 1989, 250, 211–214. [Google Scholar] [CrossRef]
- Katsel, P.L.; Tagliente, T.M.; Schwarz, T.E.; Craddock-Royal, B.D.; Patel, N.D.; Maayani, S. Molecular and biochemical evidence for the presence of type III adenylyl cyclase in human platelets. Platelets 2003, 14, 21–33. [Google Scholar] [CrossRef]
- Guan, B.; Gao, J.; Tan, Y.; Ma, X.; Shi, D. Antiplatelet Activity of Tetramethylpyrazine via Regulation of the P2Y12 Receptor Downstream Signaling Pathway. Evid. Based Complement. Altern. Med. 2022, 2022, 7941039. [Google Scholar] [CrossRef]
- Josefsson, E.C.; Ramström, S.; Thaler, J.; Lordkipanidzé, M.; COAGAPO Study Group. Consensus report on markers to distinguish procoagulant platelets from apoptotic platelets: Communication from the Scientific and Standardization Committee of the ISTH. J. Thromb. Haemost. 2023, 21, 2291–2299. [Google Scholar] [CrossRef]
- Misztal, T.; Rusak, T.; Brańska-Januszewska, J.; Ostrowska, H.; Tomasiak, M. Peroxynitrite may affect fibrinolysis via the reduction of platelet-related fibrinolysis resistance and alteration of clot structure. Free Radic. Biol. Med. 2015, 89, 533–547. [Google Scholar] [CrossRef]
- Harris, D.N.; Asaad, M.M.; Phillips, M.B.; Goldenberg, H.J.; Antonaccio, M.J. Inhibition of adenylate cyclase in human blood platelets by 9-substituted adenine derivatives. J. Cyclic Nucleotide Res. 1979, 5, 125–134. [Google Scholar]
- Graber, S.E.; Hawiger, J. Evidence that changes in platelet cyclic AMP levels regulate the fibrinogen receptor on human platelets. J. Biol. Chem. 1982, 257, 14606–14609. [Google Scholar] [CrossRef]
- Misztal, T.; Golaszewska, A.; Branska-Januszewska, J.; Marcinczyk, N.; Chabielska, E.; Tomasiak, M.; Rusak, T. HAuCl4, Putative General Aquaporins Blocker, Reduces Platelet Spreading, Filopodia Formation, Procoagulant Response, and Thrombus Formation under Flow. Front. Physiol. 2020, 11, 1025. [Google Scholar] [CrossRef]
- Olbrich, C.; Aepfelbacher, M.; Siess, W. Epinephrine potentiates calcium mobilization and activation of protein kinases in platelets stimulated by ADP through a mechanism unrelated to phospholipase C. Cell. Signal. 1989, 1, 483–492. [Google Scholar] [CrossRef]
- Figures, W.R.; Scearce, L.M.; Wachtfogel, Y.; Chen, J.; Colman, R.F.; Colman, R.W. Platelet ADP receptor and alpha 2-adrenoreceptor interaction. Evidence for an ADP requirement for epinephrine-induced platelet activation and an influence of epinephrine on ADP binding. J. Biol. Chem. 1986, 261, 5981–5986. [Google Scholar] [CrossRef]
- Plow, E.F.; Marguerie, G.A. Induction of the fibrinogen receptor on human platelets by epinephrine and the combination of epinephrine and ADP. J. Biol. Chem. 1980, 255, 10971–10977. [Google Scholar] [CrossRef]
- Lanza, F.; Cazenave, J.P.; Beretz, A.; Sutter-Bay, A.; Kretz, J.G.; Kieny, R. Potentiation by adrenaline of human platelet activation and the inhibition by the alpha-adrenergic antagonist nicergoline of platelet adhesion, secretion and aggregation. Agents Actions 1986, 18, 586–595. [Google Scholar] [CrossRef]
- Roberts, D.E.; Matsuda, T.; Bose, R. Molecular and functional characterization of the human platelet Na(+)/Ca(2+) exchangers. Br. J. Pharmacol. 2012, 165, 922–936. [Google Scholar] [CrossRef]
- Taylor, K.A.; Mahaut-Smith, M.P. Ion channels and ion homeostasis in the platelet and megakaryocyte. Platelets 2021, 32, 853–854. [Google Scholar] [CrossRef]
- Roberts, D.E.; McNicol, A.; Bose, R. Mechanism of collagen activation in human platelets. J. Biol. Chem. 2004, 279, 19421–19430. [Google Scholar] [CrossRef]
