

Inhibition of PRMT1 Suppresses the Growth of U87MG-Derived Glioblastoma Stem Cells by Blocking the STAT3 Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
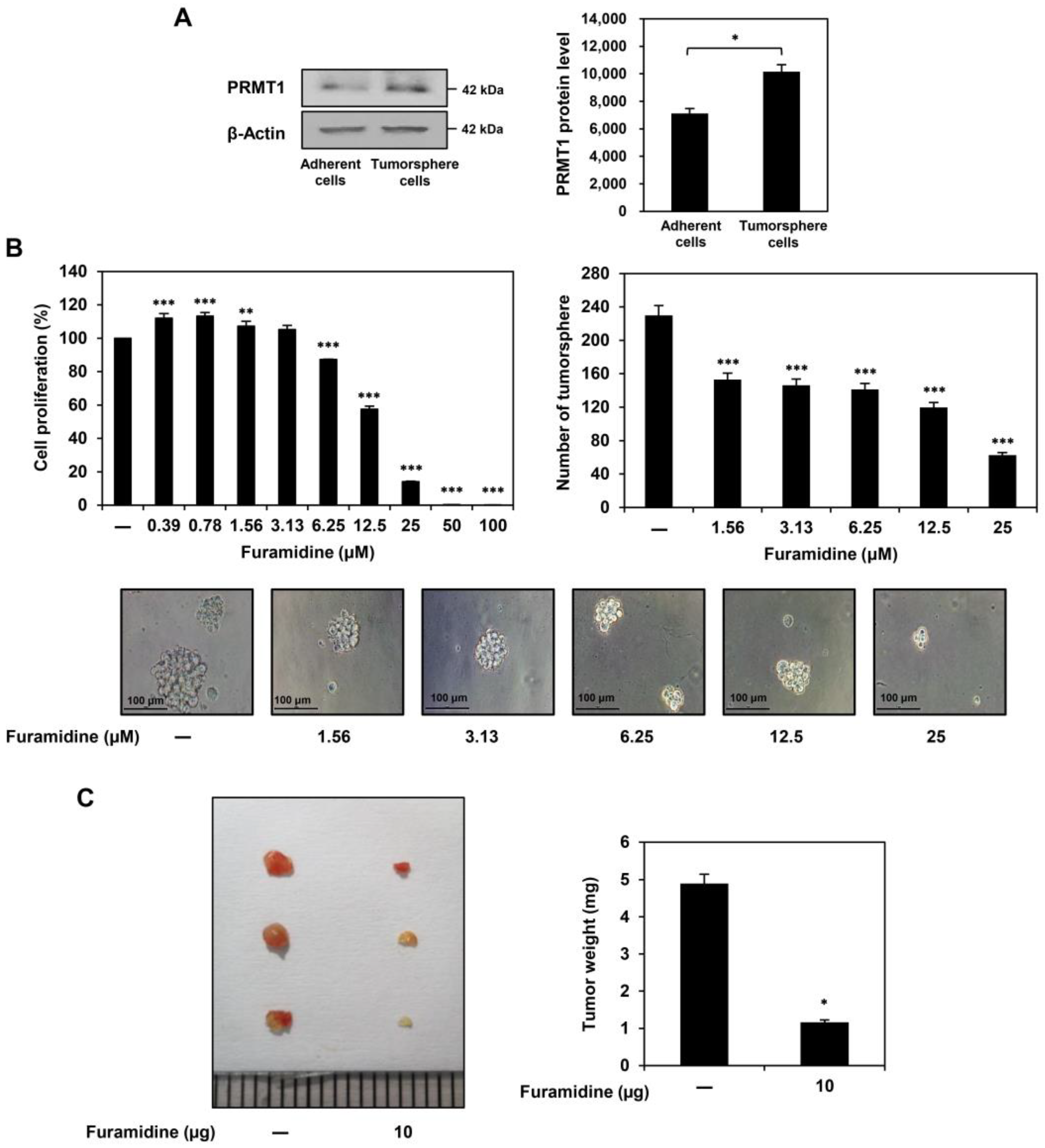
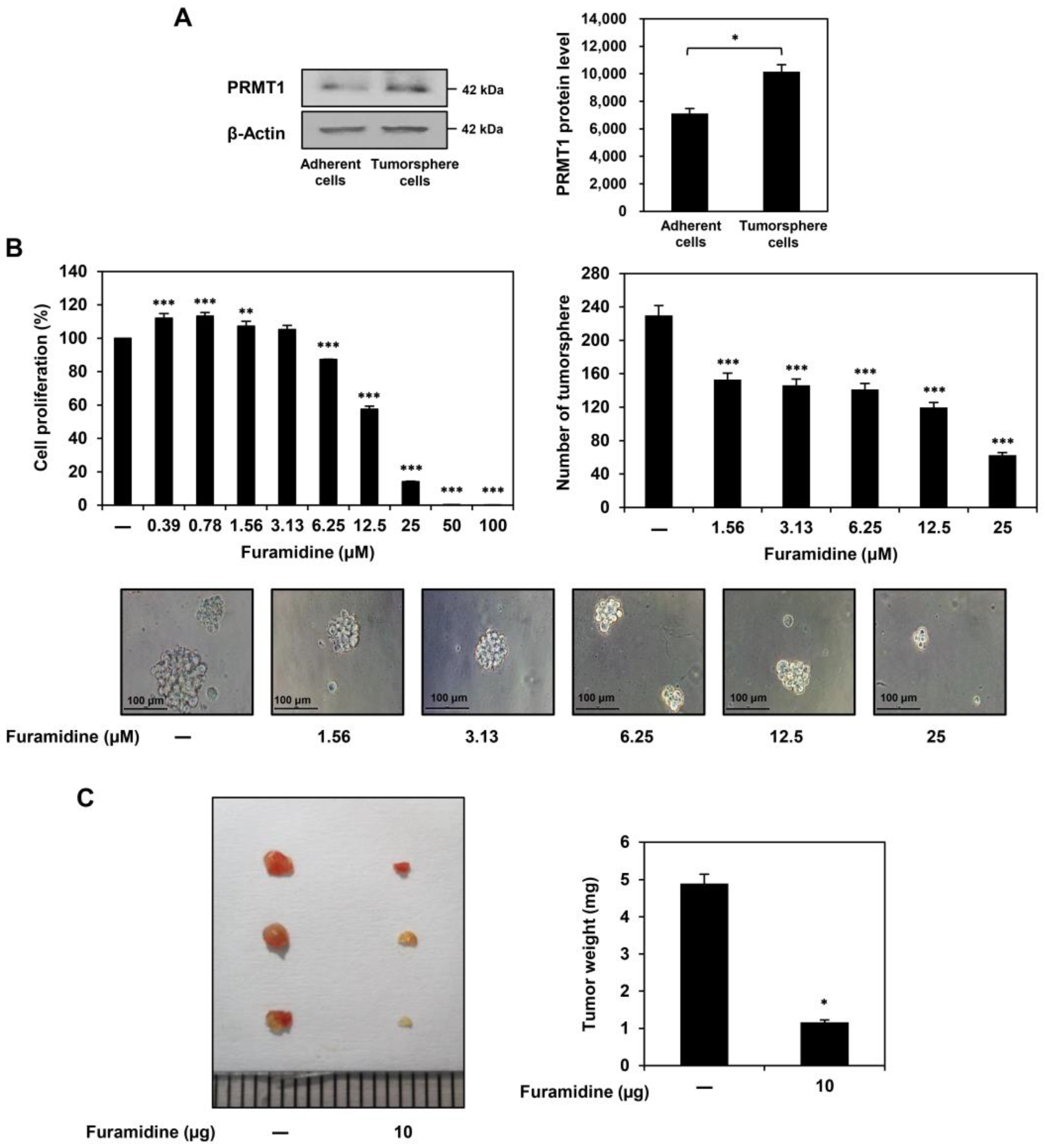
2.1. Furamidine Inhibits the Proliferation of U87MG-Derived GSCs Both In Vitro and In Vivo
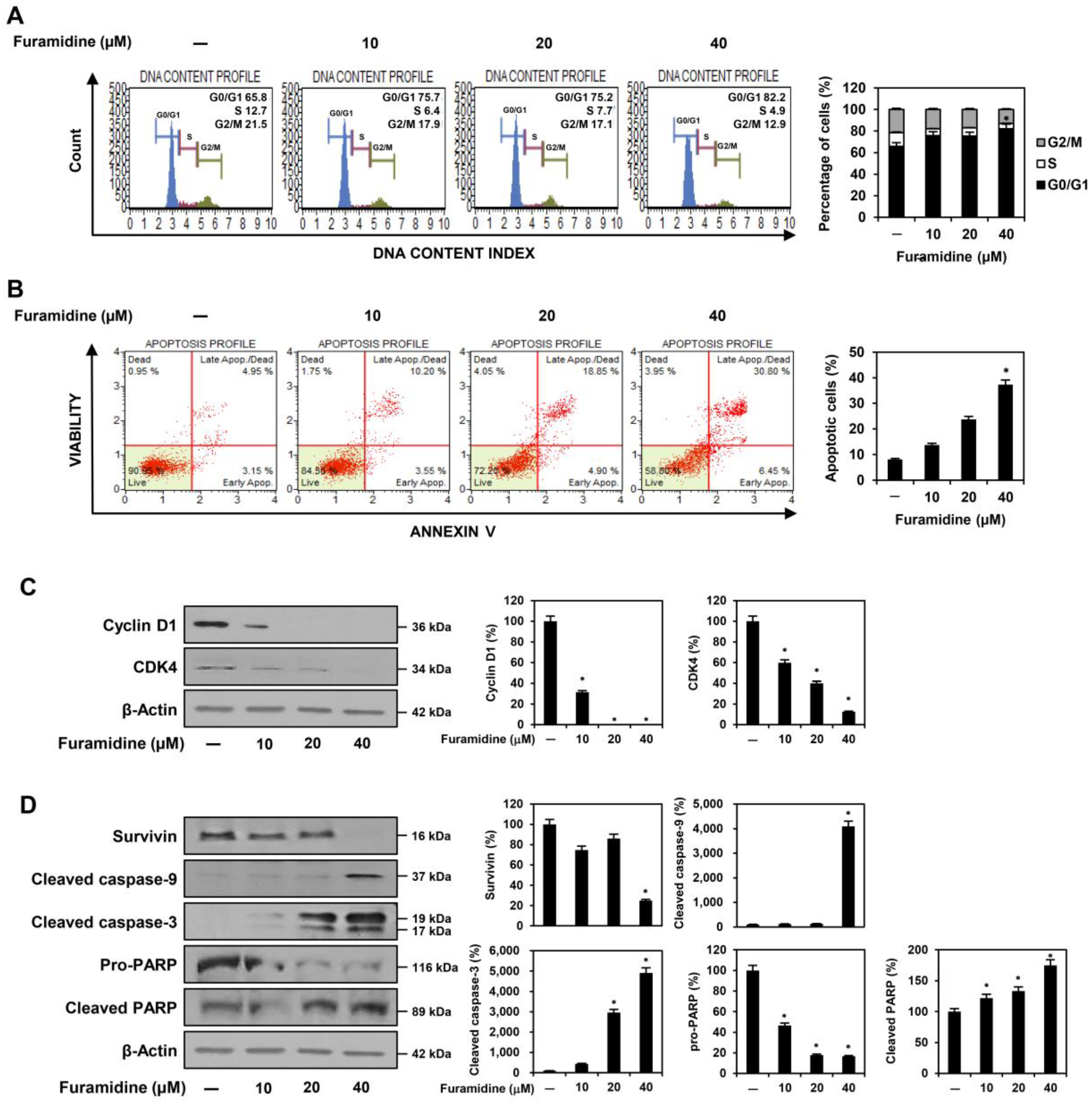
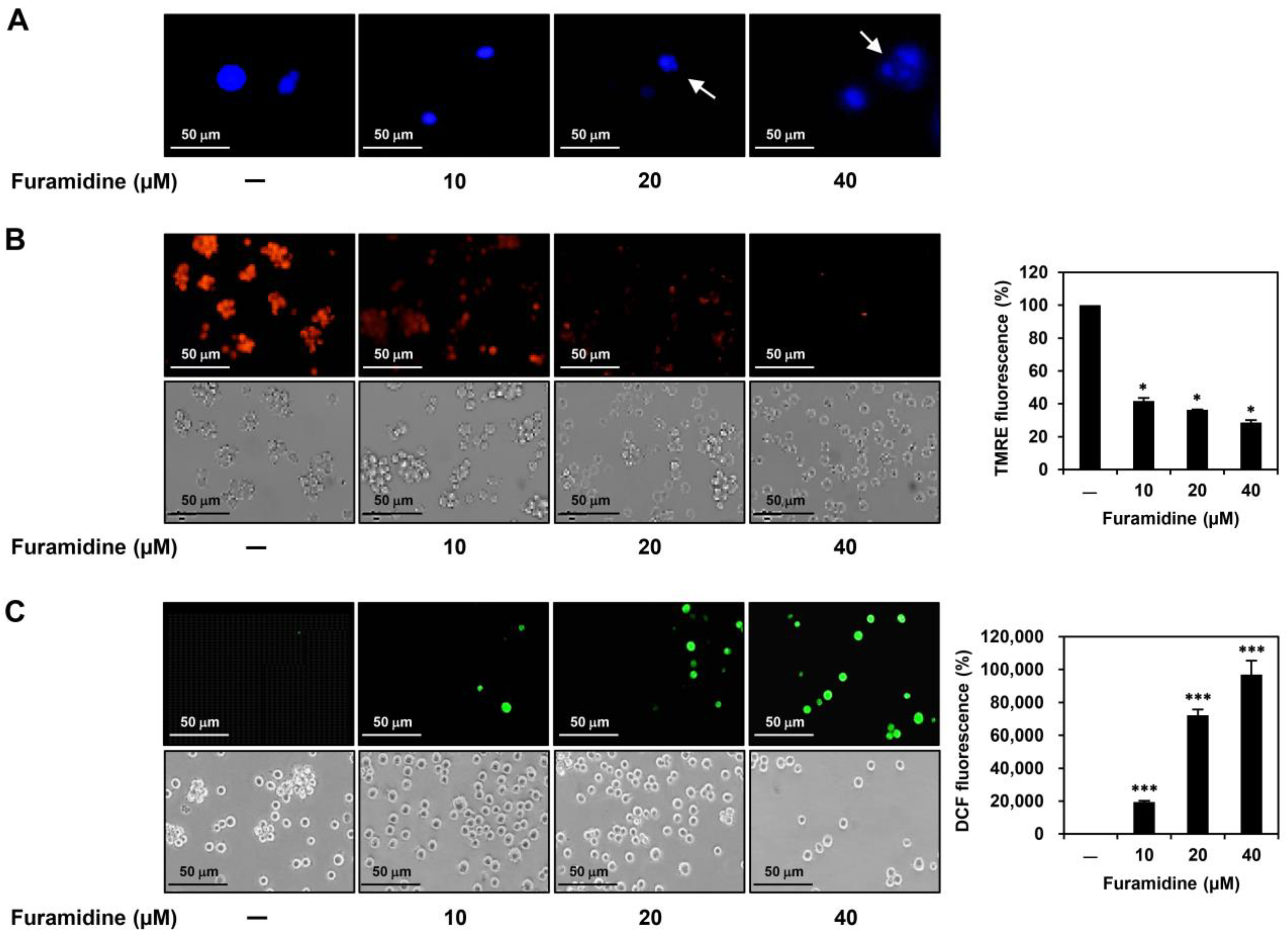
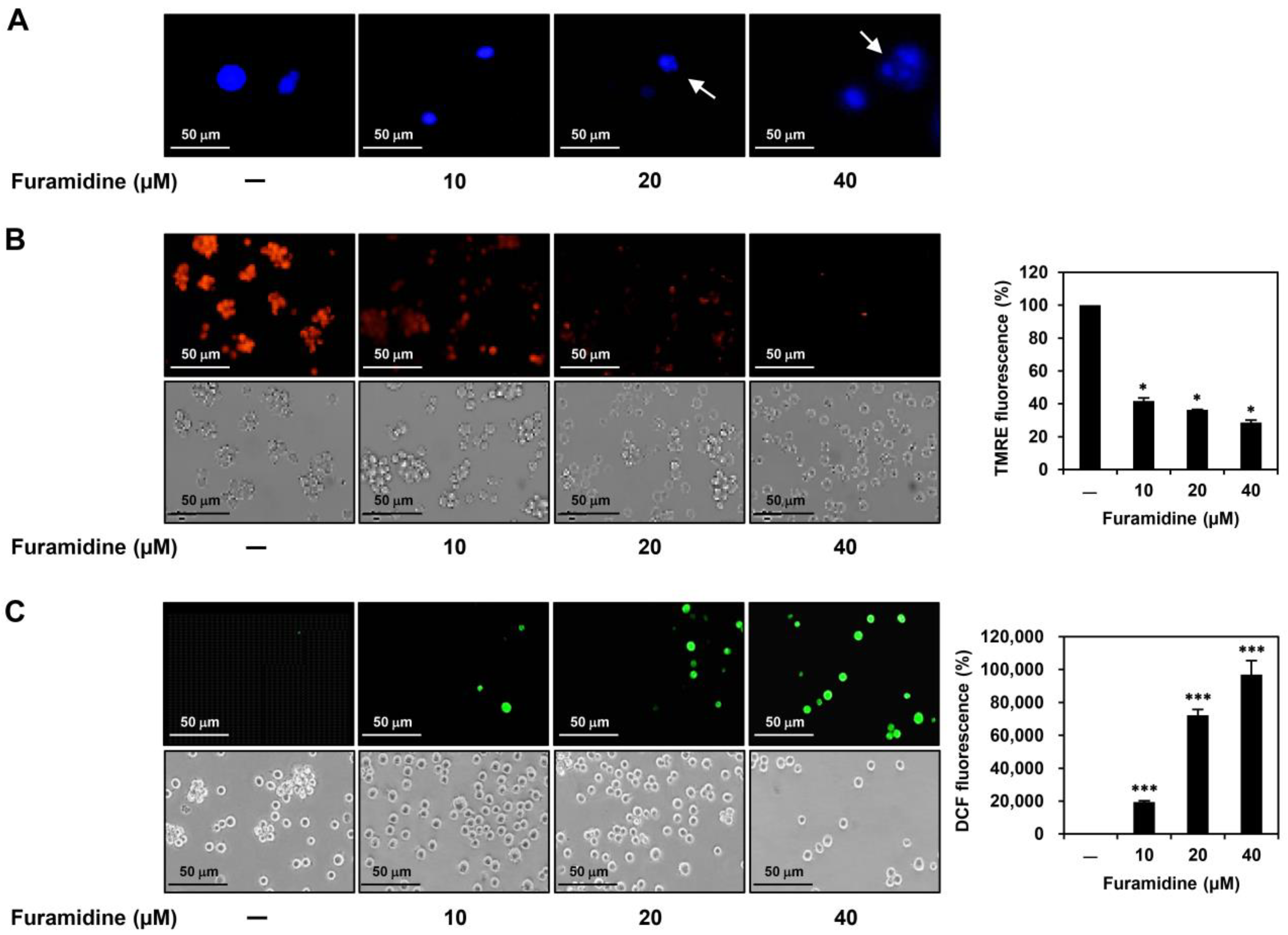
2.2. Furamidine Induces Cell Cycle Arrest and Apoptosis in U87MG-Derived GSCs
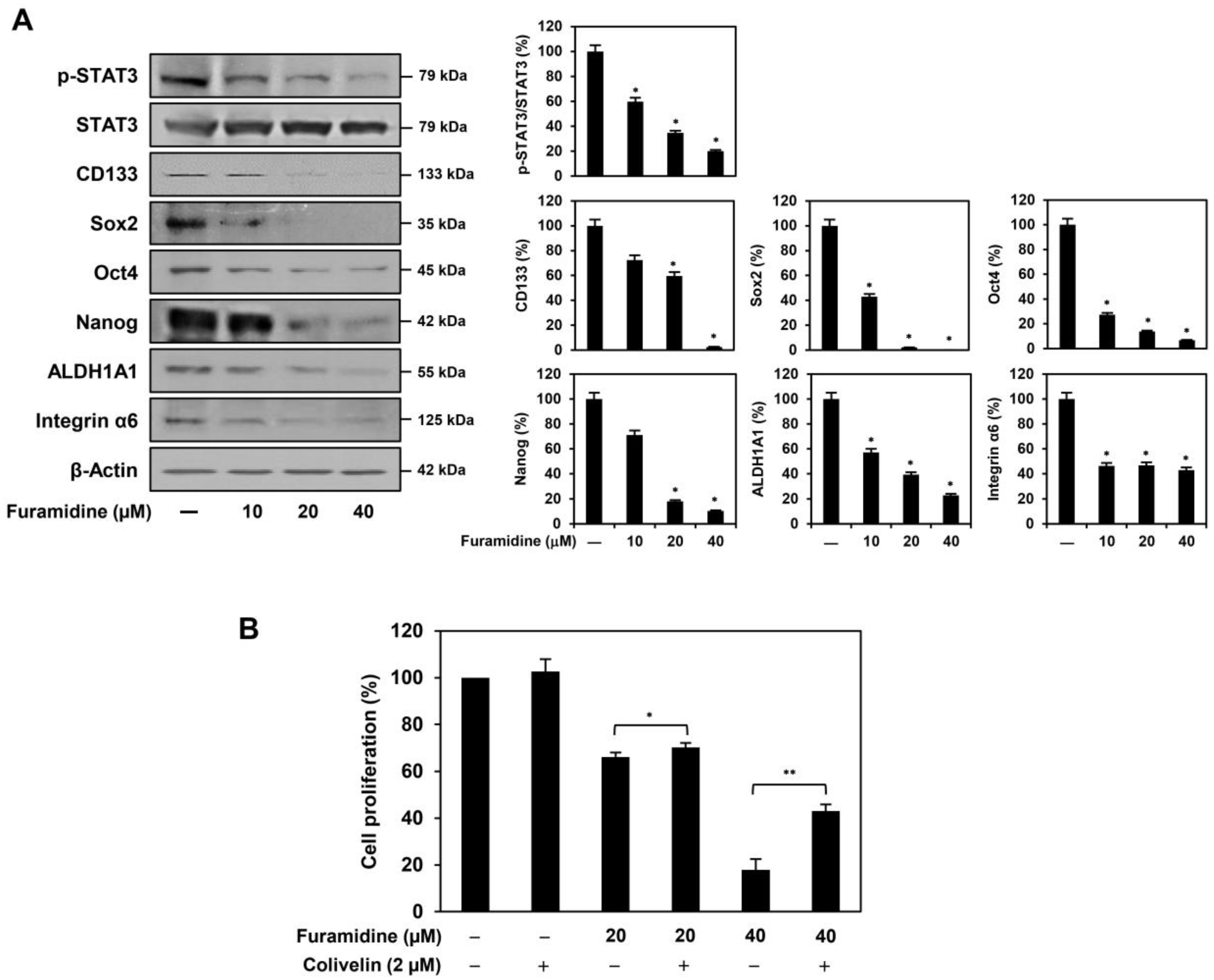
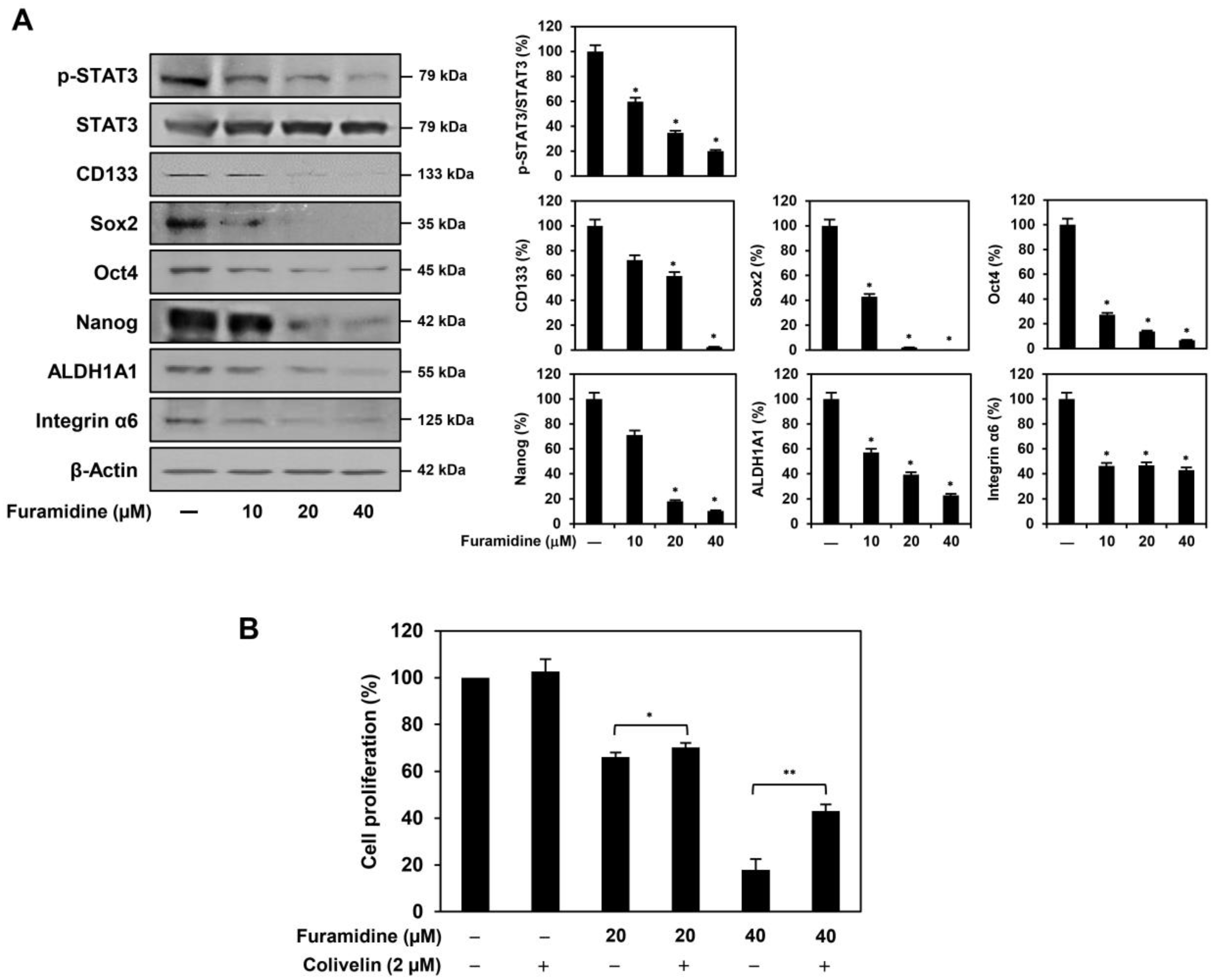
2.3. Furamidine Inhibits STAT3 Activation and Stemness Marker Expression in U87MG-Derived GSCs
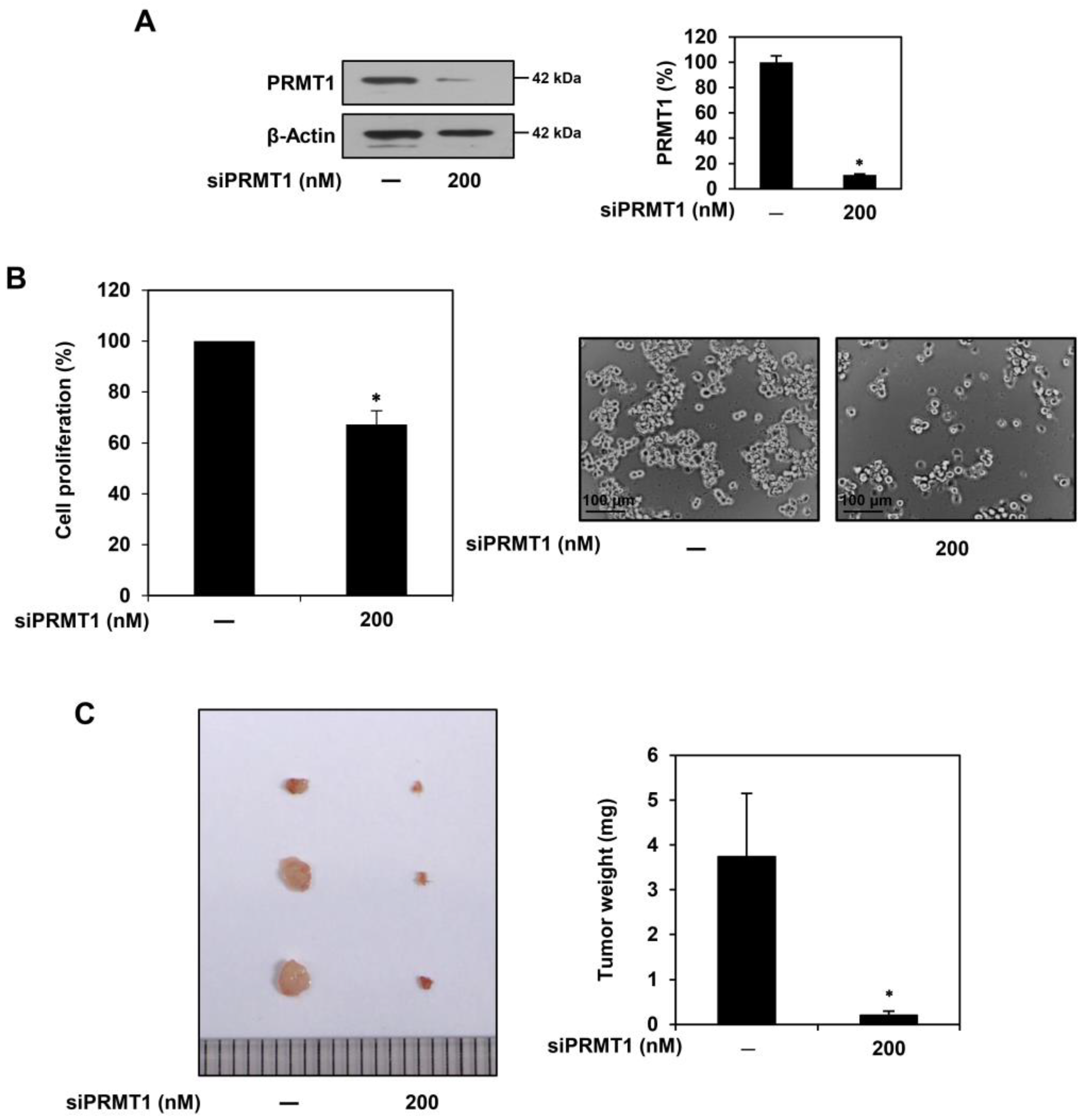
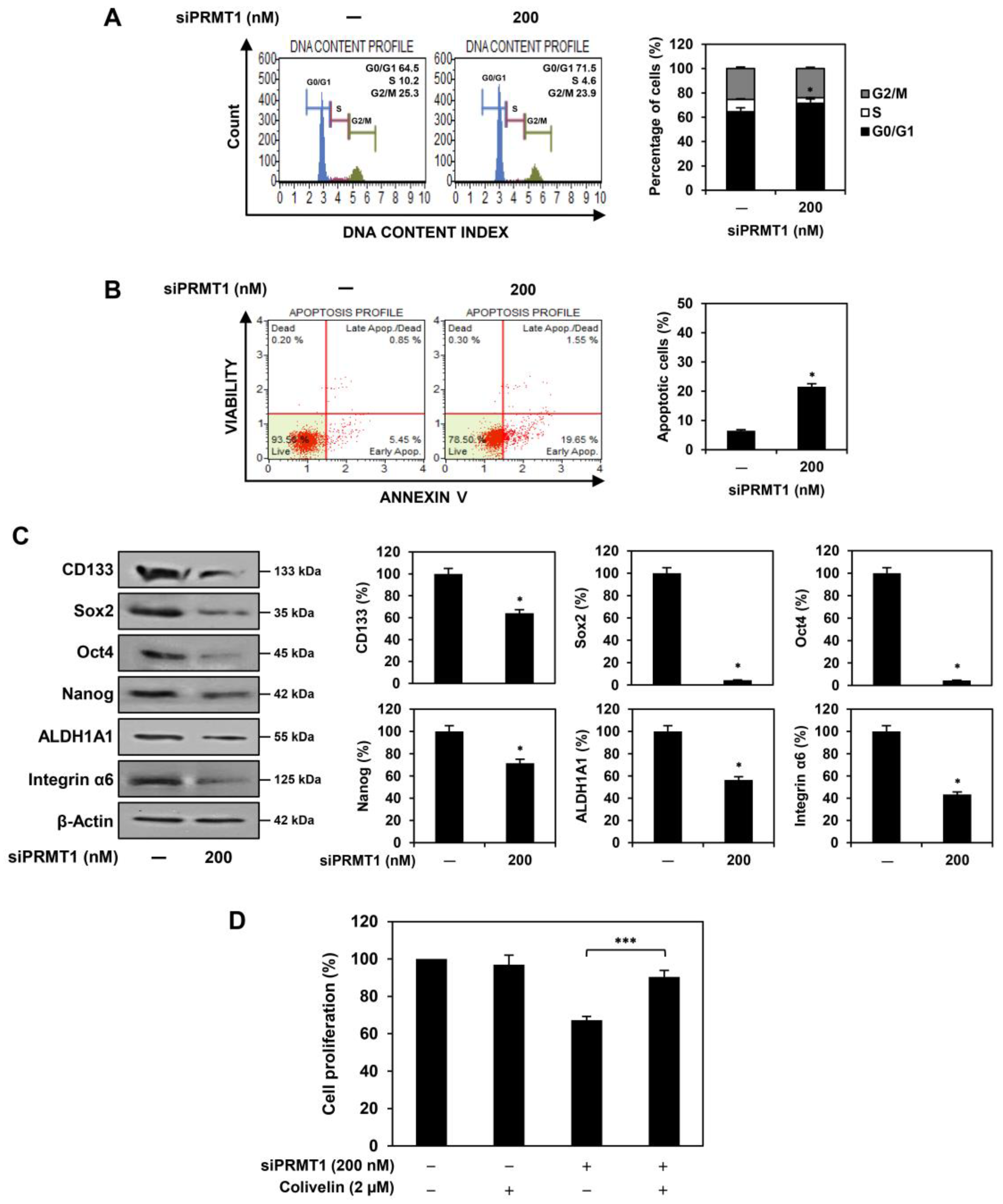
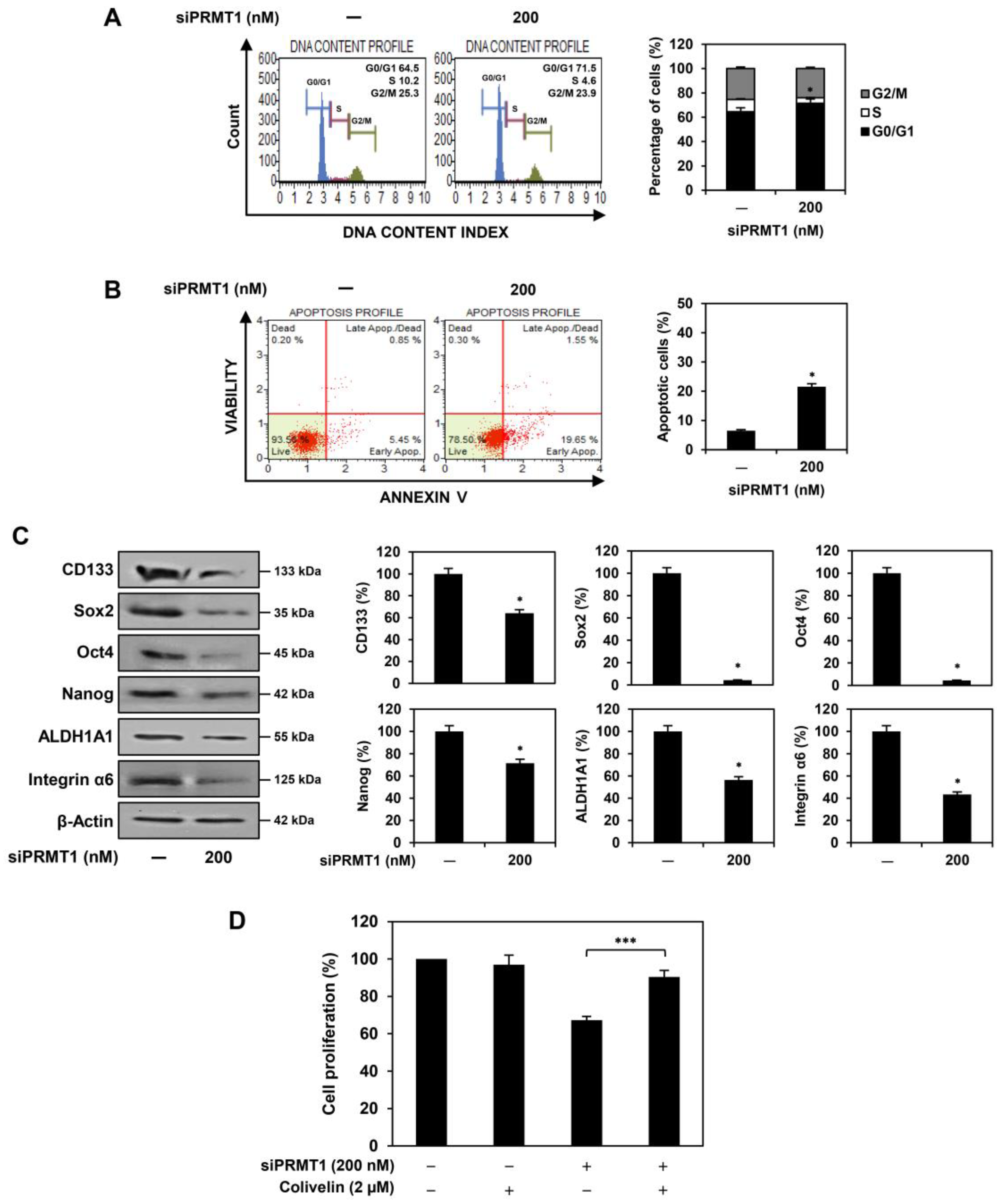
2.4. PRMT1 Knockdown Suppresses Proliferation of U87MG-Derived GSCs Both In Vitro and In Vivo
2.5. PRMT1 Knockdown Downregulates GBM Stemness Regulators in U87MG-Derived GSCs
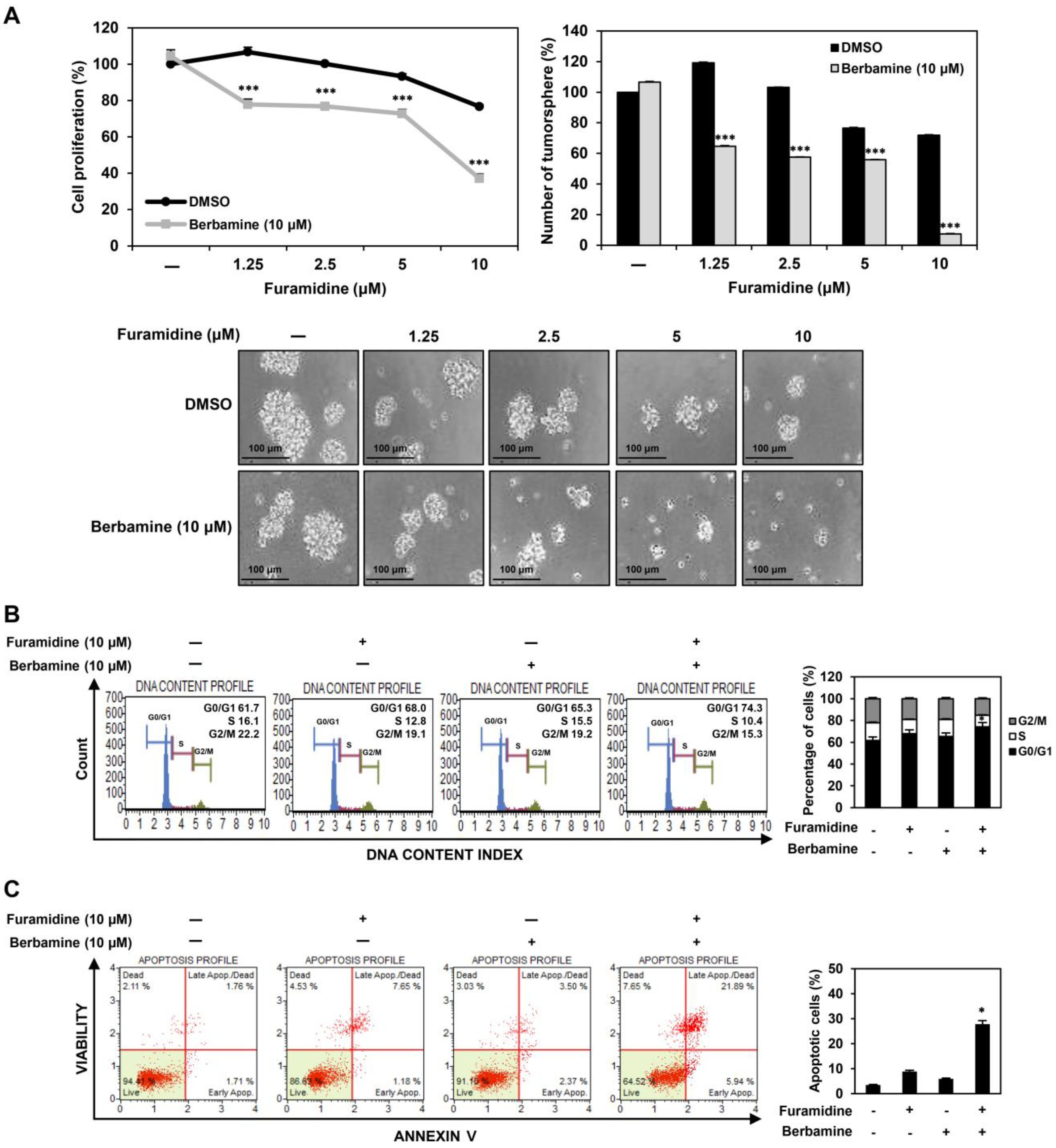
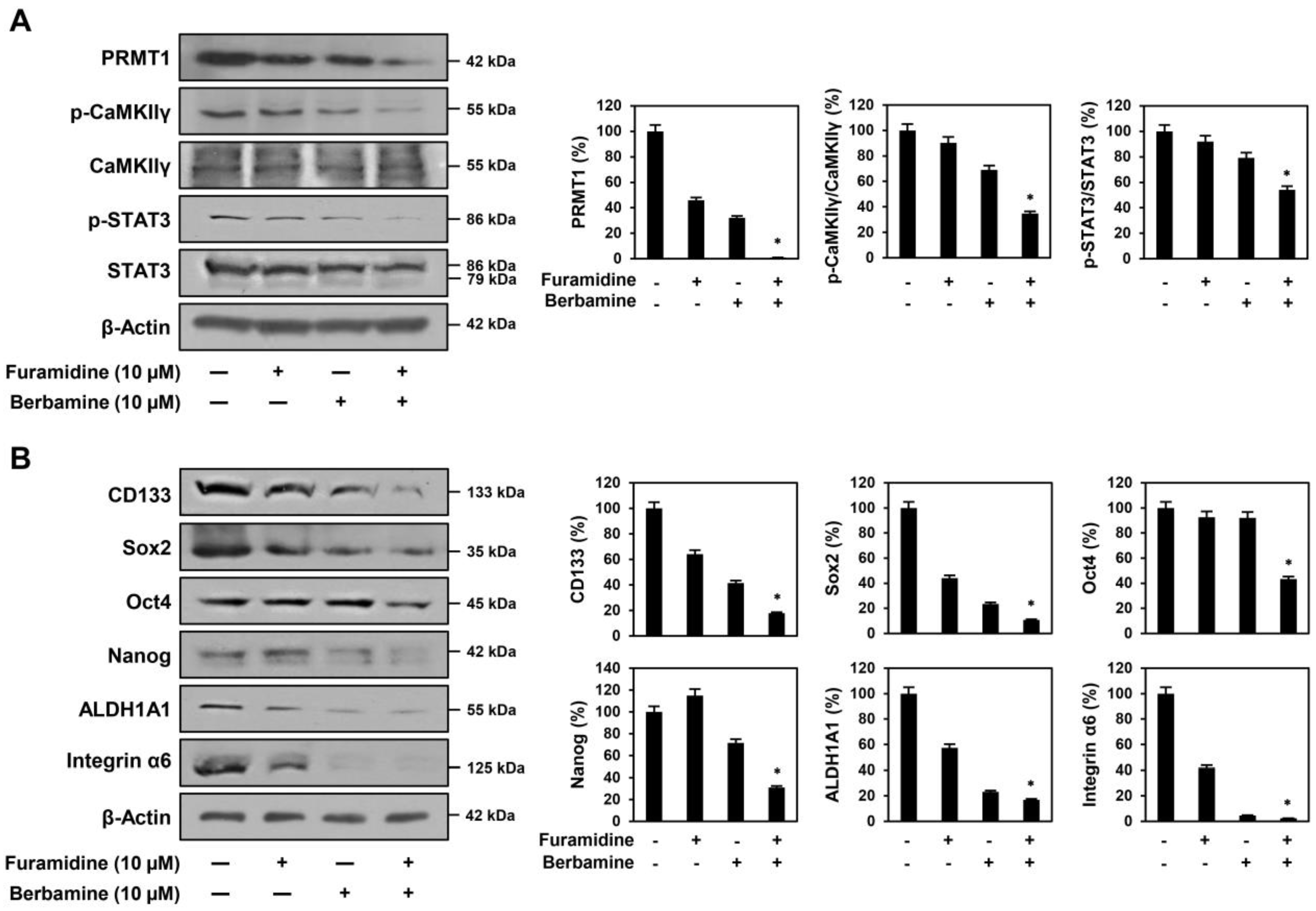
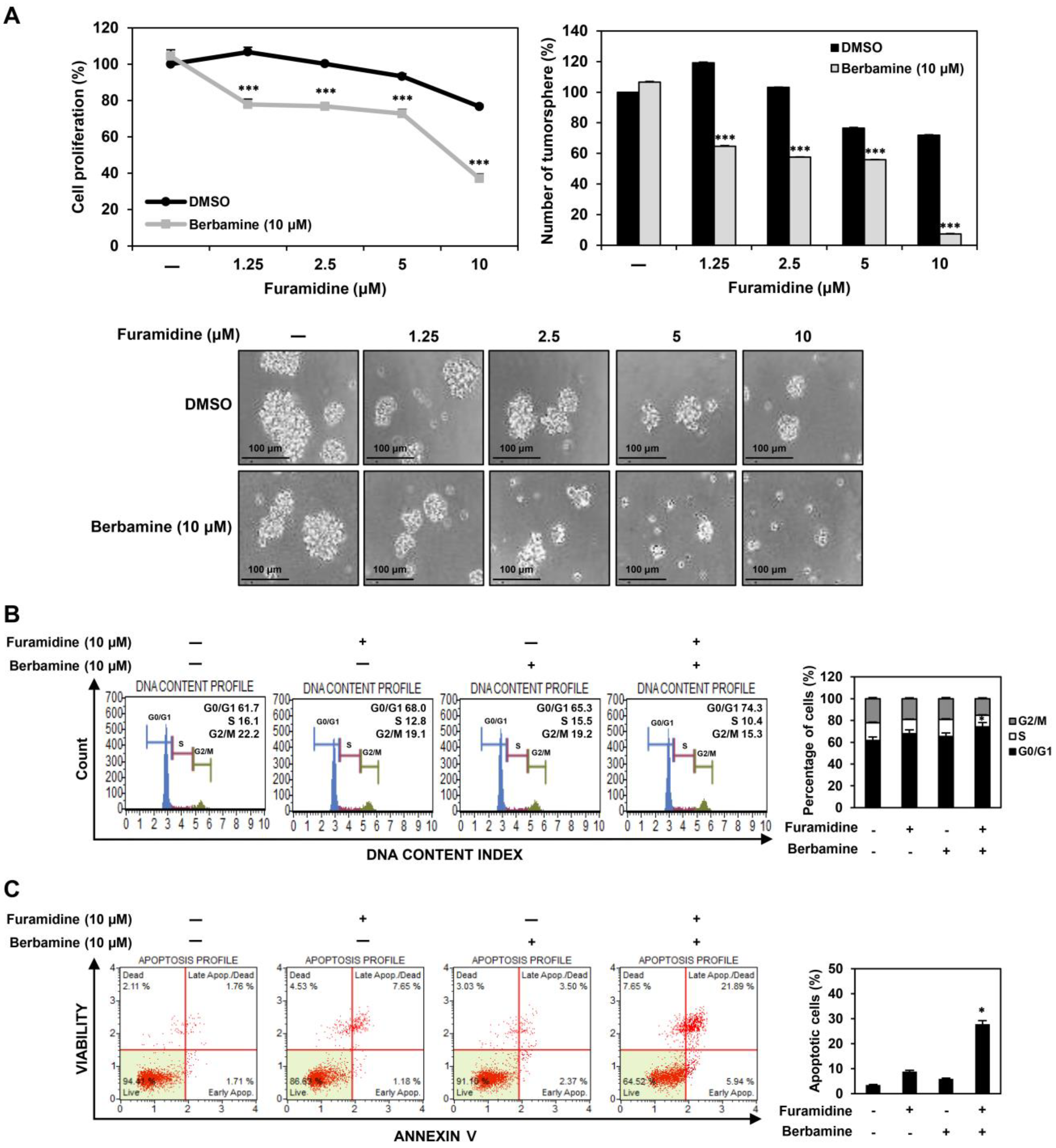
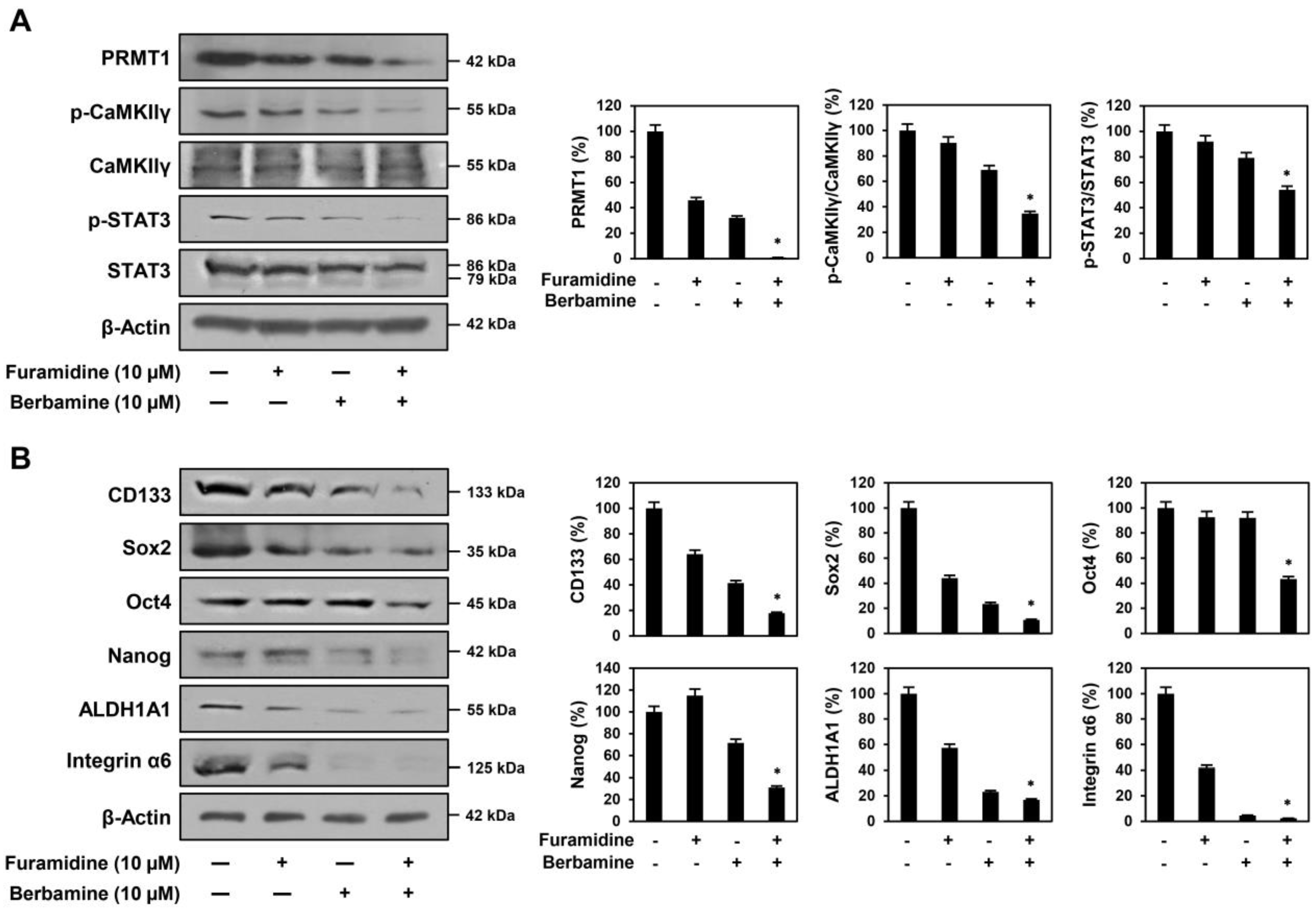
2.6. Combined Treatment with Furamidine and Berbamine Increases Lethality of U87MG-Derived GSCs
