

Early Pulmonary Fibrosis-like Changes in the Setting of Heat Exposure: DNA Damage and Cell Senescence

Abstract
1. Introduction
2. Results
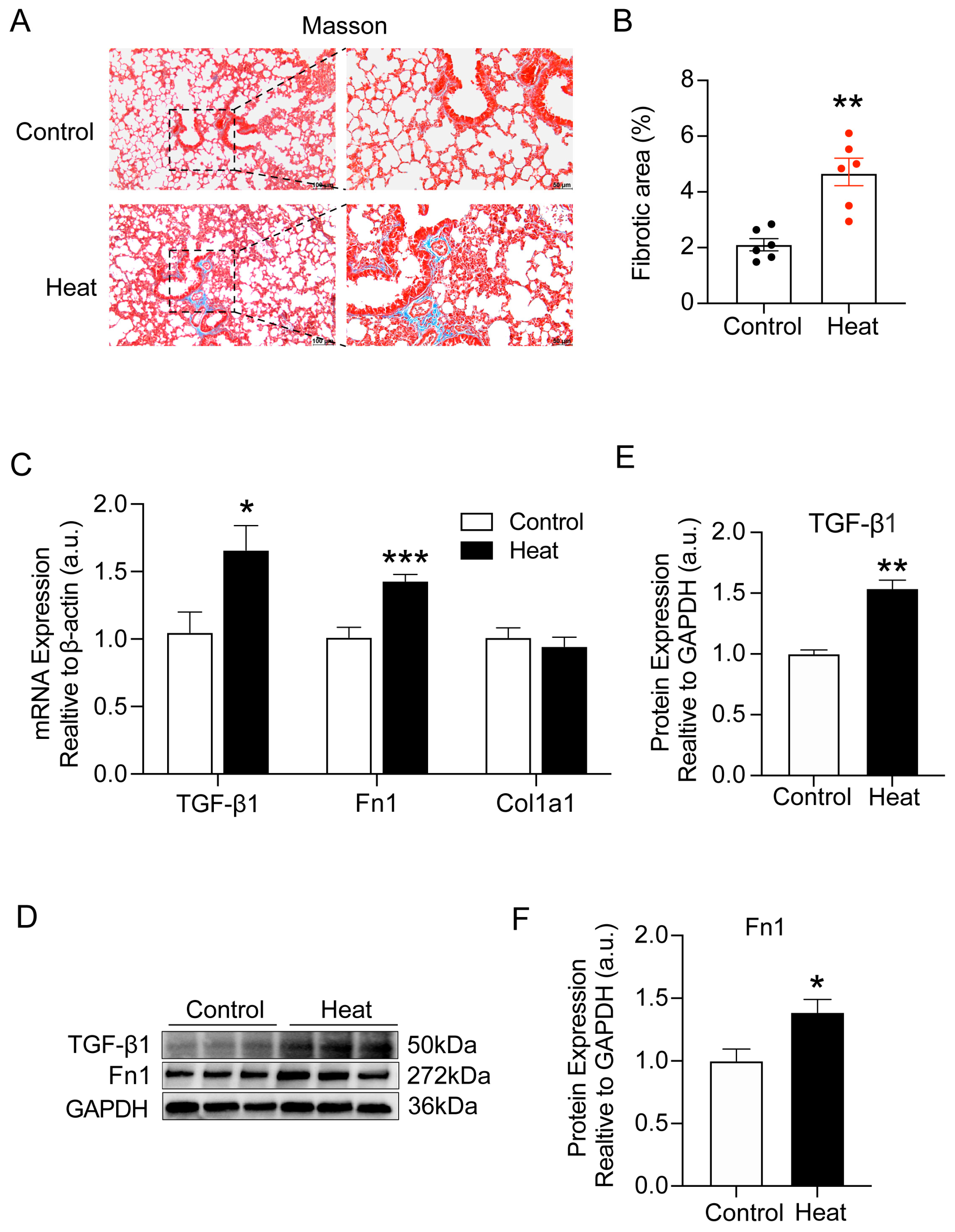
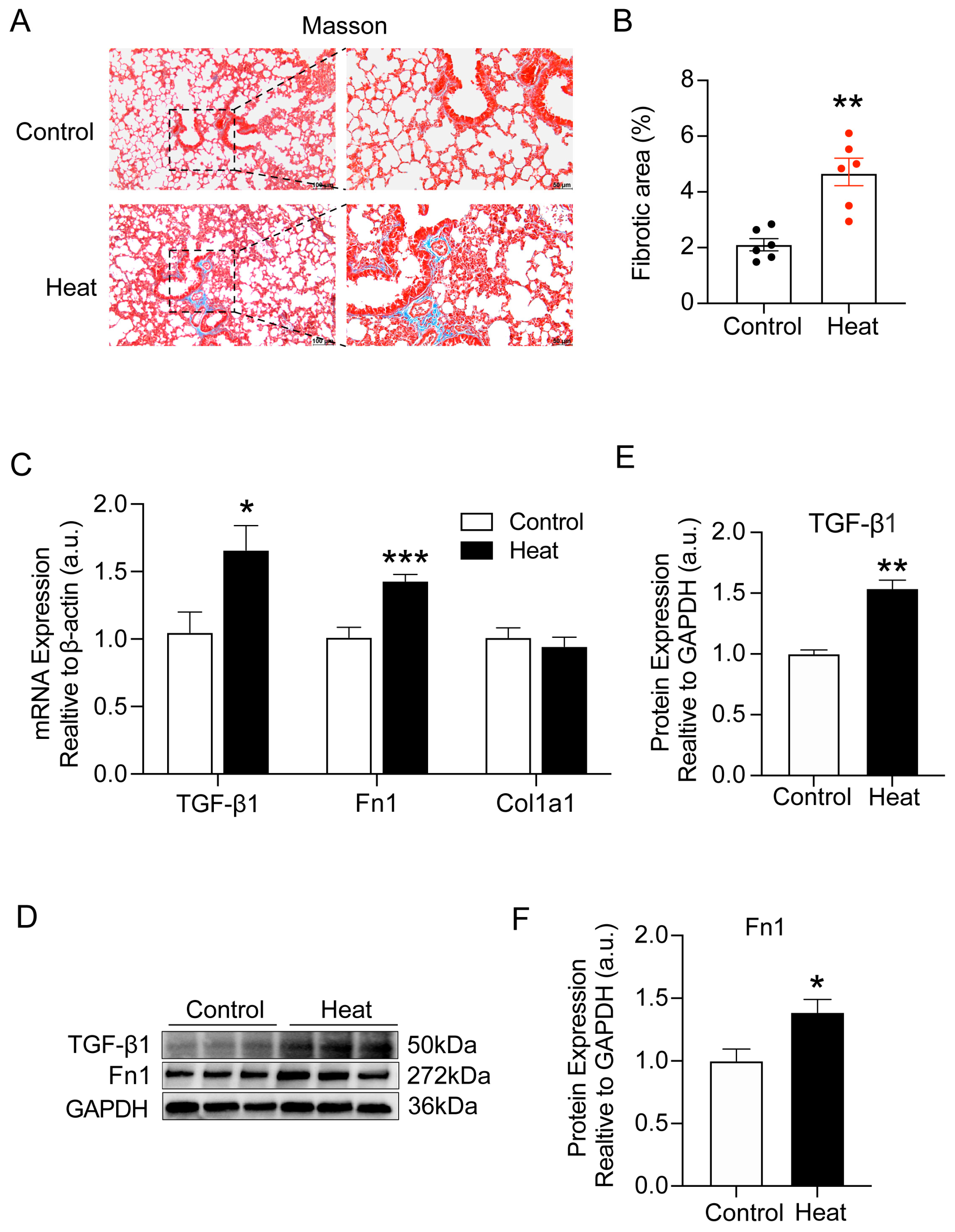
2.1. Heat Exposure-Induced Early Pulmonary Fibrosis-like Changes in Mice
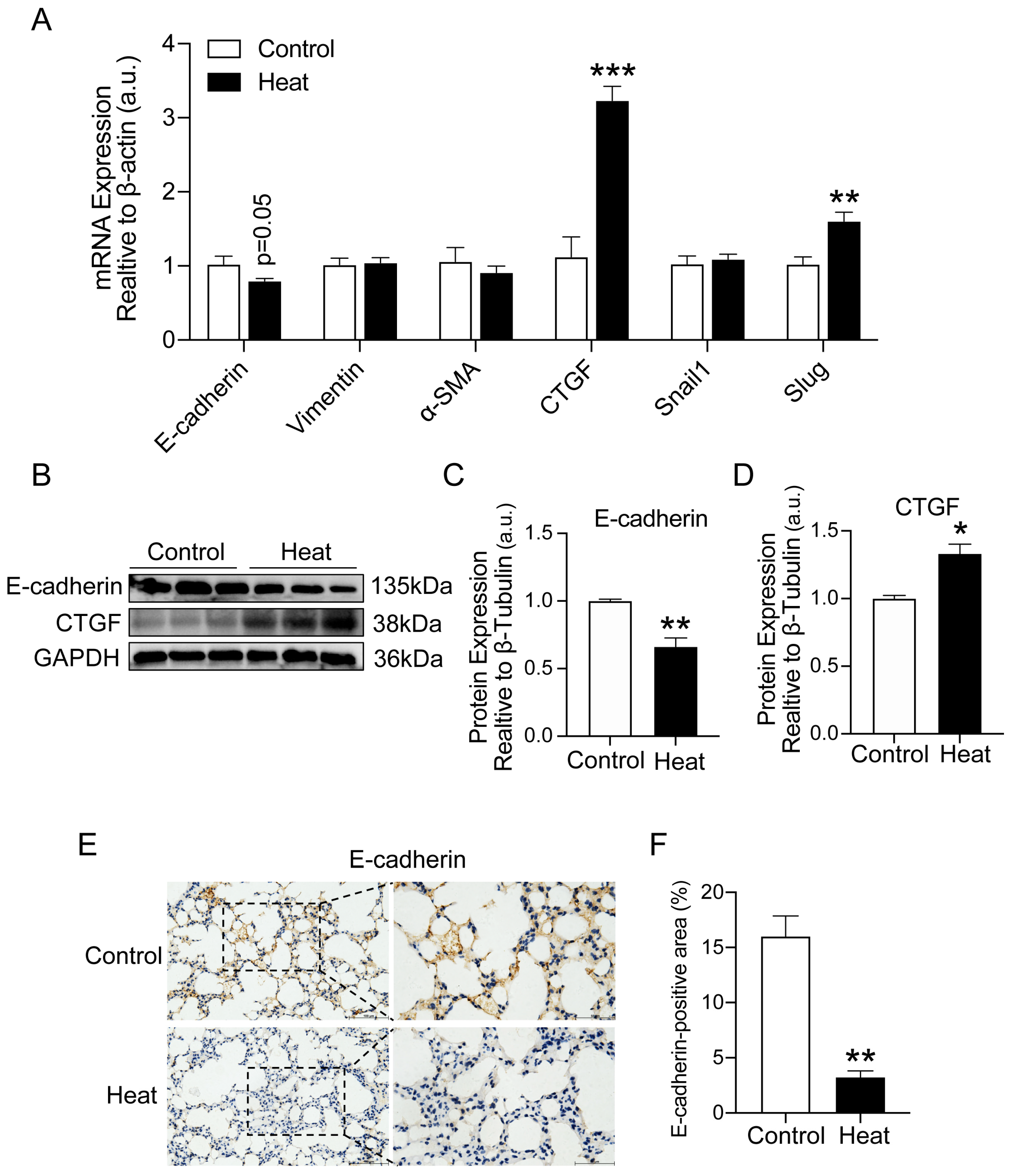
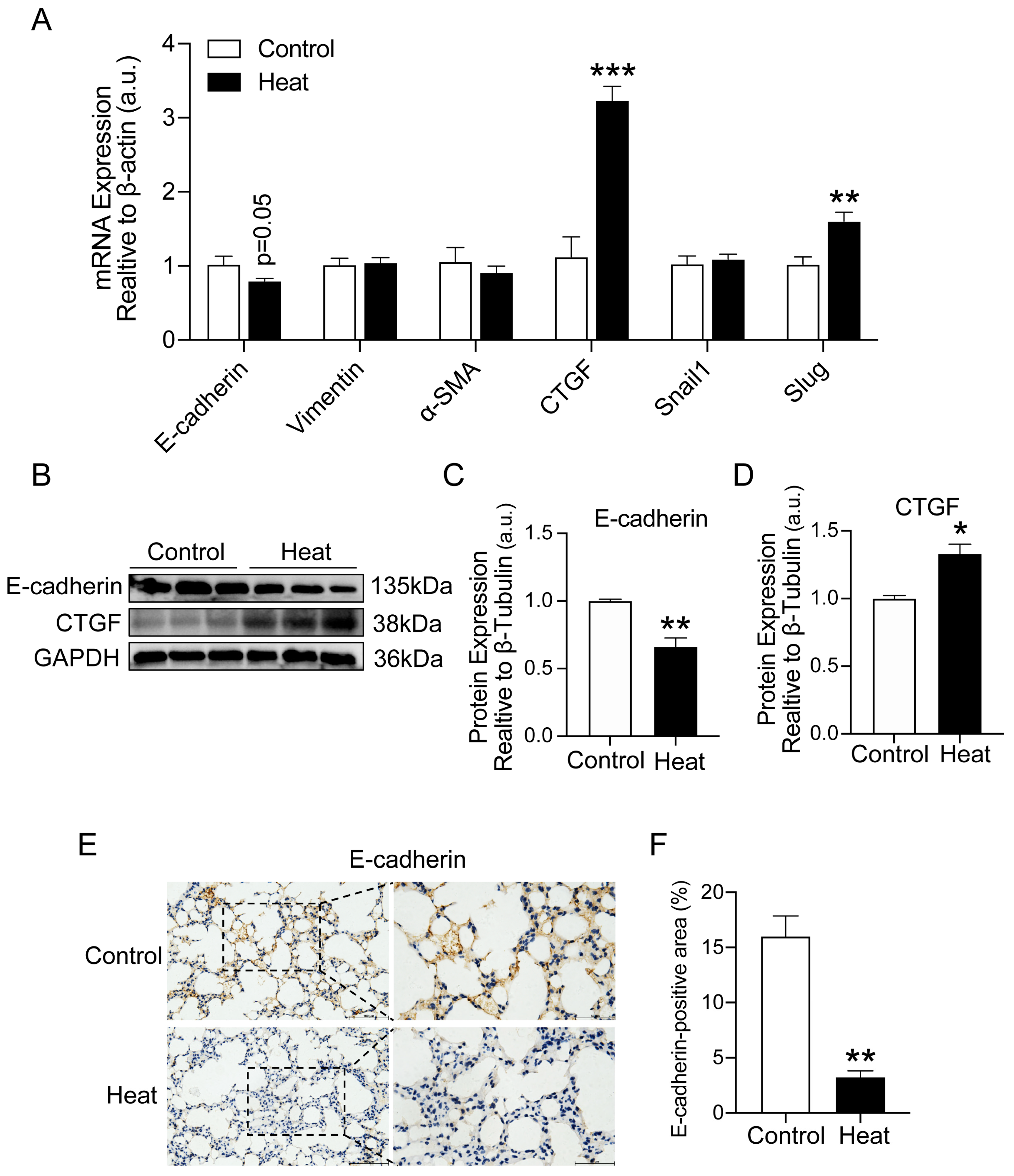
2.2. Heat Exposure-Induced EMT in Lung Tissue of Mice
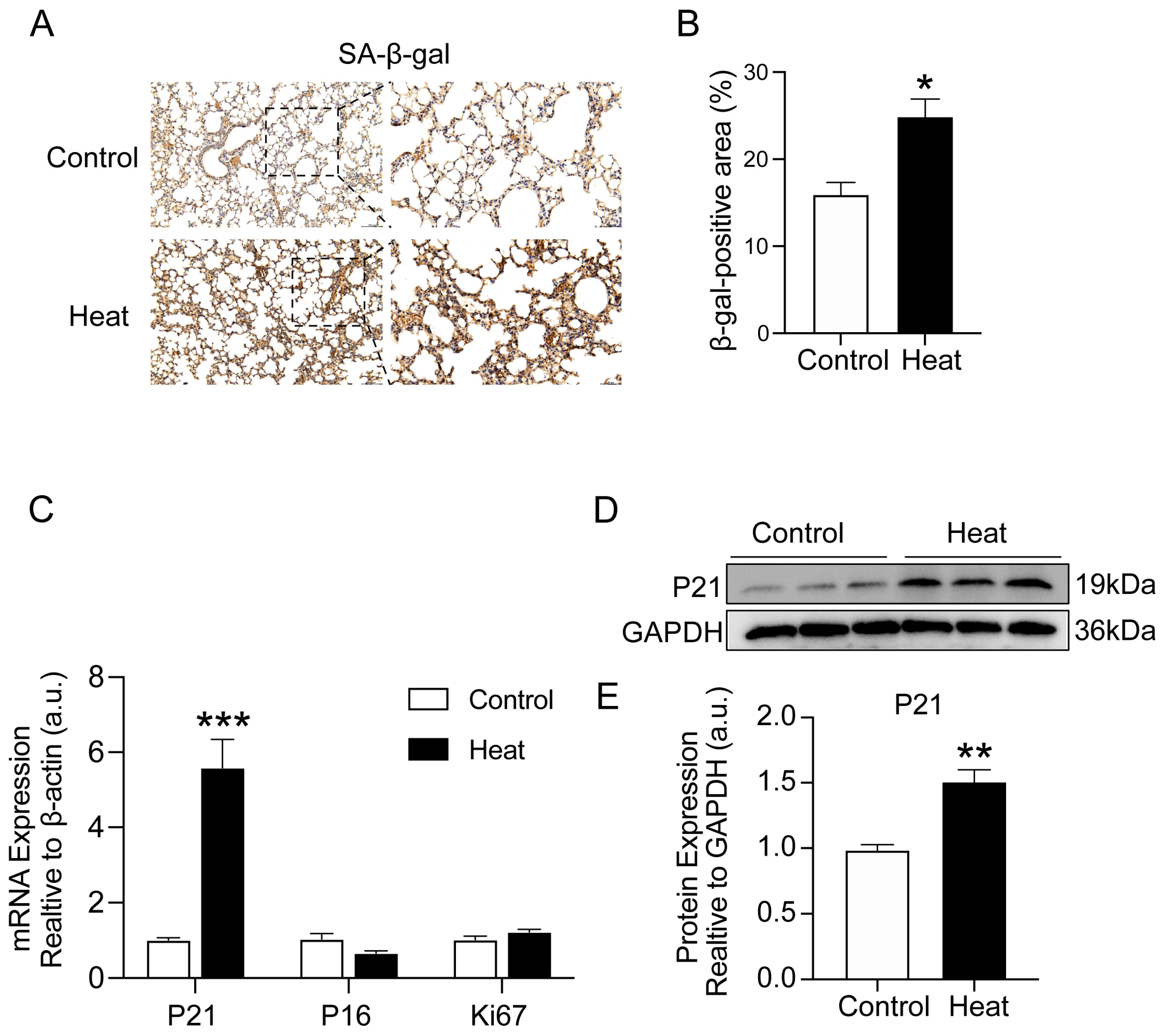
2.3. Heat Exposure-Induced Lung Cellular Senescence in Mice
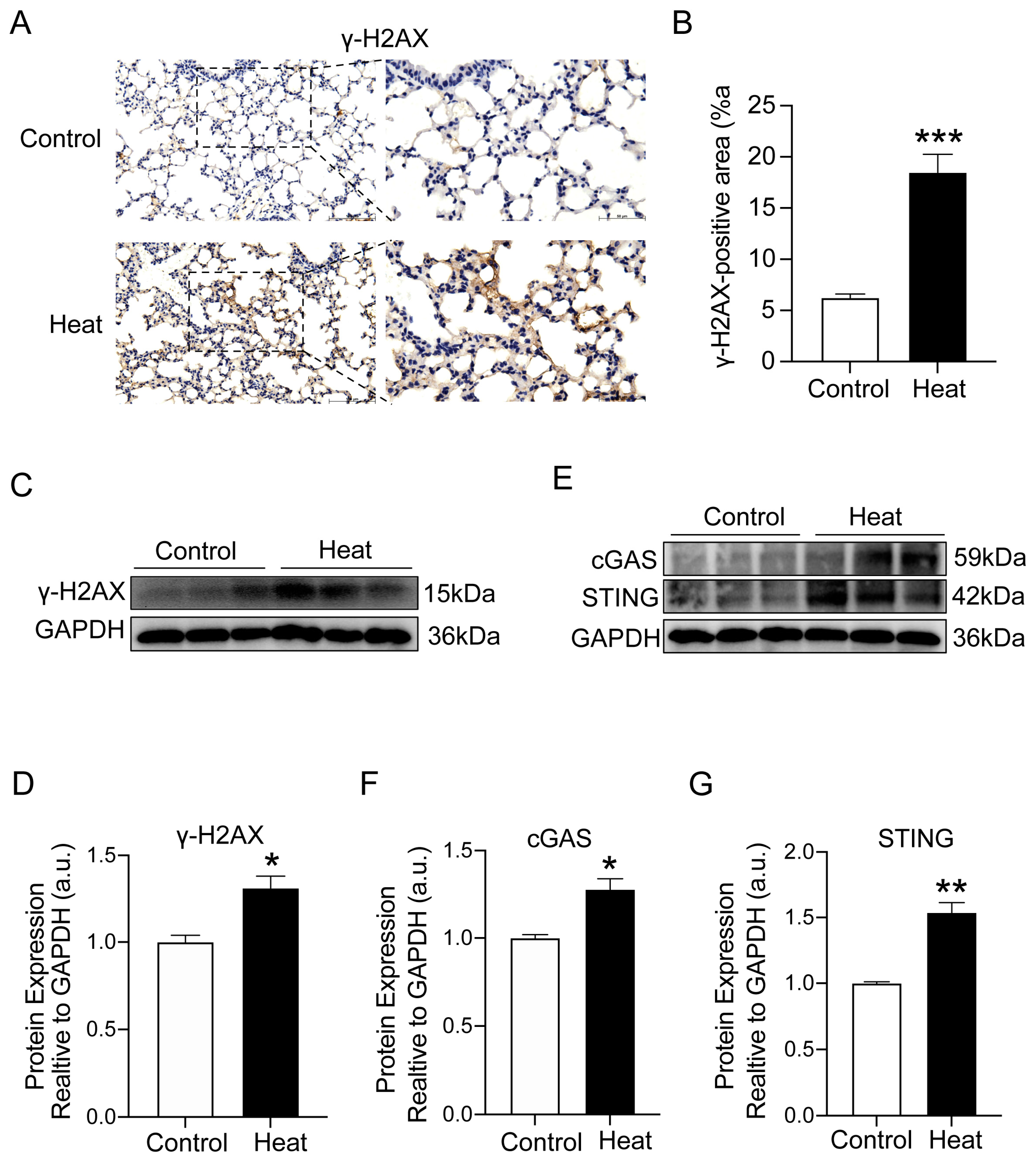
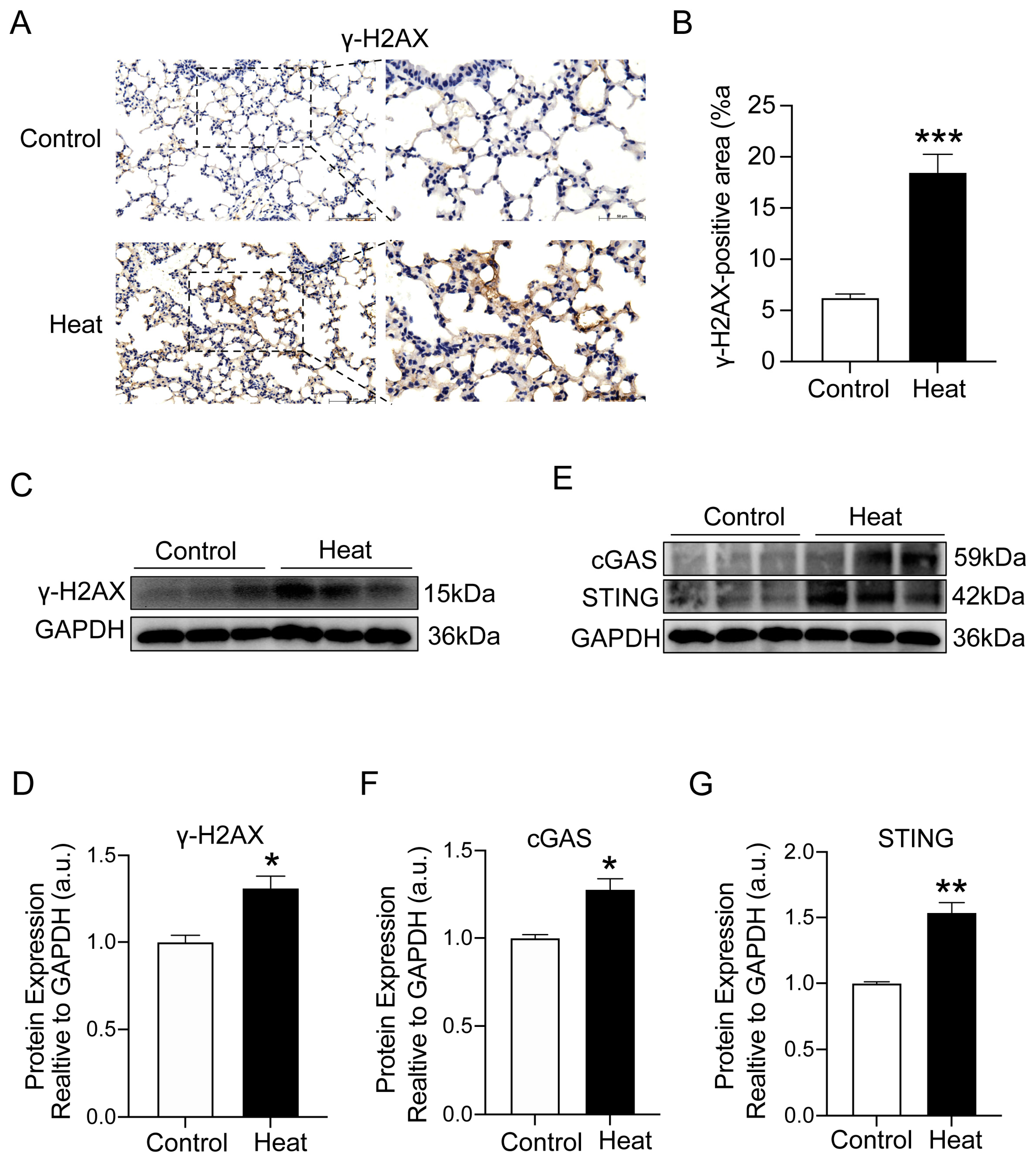
2.4. Heat Exposure-Induced DNA Damage and cGAS–STING Pathway Activation
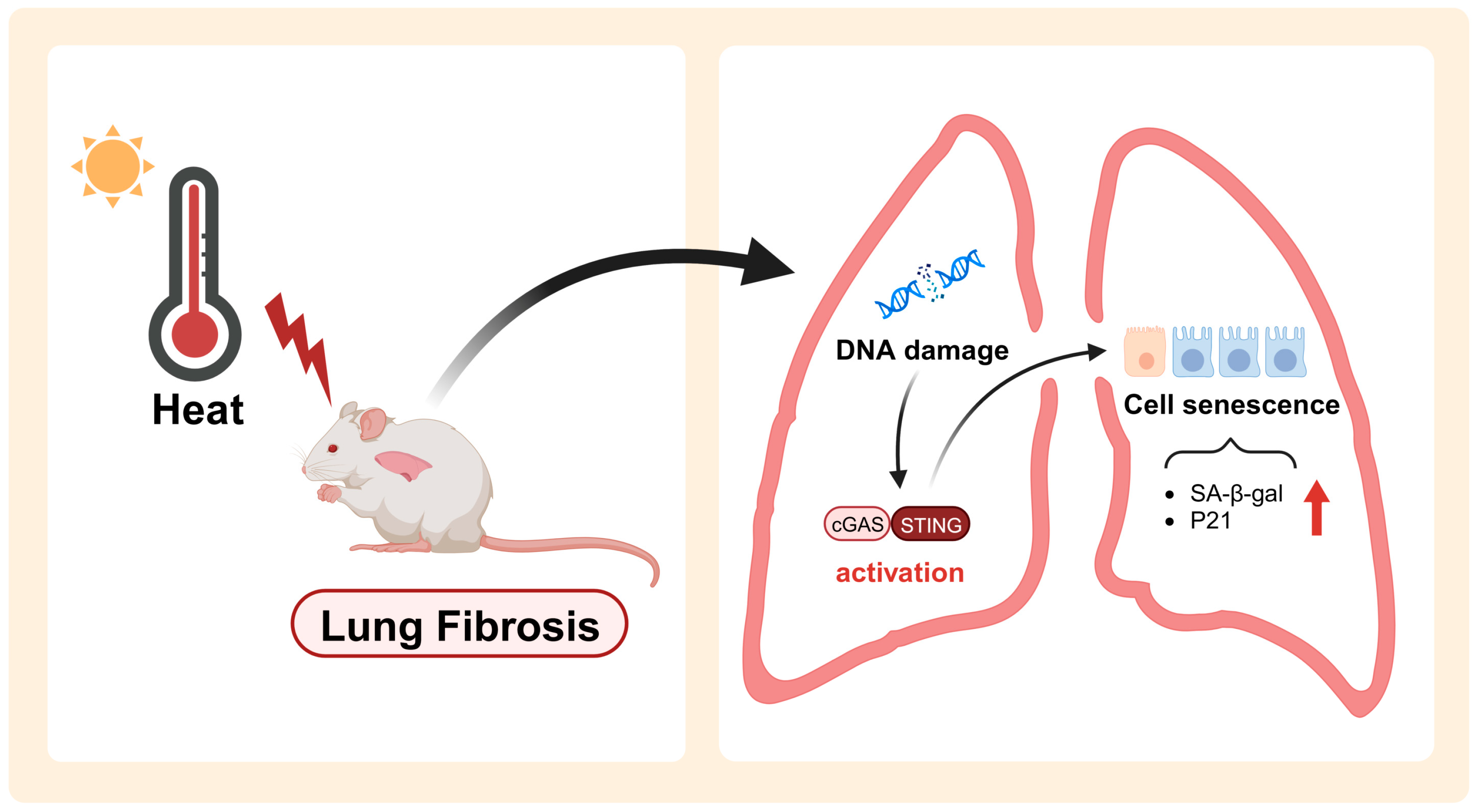
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Design
4.3. Masson’s Staining
4.4. Immunohistochemistry (IHC)
4.5. Quantitative Real-Time PCR
4.6. Western Blot
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bongioanni, P.; Del Carratore, R.; Corbianco, S.; Diana, A.; Cavallini, G.; Masciandaro, S.M.; Dini, M.; Buizza, R. Climate change and neurodegenerative diseases. Environ. Res. 2021, 201, 111511. [Google Scholar] [CrossRef]
- Armstrong McKay, D.I.; Staal, A.; Abrams, J.F.; Winkelmann, R.; Sakschewski, B.; Loriani, S.; Fetzer, I.; Cornell, S.E.; Rockström, J.; Lenton, T.M. Exceeding 1.5 °C global warming could trigger multiple climate tipping points. Science 2022, 377, eabn7950. [Google Scholar] [CrossRef]
- Covert, H.H.; Abdoel Wahid, F.; Wenzel, S.E.; Lichtveld, M.Y. Climate change impacts on respiratory health: Exposure, vulnerability, and risk. Physiol. Rev. 2023, 103, 2507–2522. [Google Scholar] [CrossRef]
- Cramer, M.N.; Gagnon, D.; Laitano, O.; Crandall, C.G. Human temperature regulation under heat stress in health, disease, and injury. Physiol. Rev. 2022, 102, 1907–1989. [Google Scholar] [CrossRef]