- Nesbitt, W.S.; Giuliano, S.; Kulkarni, S.; Dopheide, S.M.; Harper, I.S.; Jackson, S.P. Intercellular calcium communication regulates platelet aggregation and thrombus growth. J. Cell Biol. 2003, 160, 1151–1161. [Google Scholar] [CrossRef]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef]
- Agbani, E.O.; van den Bosch, M.T.; Brown, E.; Williams, C.M.; Mattheij, N.J.; Cosemans, J.M.; Collins, P.W.; Heemskerk, J.W.; Hers, I.; Poole, A.W. Coordinated Membrane Ballooning and Procoagulant Spreading in Human Platelets. Circulation 2015, 132, 1414–1424. [Google Scholar] [CrossRef]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef]
- Yang, H.; Kim, A.; David, T.; Palmer, D.; Jin, T.; Tien, J.; Huang, F.; Cheng, T.; Coughlin, S.R.; Jan, Y.N.; et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012, 151, 111–122. [Google Scholar] [CrossRef]
- Fowajuh, A.-D.N.; Mukaz, D.K.; Li, D.; Apostolidis, P.A.; Khan, A.; Caplan, J.; Vortkamp, A.; Woulfe, D.S. TMEM16F Plays a Vital Role in Platelet Microparticle Generation and Thrombosis. Blood 2013, 122, 2294. [Google Scholar] [CrossRef]
- Mattheij, N.J.; Braun, A.; van Kruchten, R.; Castoldi, E.; Pircher, J.; Baaten, C.C.; Wülling, M.; Kuijpers, M.J.; Köhler, R.; Poole, A.W.; et al. Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling, and protein cleavage. FASEB J. 2016, 30, 727–737. [Google Scholar] [CrossRef]
- Steen, V.M.; Holmsen, H.; Aarbakke, G. The platelet-stimulating effect of adrenaline through alpha 2-adrenergic receptors requires simultaneous activation by a true stimulatory platelet agonist. Evidence that adrenaline per se does not induce human platelet activation in vitro. Thromb. Haemost. 1993, 70, 506–513. [Google Scholar]
- Banga, H.S.; Simons, E.R.; Brass, L.F.; Rittenhouse, S.E. Activation of phospholipases A and C in human platelets exposed to epinephrine: Role of glycoproteins IIb/IIIa and dual role of epinephrine. Proc. Natl. Acad. Sci. USA 1986, 83, 9197–9201. [Google Scholar] [CrossRef]
- van der Meijden, P.E.; Schoenwaelder, S.M.; Feijge, M.A.; Cosemans, J.M.; Munnix, I.C.; Wetzker, R.; Heller, R.; Jackson, S.P.; Heemskerk, J.W. Dual P2Y 12 receptor signaling in thrombin-stimulated platelets—Involvement of phosphoinositide 3-kinase beta but not gamma isoform in Ca2+ mobilization and procoagulant activity. FEBS J. 2008, 275, 371–385. [Google Scholar] [CrossRef]
- Martin, A.C.; Zlotnik, D.; Bonete, G.P.; Baron, E.; Decouture, B.; Belleville-Rolland, T.; Le Bonniec, B.; Poirault-Chassac, S.; Alessi, M.C.; Gaussem, P.; et al. Epinephrine restores platelet functions inhibited by ticagrelor: A mechanistic approach. Eur. J. Pharmacol. 2020, 866, 172798. [Google Scholar] [CrossRef]
- Storey, R.F.; Newby, L.J.; Heptinstall, S. Effects of P2Y(1) and P2Y(12) receptor antagonists on platelet aggregation induced by different agonists in human whole blood. Platelets 2001, 12, 443–447. [Google Scholar] [CrossRef]
- Misztal, T.; Rusak, T.; Brańska-Januszewska, J.; Gąsowska, M.; Szynaka, B.; Gołaszewska, A.; Bruczko, M.; Tomasiak, M. Aquaporins in human platelets: Intracellular localization and possible role in granule and lysosome secretion. Acta Biochim. Pol. 2018, 65, 555–566. [Google Scholar] [CrossRef]
- Gumina, R.J.; Newman, P.J.; Gross, G.J. Effect on ex vivo platelet aggregation and in vivo cyclic flow with Na+/H+ exchange inhibition: Gumina, NHE-1 inhibition and platelet aggregation. J. Thromb. Thrombolysis 2011, 31, 431–435. [Google Scholar] [CrossRef]
- Chang, H.B.; Gao, X.; Nepomuceno, R.; Hu, S.; Sun, D. Na(+)/H(+) exchanger in the regulation of platelet activation and paradoxical effects of cariporide. Exp. Neurol. 2015, 272, 11–16. [Google Scholar] [CrossRef]