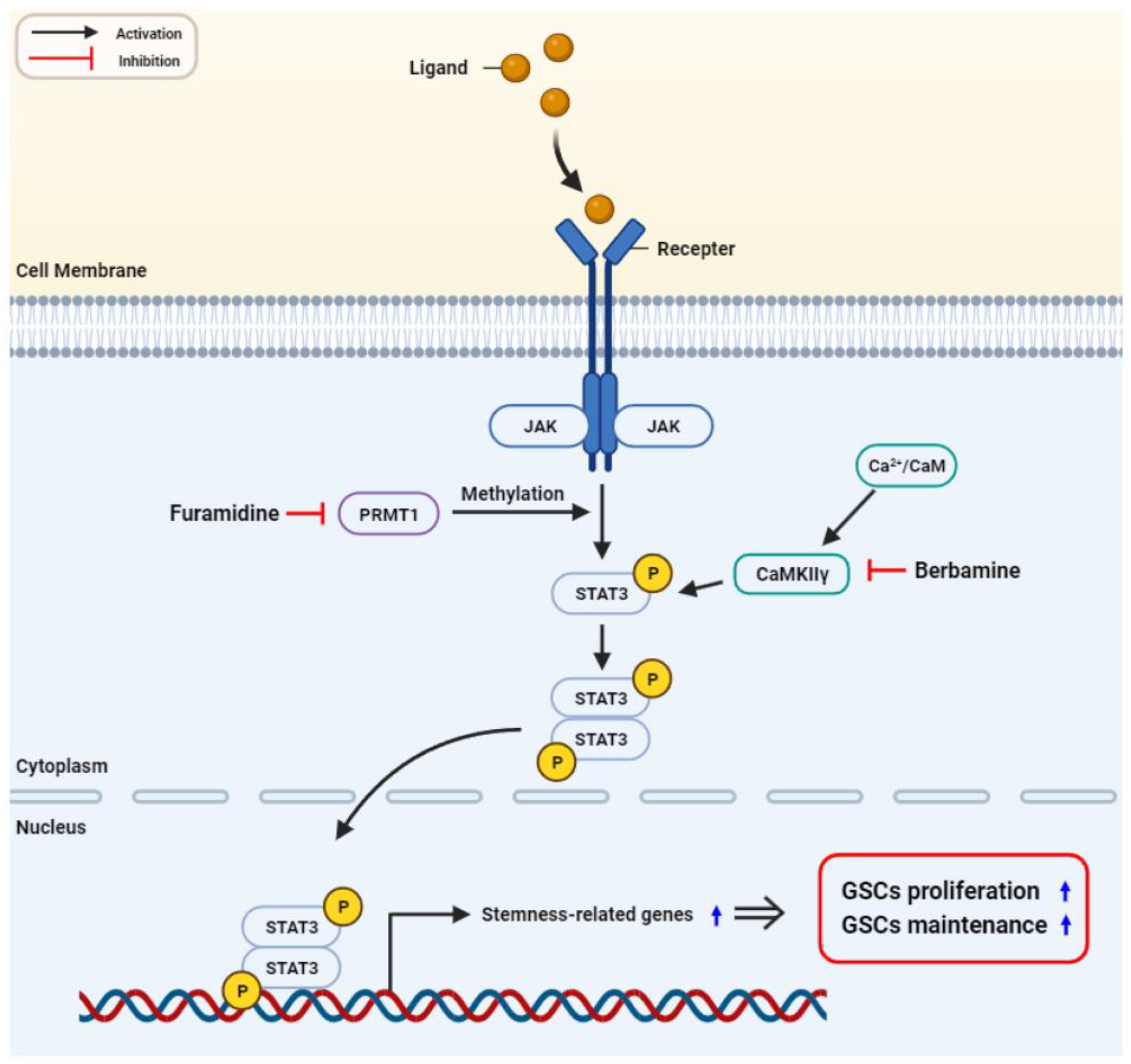
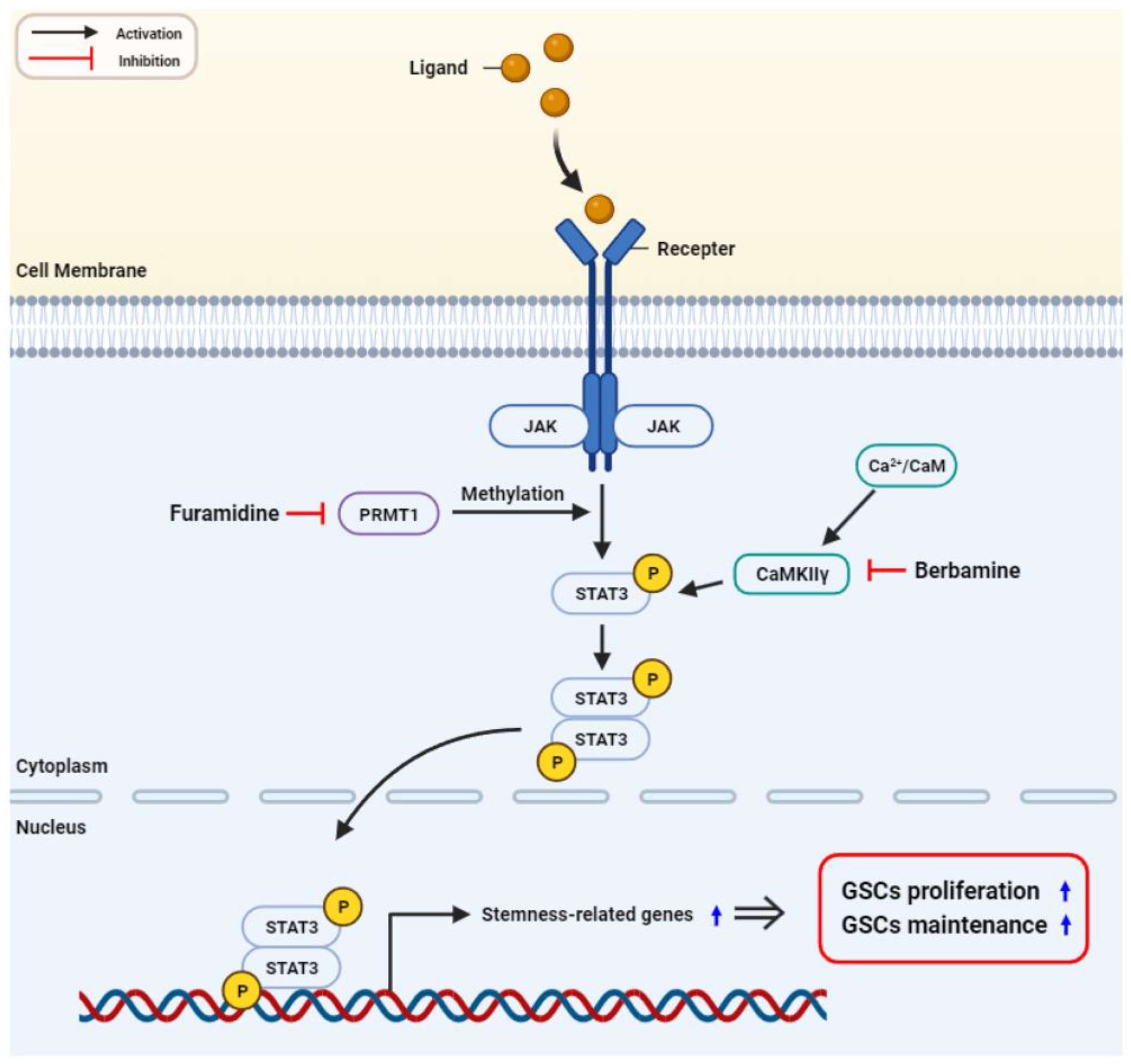
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Tumorsphere Formation Assay
4.5. CAM Assay
4.6. Cell Cycle Analysis
4.7. Apoptosis Analysis
4.8. Nuclear Fluorescent Staining with DAPI
4.9. Measurement of MMP
4.10. Measurement of Intracellular ROS
4.11. PRMT1-Directed RNA Interference
4.12. Western Blot
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Farah, P.; Ondracek, A.; Chen, Y.; Wolinsky, Y.; Stroup, N.E.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013, 15, ii1–ii56. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef]
- Lutterbach, J.; Guttenberger, R.; Pagenstecher, A. Gliosarcoma: A clinical study. Radiother. Oncol. 2001, 61, 57–64. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy; Exon Publications: Brisbane, Australia, 2017; pp. 197–241. [Google Scholar]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Soltysova, A.; Altanerova, V.; Altaner, C. Cancer stem cells. Neoplasma 2005, 52, 435–440. [Google Scholar] [PubMed]
- Finocchiaro, G. TLRgeting evasion of immune pathways in glioblastoma. Cell Stem Cell 2017, 20, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.L.V.; Gomes, I.N.F.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Kalkan, R. Glioblastoma stem cells as a new therapeutic target for glioblastoma. Clin. Med. Insights Oncol. 2015, 9, 95–103. [Google Scholar] [CrossRef]