- Bouchama, A.; Abuyassin, B.; Lehe, C.; Laitano, O.; Jay, O.; O’Connor, F.G.; Leon, L.R. Classic and exertional heatstroke. Nat. Rev. Dis. Primers 2022, 8, 8. [Google Scholar] [CrossRef]
- Miao, J.; Feng, S.; Wang, M.; Jiang, N.; Yu, P.; Wu, Y.; Ye, T.; Wen, B.; Lu, P.; Li, S.; et al. Life-time summer heat exposure and lung function in young adults: A retrospective cohort study in Shandong China. Environ. Int. 2022, 160, 107058. [Google Scholar] [CrossRef]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127. [Google Scholar] [CrossRef]
- Braga, A.L.; Zanobetti, A.; Schwartz, J. The effect of weather on respiratory and cardiovascular deaths in 12 U.S. cities. Environ. Health Perspect. 2002, 110, 859–863. [Google Scholar] [CrossRef]
- Anderson, G.B.; Dominici, F.; Wang, Y.; McCormack, M.C.; Bell, M.L.; Peng, R.D. Heat-related emergency hospitalizations for respiratory diseases in the Medicare population. Am. J. Respir. Crit. Care Med. 2013, 187, 1098–1103. [Google Scholar] [CrossRef]
- Koudstaal, T.; Funke-Chambour, M.; Kreuter, M.; Molyneaux, P.L.; Wijsenbeek, M.S. Pulmonary fibrosis: From pathogenesis to clinical decision-making. Trends Mol. Med. 2023, 29, 1076–1087. [Google Scholar] [CrossRef]
- Savin, I.A.; Zenkova, M.A.; Sen’kova, A.V. Pulmonary Fibrosis as a Result of Acute Lung Inflammation: Molecular Mechanisms, Relevant In Vivo Models, Prognostic and Therapeutic Approaches. Int. J. Mol. Sci. 2022, 23, 14959. [Google Scholar] [CrossRef]
- Geng, Y.; Li, R.; He, S.X.; Yang, H.H.; Deng, Q.T.; Shao, X.Y.; Wu, Y.S.; Xu, W.W.; Ma, Q. Dexmedetomidine Attenuates Acute Lung Injury Induced by Heatstroke and Improve Outcome. Shock 2019, 52, 532–539. [Google Scholar] [CrossRef]
