
The Role of Glutathione in the Management of Cell-Mediated Immune Responses in Individuals with HIV
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
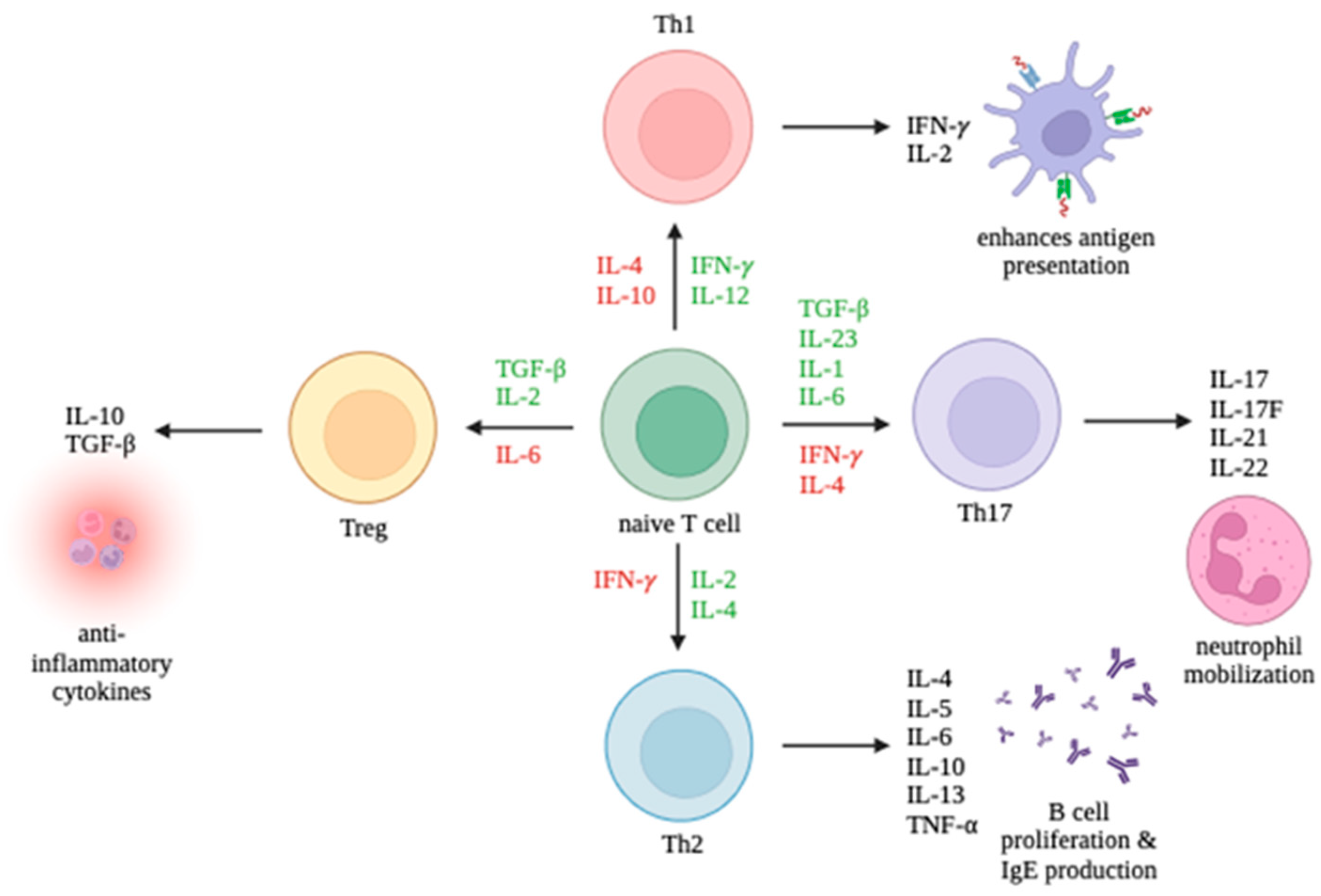
3. Overview of Cell-Mediated Immunity
4. HIV’s Effect on Inflammation
5. Glutathione: Inflammation and Immune Response
6. Glutathione: Mechanisms
7. Glutathione: Treating HIV- and AIDS-Related Diseases
7.1. Effects of Glutathione on Metabolic Health and Overall Survival in Patients with HIV
7.2. Effects of Glutathione on HIV-Associated Neurodegenerative Diseases
7.3. Effects of Glutathione on HIV and Th17 Depletion in GI Tract
7.4. Effects of Glutathione on HIV and Tuberculosis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kazer, S.W.; Walker, B.D.; Shalek, A.K. Evolution and Diversity of Immune Responses during Acute HIV Infection. Immunity 2020, 53, 908–924. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Chomont, N.; Douek, D.C.; Deeks, S.G. Immune Activation and HIV Persistence: Implications for Curative Approaches to HIV Infection. Immunol. Rev. 2013, 254, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Ipp, H.; Zemlin, A. The Paradox of the Immune Response in HIV Infection: When Inflammation Becomes Harmful. Clin. Chim. Acta 2013, 416, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, A.; Munshi, M.; Khan, S.Z.; Fatima, S.; Arya, R.; Jameel, S.; Singh, A. Measuring Glutathione Redox Potential of HIV-1-Infected Macrophages. J. Biol. Chem. 2015, 290, 1020–1038. [Google Scholar] [CrossRef]
- Jariwalla, R.J.; Lalezari, J.; Cenko, D.; Mansour, S.E.; Kumar, A.; Gangapurkar, B.; Nakamura, D. Restoration of Blood Total Glutathione Status and Lymphocyte Function Following Alpha-Lipoic Acid Supplementation in Patients with HIV Infection. J. Altern. Complement. Med. 2008, 14, 139–146. [Google Scholar] [CrossRef]
- Morris, D.; Gonzalez, B.; Khurasany, M.; Kassissa, C.; Luong, J.; Kasko, S.; Pandya, S.; Chu, M.; Chi, P.-T.; Bui, S.; et al. Characterization of Dendritic Cell and Regulatory T Cell Functions against Mycobacterium Tuberculosis Infection. Biomed. Res. Int. 2013, 2013, 402827. [Google Scholar] [CrossRef]
- Abnousian, A.; Vasquez, J.; Sasaninia, K.; Kelley, M.; Venketaraman, V. Glutathione Modulates Efficacious Changes in the Immune Response against Tuberculosis. Biomedicines 2023, 11, 1340. [Google Scholar] [CrossRef]
- Ly, J.; Lagman, M.; Saing, T.; Singh, M.K.; Tudela, E.V.; Morris, D.; Anderson, J.; Daliva, J.; Ochoa, C.; Patel, N.; et al. Liposomal Glutathione Supplementation Restores TH1 Cytokine Response to Mycobacterium Tuberculosis Infection in HIV-Infected Individuals. J. Interferon Cytokine Res. 2015, 35, 875–887. [Google Scholar] [CrossRef]
- Le Hingrat, Q.; Sereti, I.; Landay, A.L.; Pandrea, I.; Apetrei, C. The Hitchhiker Guide to CD4+ T-Cell Depletion in Lentiviral Infection. A Critical Review of the Dynamics of the CD4+ T Cells in SIV and HIV Infection. Front. Immunol. 2021, 12, 695674. [Google Scholar] [CrossRef]