- Eram, M.S.; Shen, Y.; Szewczyk, M.; Wu, H.; Senisterra, G.; Li, F.; Butler, K.V.; Kaniskan, H.Ü.; Speed, B.A.; Dela Seña, C.; et al. A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. [Google Scholar] [CrossRef]
- Bryant, J.; Heiss, J.; Banasavadi-Siddegowda, Y.K. Arginine methylation in brain tumors: Tumor biology and therapeutic strategies. Cells 2021, 10, 124. [Google Scholar] [CrossRef]
- Tang, J.; Frankel, A.; Cook, R.J.; Kim, S.; Paik, W.K.; Williams, K.R.; Clarke, S.; Herschman, H.R. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000, 275, 7723–7730. [Google Scholar] [CrossRef]
- Thiebaut, C.; Eve, L.; Poulard, C.; Le Romancer, M. Structure, activity, and function of PRMT1. Life 2021, 11, 1147. [Google Scholar] [CrossRef]
- Avasarala, S.; Van Scoyk, M.; Karuppusamy Rathinam, M.K.; Zerayesus, S.; Zhao, X.; Zhang, W.; Pergande, M.R.; Borgia, J.A.; DeGregori, J.; Port, J.D.; et al. PRMT1 is a novel regulator of epithelial-mesenchymal-transition in non-small cell lung cancer. J. Biol. Chem. 2015, 290, 13479–13489. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Takeichi, K.; Murata, K.; Kozakai, A.; Yagi, A.; Ishikawa, K.; Suzuki-Nakagawa, C.; Kasuya, Y.; Fukamizu, A.; Nakagawa, T. Regulation of neural stem cell proliferation and survival by protein arginine methyltransferase 1. Front. Neurosci. 2022, 16, 948517. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, M.; Fang, L.; Yang, X.; Cao, N.; Xu, L.; Shi, L.; Cao, Y. Coordinated regulation of the ribosome and proteasome by PRMT1 in the maintenance of neural stemness in cancer cells and neural stem cells. J. Biol. Chem. 2021, 297, 101275. [Google Scholar] [CrossRef]
- Kim, B.; Jung, N.; Lee, S.; Sohng, J.K.; Jung, H.J. Apigenin inhibits cancer stem cell-like phenotypes in human glioblastoma cells via suppression of c-Met signaling. Phytother. Res. 2016, 30, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Kwon, H.J.; Jung, H.J. Downregulation of mitochondrial UQCRB inhibits cancer stem cell-like properties in glioblastoma. Int. J. Oncol. 2018, 52, 241–251. [Google Scholar] [CrossRef]
- He, Y.; Luo, M.; Lei, S.; Zeng, Z.; Chen, T.; Wu, Y.; Wang, D.; Wang, L.; Wang, L. Luteoloside Induces G0/G1 phase arrest of neuroblastoma cells by targeting p38 MAPK. Molecules 2023, 28, 1748. [Google Scholar] [CrossRef]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular pathways: Targeting the