- Habibi, P.; Ostad, S.N.; Heydari, A.; Aliebrahimi, S.; Montazeri, V.; Foroushani, A.R.; Monazzam, M.R.; Ghazi-Khansari, M.; Golbabaei, F. Effect of heat stress on DNA damage: A systematic literature review. Int. J. Biometeorol. 2022, 66, 2147–2158. [Google Scholar] [CrossRef]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, L.; Gao, H.; Kuang, X.; Xiao, H.; Yang, C.; Cheng, Y.; Zhang, L.; Guo, X.; Zhong, Y.; et al. APE1 promotes non-homologous end joining by initiating DNA double-strand break formation and decreasing ubiquitination of artemis following oxidative genotoxic stress. J. Transl. Med. 2023, 21, 183. [Google Scholar] [CrossRef]
- Chen, C.; Xu, P. Cellular functions of cGAS-STING signaling. Trends Cell Biol. 2023, 33, 630–648. [Google Scholar] [CrossRef]
- Pan, J.; Fei, C.J.; Hu, Y.; Wu, X.Y.; Nie, L.; Chen, J. Current understanding of the cGAS-STING signaling pathway: Structure, regulatory mechanisms, and related diseases. Zool. Res. 2023, 44, 183–218. [Google Scholar] [CrossRef]
- Patel, D.J.; Yu, Y.; Xie, W. cGAMP-activated cGAS-STING signaling: Its bacterial origins and evolutionary adaptation by metazoans. Nat. Struct. Mol. Biol. 2023, 30, 245–260. [Google Scholar] [CrossRef]
- Schmitz, C.R.R.; Maurmann, R.M.; Guma, F.; Bauer, M.E.; Barbé-Tuana, F.M. cGAS-STING pathway as a potential trigger of immunosenescence and inflammaging. Front. Immunol. 2023, 14, 1132653. [Google Scholar] [CrossRef]
- Kessler, N.; Viehmann, S.F.; Krollmann, C.; Mai, K.; Kirschner, K.M.; Luksch, H.; Kotagiri, P.; Böhner, A.M.C.; Huugen, D.; de Oliveira Mann, C.C.; et al. Monocyte-derived macrophages aggravate pulmonary vasculitis via cGAS/STING/IFN-mediated nucleic acid sensing. J. Exp. Med. 2022, 219, e20220759. [Google Scholar] [CrossRef]
- Zhao, Y.; Simon, M.; Seluanov, A.; Gorbunova, V. DNA damage and repair in age-related inflammation. Nat. Rev. Immunol. 2023, 23, 75–89. [Google Scholar] [CrossRef]
- Ogrodnik, M. Cellular aging beyond cellular senescence: Markers of senescence prior to cell cycle arrest in vitro and in vivo. Aging Cell 2021, 20, e13338. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef]