- Okoye, A.A.; Picker, L.J. CD4(+) T-Cell Depletion in HIV Infection: Mechanisms of Immunological Failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef]
- Lv, T.; Cao, W.; Li, T. HIV-Related Immune Activation and Inflammation: Current Understanding and Strategies. J. Immunol. Res. 2021, 2021, 7316456. [Google Scholar] [CrossRef]
- Gorenec, L.; Zidovec Lepej, S.; Grgic, I.; Planinic, A.; Iscic Bes, J.; Vince, A.; Begovac, J. The Comparison of Th1, Th2, Th9, Th17 and Th22 Cytokine Profiles in Acute and Chronic HIV-1 Infection. Microb. Pathog. 2016, 97, 125–130. [Google Scholar] [CrossRef]
- Keating, S.M.; Jacobs, E.S.; Norris, P.J. Soluble Mediators of Inflammation in HIV and Their Implications for Therapeutics and Vaccine Development. Cytokine Growth Factor Rev. 2012, 23, 193–206. [Google Scholar] [CrossRef]
- Saing, T.; Valdivia, A.; Hussain, P.; Ly, J.; Gonzalez, L.; Guilford, F.T.; Pearce, D.; Venketaraman, V. Data on Pro-Inflammatory Cytokines IL-1β, IL-17, and IL-6 in the Peripheral Blood of HIV-Infected Individuals. Data Brief 2016, 8, 1044–1047. [Google Scholar] [CrossRef][Green Version]
- Klein, S.A.; Dobmeyer, J.M.; Dobmeyer, T.S.; Pape, M.; Ottmann, O.G.; Helm, E.B.; Hoelzer, D.; Rossol, R. Demonstration of the Th1 to Th2 Cytokine Shift during the Course of HIV-1 Infection Using Cytoplasmic Cytokine Detection on Single Cell Level by Flow Cytometry. AIDS 1997, 11, 1111–1118. [Google Scholar] [CrossRef]
- Theron, A.J.; Anderson, R.; Rossouw, T.M.; Steel, H.C. The Role of Transforming Growth Factor Beta-1 in the Progression of HIV/AIDS and Development of Non-AIDS-Defining Fibrotic Disorders. Front. Immunol. 2017, 8, 1461. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, A.; Ly, J.; Gonzalez, L.; Hussain, P.; Saing, T.; Islamoglu, H.; Pearce, D.; Ochoa, C.; Venketaraman, V. Restoring Cytokine Balance in HIV-Positive Individuals with Low CD4 T Cell Counts. AIDS Res. Hum. Retroviruses 2017, 33, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Pizzorno, J. Glutathione! Integr. Med. 2014, 13, 8–12. [Google Scholar]
- Chen, Y.; Yang, Y.; Miller, M.L.; Shen, D.; Shertzer, H.G.; Stringer, K.F.; Wang, B.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Hepatocyte-Specific Gclc Deletion Leads to Rapid Onset of Steatosis with Mitochondrial Injury and Liver Failure. Hepatology 2007, 45, 1118–1128. [Google Scholar] [CrossRef]
- Loguercio, C.; Di Pierro, M. The Role of Glutathione in the Gastrointestinal Tract: A Review. Ital. J. Gastroenterol. Hepatol. 1999, 31, 401–407. [Google Scholar]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, Function, and Post-Translational Regulation of the Catalytic and Modifier Subunits of Glutamate Cysteine Ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef]