- Sweatt, J.D.; Connolly, T.M.; Cragoe, E.J.; Limbird, L.E. Evidence that Na+/H+ exchange regulates receptor-mediated phospholipase A2 activation in human platelets. J. Biol. Chem. 1986, 261, 8667–8673. [Google Scholar] [CrossRef]
- Bucki, R.; Pastore, J.J.; Giraud, F.; Janmey, P.A.; Sulpice, J.C. Involvement of the Na+/H+ exchanger in membrane phosphatidylserine exposure during human platelet activation. Biochim. Biophys. Acta 2006, 1761, 195–204. [Google Scholar] [CrossRef][Green Version]
- Samson, J.; Stelmach, H.; Tomasiak, M. The importance of Na+/H+ exchanger for the generation of procoagulant activity by porcine blood platelets. Platelets 2001, 12, 436–442. [Google Scholar] [CrossRef]
- Huber, J.D.; Bentzien, J.; Boyer, S.J.; Burke, J.; De Lombaert, S.; Eickmeier, C.; Guo, X.; Haist, J.V.; Hickey, E.R.; Kaplita, P.; et al. Identification of a potent sodium hydrogen exchanger isoform 1 (NHE1) inhibitor with a suitable profile for chronic dosing and demonstrated cardioprotective effects in a preclinical model of myocardial infarction in the rat. J. Med. Chem. 2012, 55, 7114–7140. [Google Scholar] [CrossRef]
- Tomasiak, M.M.; Stelmach, H.; Bodzenta-Łukaszyk, A.; Tomasiak, M. Involvement of Na+/H+ exchanger in desmopressin-induced platelet procoagulant response. Acta Biochim. Pol. 2004, 51, 773–788. [Google Scholar] [CrossRef]
- Tomasiak, M.; Stelmach, H.; Rusak, T.; Ciborowski, M.; Radziwon, P. Vasopressin acts on platelets to generate procoagulant activity. Blood Coagul. Fibrinolysis 2008, 19, 615–624. [Google Scholar] [CrossRef]
- Shiraga, M.; Tomiyama, Y.; Honda, S.; Suzuki, H.; Kosugi, S.; Tadokoro, S.; Kanakura, Y.; Tanoue, K.; Kurata, Y.; Matsuzawa, Y. Involvement of Na+/Ca2+ exchanger in inside-out signaling through the platelet integrin IIbbeta3. Blood 1998, 92, 3710–3720. [Google Scholar] [CrossRef]
- Shiraga, M.; Tomiyama, Y.; Honda, S.; Kashiwagi, H.; Kosugi, S.; Handa, M.; Ikeda, Y.; Kanakura, Y.; Kurata, Y.; Matsuzawa, Y. Affinity modulation of the platelet integrin alpha IIb beta 3 by alpha-chymotrypsin: A possible role for Na+/Ca2+ exchanger. Blood 1996, 88, 2594–2602. [Google Scholar] [CrossRef]
- Zou, J.; Swieringa, F.; de Laat, B.; de Groot, P.G.; Roest, M.; Heemskerk, J.W.M. Reversible platelet integrin αIIbβ3 activation and thrombus instability. Int. J. Mol. Sci. 2022, 23, 12512. [Google Scholar] [CrossRef]
- Mattheij, N.J.A.; Gilio, K.; van Kruchten, R.; Jobe, S.M.; Wieschhaus, A.J.; Chishti, A.H.; Collins, P.; Heemskerk, J.W.; Cosemans, J.M. Dual mechanism of integrin αIIbβ3 closure in procoagulant platelets. J. Biol. Chem. 2013, 288, 13325036. [Google Scholar] [CrossRef]
- Liu, F.; Gamez, G.; Myers, D.R.; Clemmons, W.; Lam, W.A.; Jobe, S.M. Mitochondrially mediated integrin αIIbβ3 protein inactivation limits thrombus growth. J. Biol. Chem. 2013, 288, 30672–30681. [Google Scholar] [CrossRef]
- Topalov, N.N.; Yakimenko, A.O.; Canault, M.; Artemenko, E.O.; Zakharova, N.V.; Abaeva, A.A.; Loosveld, M.; Ataullakhanov, F.I.; Nurden, A.T.; Alessi, M.-C.; et al. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrin α(IIb)β(3). Arter. Thromb. Vasc. Biol. 2012, 32, 2475–2483. [Google Scholar] [CrossRef]
- Ilveskero, S.; Siljander, P.; Lassila, R. Procoagulant activity on platelets adhered to collagen or plasma clot. Arter. Thromb. Vasc. Biol. 2001, 21, 628–635. [Google Scholar] [CrossRef][Green Version]
- Ilveskero, S.; Lassila, R. Abciximab inhibits procoagulant activity but not the release reaction upon collagen- or clot-adherent platelets. J. Thromb. Haemost. 2003, 1, 805–813. [Google Scholar] [CrossRef]