- Chen, Y.; Yuan, S.; Cao, Y.; Kong, G.; Jiang, F.; Li, Y.; Wang, Q.; Tang, M.; Zhang, Q.; Wang, Q.; et al. Gasotransmitters: Potential Therapeutic Molecules of Fibrotic Diseases. Oxid. Med. Cell Longev. 2021, 2021, 3206982. [Google Scholar] [CrossRef]
- Liu, L.; Sun, Q.; Davis, F.; Mao, J.; Zhao, H.; Ma, D. Epithelial-mesenchymal transition in organ fibrosis development: Current understanding and treatment strategies. Burns Trauma. 2022, 10, tkac011. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef]
- Groen, K.; van der Vis, J.J.; van Batenburg, A.A.; Kazemier, K.M.; Grutters, J.C.; van Moorsel, C.H.M. Genetic Variant Overlap Analysis Identifies Established and Putative Genes Involved in Pulmonary Fibrosis. Int. J. Mol. Sci. 2023, 24, 2790. [Google Scholar] [CrossRef]
- Confalonieri, P.; Volpe, M.C.; Jacob, J.; Maiocchi, S.; Salton, F.; Ruaro, B.; Confalonieri, M.; Braga, L. Regeneration or Repair? The Role of Alveolar Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Cells 2022, 11, 2095. [Google Scholar] [CrossRef]
- Renaud, L.; Waldrep, K.M.; da Silveira, W.A.; Pilewski, J.M.; Feghali-Bostwick, C.A. First Characterization of the Transcriptome of Lung Fibroblasts of SSc Patients and Healthy Donors of African Ancestry. Int. J. Mol. Sci. 2023, 24, 3645. [Google Scholar] [CrossRef]
- Jia, Q.; Lei, Y.; Chen, S.; Liu, S.; Wang, T.; Cheng, Y. Circulating inflammatory cytokines and risk of idiopathic pulmonary fibrosis: A Mendelian randomization study. BMC Pulm. Med. 2023, 23, 369. [Google Scholar] [CrossRef]
- Burkart, K.G.; Brauer, M.; Aravkin, A.Y.; Godwin, W.W.; Hay, S.I.; He, J.; Iannucci, V.C.; Larson, S.L.; Lim, S.S.; Liu, J.; et al. Estimating the cause-specific relative risks of non-optimal temperature on daily mortality: A two-part modelling approach applied to the Global Burden of Disease Study. Lancet 2021, 398, 685–697. [Google Scholar] [CrossRef]