Cyclin D-CDK4/6 axis for cancer treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and tumorigenesis: Molecular mechanisms and therapeutic strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal. 2010, 8, 31. [Google Scholar] [CrossRef]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef]
- Xu, H.S.; Qin, X.L.; Zong, H.L.; He, X.G.; Cao, L. Cancer stem cell markers in glioblastoma—An update. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3207–3211. [Google Scholar]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Préat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef]
- Shin, H.J.; Lee, S.; Jung, H.J. A curcumin derivative hydrazinobenzoylcurcumin suppresses stem-like features of glioblastoma cells by targeting Ca2+/calmodulin-dependent protein kinase II. J. Cell. Biochem. 2019, 120, 6741–6752. [Google Scholar] [CrossRef]
- Han, J.M.; Kim, Y.J.; Jung, H.J. Discovery of a new CaMKII-targeted synthetic lethal therapy against glioblastoma stem-like cells. Cancers 2022, 14, 1315. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jung, H.J. Synergistic anticancer effect of a combination of berbamine and arcyriaflavin A against glioblastoma stem-like cells. Molecules 2022, 27, 7968. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 2018, 23, 12. [Google Scholar] [CrossRef] [PubMed]
- Stitzlein, L.M.; Adams, J.T.; Stitzlein, E.N.; Dudley, R.W.; Chandra, J. Current and future therapeutic strategies for high-grade gliomas leveraging the interplay between epigenetic regulators and kinase signaling networks. J. Exp. Clin. Cancer Res. 2024, 43, 12. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Patel, M.; Ruzevick, J.; Jackson, C.M.; Lim, M. STAT3 activation in glioblastoma: Biochemical and therapeutic implications. Cancers 2014, 6, 376–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, Y.; Zhong, C.; Ma, N.; Zhang, E.; Zhang, F.; Li, J.; Deng, Y.; Wang, K.; Xie, D.; et al. PRMT1 promoted HCC growth and metastasis in vitro and in vivo via activating the STAT3 signalling pathway. Cell. Physiol. Biochem. 2018, 47, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Honda, M.; Nakashima, K.; Katada, S. PRMT1 regulates astrocytic differentiation of embryonic neural stem/precursor cells. J. Neurochem. 2017, 142, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, T.; Meng, Z.; Gan, Y.; Xu, X.; Lou, G.; Li, H.; Gan, X.; Zhou, H.; Tang, J.; et al. CaMKII γ, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood 2012, 120, 4829–4839. [Google Scholar] [CrossRef]