- Tosic, M.; Ott, J.; Barral, S.; Bovet, P.; Deppen, P.; Gheorghita, F.; Matthey, M.-L.; Parnas, J.; Preisig, M.; Saraga, M.; et al. Schizophrenia and Oxidative Stress: Glutamate Cysteine Ligase Modifier as a Susceptibility Gene. Am. J. Hum. Genet. 2006, 79, 586–592. [Google Scholar] [CrossRef]
- Baum, L.; Chen, X.; Cheung, W.S.; Cheung, C.K.A.; Cheung, L.W.; Chiu, K.F.P.; Wen, H.M.; Poon, P.; Woo, K.S.; Ng, H.K.; et al. Polymorphisms and Vascular Cognitive Impairment after Ischemic Stroke. J. Geriatr. Psychiatry Neurol. 2007, 20, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Shephard, C.; Janer, M.; Graham, J.; McNeney, B.; Shin, J.; Zarghami, M.; Griffith, W.; Farin, F.; Kavanagh, T.J.; et al. Glutamate Cysteine Ligase Catalytic Subunit Promoter Polymorphisms and Associations with Type 1 Diabetes Age-at-Onset and GAD65 Autoantibody Levels. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2007, 115, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Polonikov, A.V.; Ivanov, V.P.; Solodilova, M.A.; Khoroshaya, I.V.; Kozhuhov, M.A.; Panfilov, V.I. The Relationship between Polymorphisms in the Glutamate Cysteine Ligase Gene and Asthma Susceptibility. Respir. Med. 2007, 101, 2422–2424. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakamura, S.; Kugiyama, K.; Sugiyama, S.; Miyamoto, S.; Koide, S.; Fukushima, H.; Honda, O.; Yoshimura, M.; Ogawa, H. Polymorphism in the 5′-Flanking Region of Human Glutamate-Cysteine Ligase Modifier Subunit Gene Is Associated with Myocardial Infarction. Circulation 2002, 105, 2968–2973. [Google Scholar] [CrossRef]
- Nakamura, S.; Sugiyama, S.; Fujioka, D.; Kawabata, K.; Ogawa, H.; Kugiyama, K. Polymorphism in Glutamate-Cysteine Ligase Modifier Subunit Gene Is Associated with Impairment of Nitric Oxide-Mediated Coronary Vasomotor Function. Circulation 2003, 108, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Carretero, J.; Obrador, E.; Anasagasti, M.J.; Martin, J.J.; Vidal-Vanaclocha, F.; Estrela, J.M. Growth-Associated Changes in Glutathione Content Correlate with Liver Metastatic Activity of B16 Melanoma Cells. Clin. Exp. Metastasis 1999, 17, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Forman, H.J. Redox Regulation of γ-Glutamyl Transpeptidase. Am. J. Respir. Cell Mol. Biol. 2009, 41, 509–515. [Google Scholar] [CrossRef]
- Brundu, S.; Palma, L.; Picceri, G.G.; Ligi, D.; Orlandi, C.; Galluzzi, L.; Chiarantini, L.; Casabianca, A.; Schiavano, G.F.; Santi, M.; et al. Glutathione Depletion Is Linked with Th2 Polarization in Mice with a Retrovirus-Induced Immunodeficiency Syndrome, Murine AIDS: Role of Proglutathione Molecules as Immunotherapeutics. J. Virol. 2016, 90, 7118–7130. [Google Scholar] [CrossRef] [PubMed]