- Vicedo-Cabrera, A.M.; Scovronick, N.; Sera, F.; Royé, D.; Schneider, R.; Tobias, A.; Astrom, C.; Guo, Y.; Honda, Y.; Hondula, D.M.; et al. The burden of heat-related mortality attributable to recent human-induced climate change. Nat. Clim. Chang. 2021, 11, 492–500. [Google Scholar] [CrossRef]
- Konstantinoudis, G.; Minelli, C.; Lam, H.C.Y.; Fuertes, E.; Ballester, J.; Davies, B.; Vicedo-Cabrera, A.M.; Gasparrini, A.; Blangiardo, M. Asthma Hospitalisations and Heat Exposure in England: A Case-Crossover Study during 2002–2019. Thorax 2023, 78, 875–881. [Google Scholar] [CrossRef]
- Konstantinoudis, G.; Minelli, C.; Vicedo-Cabrera, A.M.; Ballester, J.; Gasparrini, A.; Blangiardo, M. Ambient heat exposure and COPD hospitalisations in England: A nationwide case-crossover study during 2007–2018. Thorax 2022, 77, 1098–1104. [Google Scholar] [CrossRef]
- Scheerens, C.; Nurhussien, L.; Aglan, A.; Synn, A.J.; Coull, B.A.; Koutrakis, P.; Rice, M.B. The impact of personal and outdoor temperature exposure during cold and warm seasons on lung function and respiratory symptoms in COPD. ERJ Open Res. 2022, 8, 00574–2021. [Google Scholar] [CrossRef]
- Smirnova, N.; Shaver, A.C.; Mehta, A.J.; Philipsborn, R.; Scovronick, N. Climate Change, Air Quality, and Pulmonary Health Disparities. Clin. Chest Med. 2023, 44, 489–499. [Google Scholar] [CrossRef]
- Wang, X.; Lu, W.; Xia, X.; Zhu, Y.; Ge, C.; Guo, X.; Zhang, N.; Chen, H.; Xu, S. Selenomethionine mitigate PM2.5-induced cellular senescence in the lung via attenuating inflammatory response mediated by cGAS/STING/NF-κB pathway. Ecotoxicol. Environ. Saf. 2022, 247, 114266. [Google Scholar] [CrossRef]
- Trachalaki, A.; Irfan, M.; Wells, A.U. Pharmacological management of Idiopathic Pulmonary Fibrosis: Current and emerging options. Expert. Opin. Pharmacother. 2021, 22, 191–204. [Google Scholar] [CrossRef]