- Gauer, J.S.; Duval, C.; Xu, R.-G.; Macrae, F.L.; McPherson, H.R.; Tiede, C.; Tomlinson, D.; Watson, S.P.; Ariëns, R.A. Fibrin-glycoprotein VI interaction increases platelet procoagulant activity and impacts clot structure. J. Thromb. Haemost. 2023, 21, 667–681. [Google Scholar] [CrossRef]
- Berny-Lang, M.A.; Jakubowski, J.A.; Sugidachi, A.; Barnard, M.R.; Michelson, A.D.; Frelinger, A.R., 3rd. P2Y12 receptor blockade augments glycoprotein IIb-IIIa antagonist inhibition of platelet activation, aggregation, and procoagulant activity. J. Am. Heart Assoc 2013, 2, e000026. [Google Scholar] [CrossRef]
- Tanaka, K.A.; Katori, N.; Szlam, F.; Sato, N.; Kelly, A.B.; Levy, J.H. Effects of tirofiban on haemostatic activation in vitro. Br. J. Anaesth. 2004, 93, 263–269. [Google Scholar] [CrossRef]
- Li, Y.; Spencer, F.A.; Ball, S.; Becker, R.C. Inhibition of platelet-dependent prothrombinase activity and thrombin generation by glycoprotein IIb/IIIa receptor-directed antagonists: Potential contributing mechanism of benefit in acute coronary syndromes. J. Thromb. Thrombolysis 2000, 10, 69–76. [Google Scholar] [CrossRef]
- Spalding, A.; Vaitkevicius, H.; Dill, S.; MacKenzie, S.; Schmaier, A.; Lockette, W. Mechanism of epinephrine-induced platelet aggregation. Hypertension 1998, 31, 603–607. [Google Scholar] [CrossRef]
- Setiabakti, N.M.; Larsson, P.; Hamilton, J.R. Phosphoinositide 3-Kinases as Potential Targets for Thrombosis Prevention. Int. J. Mol Sci. 2022, 23, 4840. [Google Scholar] [CrossRef]
- Valet, C.; Chicanne, G.; Severac, C.; Chaussade, C.; Whitehead, M.A.; Cabou, C.; Gratacap, M.P.; Gaits-Iacovoni, F.; Vanhaesebroeck, B.; Payrastre, B.; et al. Essential role of class II PI3K-C2α in platelet membrane morphology. Blood 2015, 126, 1128–1137. [Google Scholar] [CrossRef]
- Ribes, A.; Oprescu, A.; Viaud, J.; Hnia, K.; Chicanne, G.; Xuereb, J.M.; Severin, S.; Gratacap, M.P.; Payrastre, B. Phosphoinositide 3-kinases in platelets, thrombosis and therapeutics. Biochem. J. 2020, 477, 4327–4342. [Google Scholar] [CrossRef]
- Gilio, K.; Munnix, I.C.; Mangin, P.; Cosemans, J.M.; Feijge, M.A.; van der Meijden, P.E.; Olieslagers, S.; Chrzanowska-Wodnicka, M.B.; Lillian, R.; Schoenwaelder, S.; et al. Non-redundant roles of phosphoinositide 3-kinase isoforms alpha and beta in glycoprotein VI-induced platelet signaling and thrombus formation. J. Biol. Chem. 2009, 284, 33750–33762. [Google Scholar] [CrossRef]
- Cosemans, J.M.; Munnix, I.C.; Wetzker, R.; Heller, R.; Jackson, S.P.; Heemskerk, J.W. Continuous signaling via PI3K isoforms beta and gamma is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood 2006, 108, 3045–3052. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Ono, A.; Sturgeon, S.; Chan, S.M.; Mangin, P.; Maxwell, M.J.; Turnbull, S.; Mulchandani, M.; Anderson, K.; Kauffenstein, G.; et al. Identification of a unique co-operative phosphoinositide 3-kinase signaling mechanism regulating integrin alpha IIb beta 3 adhesive function in platelets. J. Biol. Chem. 2007, 282, 28648–28658. [Google Scholar] [CrossRef]
- Erb, L.; Weisman, G.A. Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 789–803. [Google Scholar] [CrossRef]
- Smrcka, A.V. G protein βγ subunits: Central mediators of G protein-coupled receptor signaling. Cell Mol. Life Sci. 2008, 65, 2191–2214. [Google Scholar] [CrossRef]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signalling in the vascular system. Cardiovasc. Res. 2009, 82, 261–271. [Google Scholar] [CrossRef]