- Fraternale, A.; Zara, C.; De Angelis, M.; Nencioni, L.; Palamara, A.T.; Retini, M.; Di Mambro, T.; Magnani, M.; Crinelli, R. Intracellular Redox-Modulated Pathways as Targets for Effective Approaches in the Treatment of Viral Infection. Int. J. Mol. Sci. 2021, 22, 3603. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.; Khurasany, M.; Nguyen, T.; Kim, J.; Guilford, F.; Mehta, R.; Gray, D.; Saviola, B.; Venketaraman, V. Glutathione and Infection. Biochim. Biophys. Acta 2013, 1830, 3329–3349. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.; Guerra, C.; Donohue, C.; Oh, H.; Khurasany, M.; Venketaraman, V. Unveiling the Mechanisms for Decreased Glutathione in Individuals with HIV Infection. Clin. Dev. Immunol. 2012, 2012, 734125. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The Glutathione Peroxidase Gpx4 Prevents Lipid Peroxidation and Ferroptosis to Sustain Treg Cell Activation and Suppression of Antitumor Immunity. Cell Rep. 2021, 35, 109235. [Google Scholar] [CrossRef]
- Fox, E.S.; Brower, J.S.; Bellezzo, J.M.; Leingang, K.A. N-Acetylcysteine and Alpha-Tocopherol Reverse the Inflammatory Response in Activated Rat Kupffer Cells. J. Immunol. 1997, 158, 5418–5423. [Google Scholar] [CrossRef]
- Pena, L.R.; Hill, D.B.; McClain, C.J. Treatment with Glutathione Precursor Decreases Cytokine Activity. JPEN J. Parenter. Enter. Nutr. 1999, 23, 1–6. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, K.; Wang, D.; Yue, X.; Song, D.; Zhu, Y.; Wu, J. Nucleocapsid Protein of SARS-CoV Activates Interleukin-6 Expression through Cellular Transcription Factor NF-κB. Virology 2007, 365, 324–335. [Google Scholar] [CrossRef]
- Silvagno, F.; Vernone, A.; Pescarmona, G.P. The Role of Glutathione in Protecting against the Severe Inflammatory Response Triggered by COVID-19. Antioxidants 2020, 9, 624. [Google Scholar] [CrossRef]
- Basi, Z.; Turkoglu, V. In Vitro Effect of Oxidized and Reduced Glutathione Peptides on Angiotensin Converting Enzyme Purified from Human Plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2019, 1104, 190–195. [Google Scholar] [CrossRef]
- Venketaraman, V.; Rodgers, T.; Linares, R.; Reilly, N.; Swaminathan, S.; Hom, D.; Millman, A.C.; Wallis, R.; Connell, N.D. Glutathione and Growth Inhibition of Mycobacterium Tuberculosis in Healthy and HIV Infected Subjects. AIDS Res. Ther. 2006, 3, 5. [Google Scholar] [CrossRef]
- Venketaraman, V.; Dayaram, Y.K.; Amin, A.G.; Ngo, R.; Green, R.M.; Talaue, M.T.; Mann, J.; Connell, N.D. Role of Glutathione in Macrophage Control of Mycobacteria. Infect. Immun. 2003, 71, 1864–1871. [Google Scholar] [CrossRef]
- Franco, R.; Cidlowski, J.A. Apoptosis and Glutathione: Beyond an Antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef]
- Coco-Bassey, S.B.; Asemota, E.A.; Okoroiwu, H.U.; Etura, J.E.; Efiong, E.E.; Inyang, I.J.; Uko, E.K. Glutathione, Glutathione Peroxidase and Some Hematological Parameters of HIV-Seropositive Subjects Attending Clinic in University of Calabar Teaching Hospital, Calabar, Nigeria. BMC Infect. Dis. 2019, 19, 944. [Google Scholar] [CrossRef]