- Lingampally, A.; Jones, M.R.; Bagari, S.; Chen, C.; Rivetti, S.; Bellusci, S. Use of the Reversible Myogenic to Lipogenic Transdifferentiation Switch for the Design of Pre-clinical Drug Screening in Idiopathic Pulmonary Fibrosis. Front. Bioeng. Biotechnol. 2020, 8, 569865. [Google Scholar] [CrossRef]
- Lovisa, S. Epithelial-to-Mesenchymal Transition in Fibrosis: Concepts and Targeting Strategies. Front. Pharmacol. 2021, 12, 737570. [Google Scholar] [CrossRef]
- Recouvreux, M.V.; Moldenhauer, M.R.; Galenkamp, K.M.O.; Jung, M.; James, B.; Zhang, Y.; Lowy, A.; Bagchi, A.; Commisso, C. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J. Exp. Med. 2020, 217, e20200388. [Google Scholar] [CrossRef] [PubMed]
- Mundhara, N.; Majumder, A.; Panda, D. Hyperthermia induced disruption of mechanical balance leads to G1 arrest and senescence in cells. Biochem. J. 2021, 478, 179–196. [Google Scholar] [CrossRef]
- Shimoni, C.; Goldstein, M.; Ribarski-Chorev, I.; Schauten, I.; Nir, D.; Strauss, C.; Schlesinger, S. Heat Shock Alters Mesenchymal Stem Cell Identity and Induces Premature Senescence. Front. Cell Dev. Biol. 2020, 8, 565970. [Google Scholar] [CrossRef]
- Nir, D.; Ribarski-Chorev, I.; Shimoni, C.; Strauss, C.; Frank, J.; Schlesinger, S. Antioxidants Attenuate Heat Shock Induced Premature Senescence of Bovine Mesenchymal Stem Cells. Int. J. Mol. Sci. 2022, 23, 5750. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Okuda, R.; Aoshiba, K.; Matsushima, H.; Ogura, T.; Okudela, K.; Ohashi, K. Cellular senescence and senescence-associated secretory phenotype: Comparison of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung disease, and chronic obstructive pulmonary disease. J. Thorac. Dis. 2019, 11, 857–864. [Google Scholar] [CrossRef]
- Chin, C.; Ravichandran, R.; Sanborn, K.; Fleming, T.; Wheatcroft, S.B.; Kearney, M.T.; Tokman, S.; Walia, R.; Smith, M.A.; Flint, D.J.; et al. Loss of IGFBP2 mediates alveolar type 2 cell senescence and promotes lung fibrosis. Cell Rep. Med. 2023, 4, 100945. [Google Scholar] [CrossRef]