- Mamaeva, O.A.; Kim, J.; Feng, G.; McDonald, J.M. Calcium/calmodulin-dependent kinase II regulates Notch-1 signaling in prostate cancer cells. J. Cell. Biochem. 2009, 106, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Collins, S.J. Activated Ca2+/calmodulin-dependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res. 2008, 68, 3733–3742. [Google Scholar] [CrossRef]
- Chai, S.; Xu, X.; Wang, Y.; Zhou, Y.; Zhang, C.; Yang, Y.; Yang, Y.; Xu, H.; Xu, R.; Wang, K. Ca2+/calmodulin-dependent protein kinase IIγ enhances stem-like traits and tumorigenicity of lung cancer cells. Oncotarget 2015, 6, 16069–16083. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Li, T.; Ma, X.; Wang, X.; Van Ness, C.; Gan, Y.; Zhou, H.; Tang, J.; Lou, G.; Wang, Y.; et al. Berbamine inhibits the growth of liver cancer cells and cancer-initiating cells by targeting Ca2+/calmodulin-dependent protein kinase II. Mol. Cancer Ther. 2013, 12, 2067–2077. [Google Scholar] [CrossRef]
- Cho, H.J.; Jung, H.J. Cyclophilin A inhibitors suppress proliferation and induce apoptosis of MKN45 gastric cancer stem-like cells by regulating CypA/CD147-mediated signaling pathway. Int. J. Mol. Sci. 2023, 24, 4734. [Google Scholar] [CrossRef]