- Zhang, J.; Banfić, H.; Straforini, F.; Tosi, L.; Volinia, S.; Rittenhouse, S.E. A type II phosphoinositide 3-kinase is stimulated via activated integrin in platelets. A source of phosphatidylinositol 3-phosphate. J. Biol. Chem. 1998, 273, 14081–14084. [Google Scholar] [CrossRef]
- Guidetti, G.F.; Canobbio, I.; Torti, M. PI3K/Akt in platelet integrin signaling and implications in thrombosis. Adv. Biol. Regul. 2015, 59, 36–52. [Google Scholar] [CrossRef]
- Martin, V.; Guillermet-Guibert, J.; Chicanne, G.; Cabou, C.; Jandrot-Perrus, M.; Plantavid, M.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.P. Deletion of the p110beta isoform of phosphoinositide 3-kinase in platelets reveals its central role in Akt activation and thrombus formation in vitro and in vivo. Blood 2010, 115, 2008–2013. [Google Scholar] [CrossRef]
- Canobbio, I.; Stefanini, L.; Cipolla, L.; Ciraolo, E.; Gruppi, C.; Balduini, C.; Hirsch, E.; Torti, M. Genetic evidence for a predominant role of PI3Kbeta catalytic activity in ITAM- and integrin-mediated signaling in platelets. Blood 2009, 114, 2193–2196. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.E.; Dopheide, S.M.; Yuan, Y.; et al. PI 3-kinase p110beta: A new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef]
- Laurent, P.A.; Severin, S.; Gratacap, M.P.; Payrastre, B. Class I PI 3-kinases signaling in platelet activation and thrombosis: PDK1/Akt/GSK3 axis and impact of PTEN and SHIP1. Adv. Biol. Regul. 2014, 54, 162–174. [Google Scholar] [CrossRef]
- Laurent, P.A.; Séverin, S.; Hechler, B.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.P. Platelet PI3Kβ and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888. [Google Scholar] [CrossRef]
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Transl. Med. 2020, 9, 8. [Google Scholar] [CrossRef]
- Adamski, P.; Skonieczny, G.; Hajdukiewicz, T.; Kern, A.; Kubica, J. Reversal of Platelet Inhibition in Patients Receiving Ticagrelor. Rev. Cardiovasc. Med. 2022, 23, 300. [Google Scholar] [CrossRef]
- Sakariassen, K.S.; Orning, L.; Turitto, V.T. The impact of blood shear rate on arterial thrombus formation. Future Sci. OA 2015, 1, FSO30. [Google Scholar] [CrossRef]
- Siffert, W.; Siffert, G.; Scheid, P.; Akkerman, J.W. Activation of Na+/H+ exchange and Ca2+ mobilization start simultaneously in thrombin-stimulated platelets. Evidence that platelet shape change disturbs early rises of BCECF fluorescence which causes an underestimation of actual cytosolic alkalinization. Biochem. J. 1989, 258, 521–527. [Google Scholar] [CrossRef]
- Born, G.V.; Cross, M.J. The aggregation of blood platelets. J. Physiol. 1963, 168, 178–195. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gołaszewska, A.; Misztal, T.; Kazberuk, A.; Rusak, T. Study on the Mechanism of the Adrenaline-Evoked Procoagulant Response in Human Platelets. Int. J. Mol. Sci. 2024, 25, 2997. https://doi.org/10.3390/ijms25052997
Gołaszewska A, Misztal T, Kazberuk A, Rusak T. Study on the Mechanism of the Adrenaline-Evoked Procoagulant Response in Human Platelets. International Journal of Molecular Sciences. 2024; 25(5):2997. https://doi.org/10.3390/ijms25052997
Chicago/Turabian StyleGołaszewska, Agata, Tomasz Misztal, Adam Kazberuk, and Tomasz Rusak. 2024. "Study on the Mechanism of the Adrenaline-Evoked Procoagulant Response in Human Platelets" International Journal of Molecular Sciences 25, no. 5: 2997. https://doi.org/10.3390/ijms25052997
APA StyleGołaszewska, A., Misztal, T., Kazberuk, A., & Rusak, T. (2024). Study on the Mechanism of the Adrenaline-Evoked Procoagulant Response in Human Platelets. International Journal of Molecular Sciences, 25(5), 2997. https://doi.org/10.3390/ijms25052997