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173. [Google Scholar] [CrossRef]
- Pearce, K.M.; Okon, I.; Watson-Wright, C. Induction of Oxidative DNA Damage and Epithelial Mesenchymal Transitions in Small Airway Epithelial Cells Exposed to Cosmetic Aerosols. Toxicol. Sci. 2020, 177, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The central role of DNA damage in the ageing process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wu, X.; Zhao, D.; Liu, Y.; Du, Q.; Li, Y.; Xu, Y.; Li, Y.; Qiu, Y.; Yang, Y. Fluvoxamine alleviates bleomycin-induced lung fibrosis via regulating the cGAS-STING pathway. Pharmacol. Res. 2023, 187, 106577. [Google Scholar] [CrossRef]
- Wang, L.; Ye, B.; Liu, Y.; Li, J.; Li, C.; Wen, M.; Li, H. Xuebijing Injection Attenuates Heat Stroke-Induced Brain Injury through Oxidative Stress Blockage and Parthanatos Modulation via PARP-1/AIF Signaling. ACS Omega 2023, 8, 33392–33402. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, J.; Guo, X.; Tang, Y.; Zhou, G.; Liu, Y.; Huang, Q.; Geng, Y.; Liu, Z.; Su, L. Xuebijing injection reduces organ injuries and improves survival by attenuating inflammatory responses and endothelial injury in heatstroke mice. BMC Complement. Altern. Med. 2015, 15, 4. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| P16 | CGCAGGTTCTTGGTCACTGT | TGTTCACGAAAGCCAGAGCG |
| Ki67 | ATCATTGACCGCTCCTTTAGGT | GCTCGCCTTGATGGTTCCT |
| Snail1 | CACACGCTGCCTTGTGTCT | GGTCAGCAAAAGCACGGTT |
| Slug | TGGTCAAGAAACATTTCAACGCC | GGTGAGGATCTCTGGTTTGGTA |
| CTGF | GGGCCTCTTCTGCGATTTC | ATCCAGGCAAGTGCATTGTA |
| E-cadherin | CAGGTCTCCTCATGGCTTTGC | CTTCCGAAAAGAAGGCTGTCC |
| Vimentin | CGTCCACACGCACCTACAG | GGGGGATGAGGAATAGAGGCT |
| Col1a1 | GCTCCTCTTAGGGGCCACT | CCACGTCTCACCATTGGGG |
| Fn1 | ATGTGGACCCCTCCTGATAGT | GCCCAGTGATTTCAGCAAAGG |
| TGF-β1 | CTCCCGTGGCTTCTAGTGC | GCCTTAGTTTGGACAGGATCTG |
| α-SMA | GTCCCAGACATCAGGGAGTAA | TCGGATACTTCAGCGTCAGGA |