- Ribatti, D. The chick embryo chorioallantoic membrane (CAM) assay. Reprod. Toxicol. 2017, 70, 97–101. [Google Scholar] [CrossRef]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring mitochondrial transmembrane potential by TMRE staining. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087361. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuk, N.; Jung, H.J. Inhibition of PRMT1 Suppresses the Growth of U87MG-Derived Glioblastoma Stem Cells by Blocking the STAT3 Signaling Pathway. Int. J. Mol. Sci. 2024, 25, 2950. https://doi.org/10.3390/ijms25052950
Yuk N, Jung HJ. Inhibition of PRMT1 Suppresses the Growth of U87MG-Derived Glioblastoma Stem Cells by Blocking the STAT3 Signaling Pathway. International Journal of Molecular Sciences. 2024; 25(5):2950. https://doi.org/10.3390/ijms25052950
Chicago/Turabian StyleYuk, Nayeong, and Hye Jin Jung. 2024. "Inhibition of PRMT1 Suppresses the Growth of U87MG-Derived Glioblastoma Stem Cells by Blocking the STAT3 Signaling Pathway" International Journal of Molecular Sciences 25, no. 5: 2950. https://doi.org/10.3390/ijms25052950
APA StyleYuk, N., & Jung, H. J. (2024). Inhibition of PRMT1 Suppresses the Growth of U87MG-Derived Glioblastoma Stem Cells by Blocking the STAT3 Signaling Pathway. International Journal of Molecular Sciences, 25(5), 2950. https://doi.org/10.3390/ijms25052950