| P21 | CCTGGTGATGTCCGACCTG | CCATGAGCGCATCGCAATC |
| β-actin | TTCGTTGCCGGTCCACACCC | GCTTTGCACATGCCGGAGCC |
| Antibody | Dilution | Catalog Number | Vendor |
|---|---|---|---|
| E-Cadherin | 1:1000 | 3195 | Cell Signaling Technology |
| P21 | 1:1000 | 64016 | Cell Signaling Technology |
| CTGF | 1:1000 | A11067 | ABclonal |
| Fn1 | 1:1000 | A16678 | ABclona |
| Beta Galactosidase Polyclonal | 1:100 | 15518-1-AP | Proteintech |
| TGF-β1 | 1:1000 | 3711 | Cell Signaling Technology |
| γ-H2AX | 1:1000 | 9718 | Cell Signaling Technology |
| cGAS | 1:1000 | A23846 | ABclonal |
| STING | 1:1000 | A21051 | ABclonal |
| GAPDH | 1:1000 | 5174 | Cell Signaling Technology |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, T.; Zhang, J.; Wang, Y.; Zhang, G.; Li, S.; Fan, W.; Li, R.; Sun, Q.; Liu, C. Early Pulmonary Fibrosis-like Changes in the Setting of Heat Exposure: DNA Damage and Cell Senescence. Int. J. Mol. Sci. 2024, 25, 2992. https://doi.org/10.3390/ijms25052992
Hou T, Zhang J, Wang Y, Zhang G, Li S, Fan W, Li R, Sun Q, Liu C. Early Pulmonary Fibrosis-like Changes in the Setting of Heat Exposure: DNA Damage and Cell Senescence. International Journal of Molecular Sciences. 2024; 25(5):2992. https://doi.org/10.3390/ijms25052992
Chicago/Turabian StyleHou, Tong, Jiyang Zhang, Yindan Wang, Guoqing Zhang, Sanduo Li, Wenjun Fan, Ran Li, Qinghua Sun, and Cuiqing Liu. 2024. "Early Pulmonary Fibrosis-like Changes in the Setting of Heat Exposure: DNA Damage and Cell Senescence" International Journal of Molecular Sciences 25, no. 5: 2992. https://doi.org/10.3390/ijms25052992
APA StyleHou, T., Zhang, J., Wang, Y., Zhang, G., Li, S., Fan, W., Li, R., Sun, Q., & Liu, C. (2024). Early Pulmonary Fibrosis-like Changes in the Setting of Heat Exposure: DNA Damage and Cell Senescence. International Journal of Molecular Sciences, 25(5), 2992. https://doi.org/10.3390/ijms25052992