- Herzenberg, L.A.; De Rosa, S.C.; Dubs, J.G.; Roederer, M.; Anderson, M.T.; Ela, S.W.; Deresinski, S.C.; Herzenberg, L.A. Glutathione Deficiency Is Associated with Impaired Survival in HIV Disease. Proc. Natl. Acad. Sci. USA 1997, 94, 1967–1972. [Google Scholar] [CrossRef]
- Ande, A.; Sinha, N.; Rao, P.S.S.; McArthur, C.P.; Ayuk, L.; Achu, P.N.; Njinda, A.; Kumar, A.; Kumar, S. Enhanced Oxidative Stress by Alcohol Use in HIV+ Patients: Possible Involvement of Cytochrome P450 2E1 and Antioxidant Enzymes. AIDS Res. Ther. 2015, 12, 29. [Google Scholar] [CrossRef]
- Kumar, P.; Liu, C.; Suliburk, J.W.; Minard, C.G.; Muthupillai, R.; Chacko, S.; Hsu, J.W.; Jahoor, F.; Sekhar, R.V. Supplementing Glycine and N-Acetylcysteine (GlyNAC) in Aging HIV Patients Improves Oxidative Stress, Mitochondrial Dysfunction, Inflammation, Endothelial Dysfunction, Insulin Resistance, Genotoxicity, Strength, and Cognition: Results of an Open-Label Clinical Trial. Biomedicines 2020, 8, 390. [Google Scholar] [CrossRef]
- Ghafouri, M.; Amini, S.; Khalili, K.; Sawaya, B.E. HIV-1 Associated Dementia: Symptoms and Causes. Retrovirology 2006, 3, 28. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Osiecki, K.; Lopez, L.; Goldstein, H.; Calderon, T.M.; Berman, J.W. CCL2/Monocyte Chemoattractant Protein-1 Mediates Enhanced Transmigration of Human Immunodeficiency Virus (HIV)-Infected Leukocytes across the Blood–Brain Barrier: A Potential Mechanism of HIV–CNS Invasion and NeuroAIDS. J. Neurosci. 2006, 26, 1098–1106. [Google Scholar] [CrossRef]
- Lehmann, M.H.; Lehmann, J.M.; Erfle, V. Nef-Induced CCL2 Expression Contributes to HIV/SIV Brain Invasion and Neuronal Dysfunction. Front. Immunol. 2019, 10, 2447. [Google Scholar] [CrossRef]
- Echevarria-Lima, J. The Role of Glutathione in Viral Diseases of the Central Nervous System. In Glutathione in Health and Disease; 2018, ISBN 978-1-78984-275-3.
- Haughey, N.J.; Cutler, R.G.; Tamara, A.; McArthur, J.C.; Vargas, D.L.; Pardo, C.A.; Turchan, J.; Nath, A.; Mattson, M.P. Perturbation of Sphingolipid Metabolism and Ceramide Production in HIV-Dementia. Ann. Neurol. 2004, 55, 257–267. [Google Scholar] [CrossRef]
- Steiner, J.; Haughey, N.; Li, W.; Venkatesan, A.; Anderson, C.; Reid, R.; Malpica, T.; Pocernich, C.; Butterfield, D.A.; Nath, A. Oxidative Stress and Therapeutic Approaches in HIV Dementia. Antioxid. Redox Signal. 2006, 8, 2089–2100. [Google Scholar] [CrossRef]
- Price, T.O.; Uras, F.; Banks, W.A.; Ercal, N. A Novel Antioxidant N-Acetylcysteine Amide Prevents Gp120- and Tat-Induced Oxidative Stress in Brain Endothelial Cells. Exp. Neurol. 2006, 201, 193–202. [Google Scholar] [CrossRef]
- Pocernich, C.B.; La Fontaine, M.; Butterfield, D.A. In-Vivo Glutathione Elevation Protects against Hydroxyl Free Radical-Induced Protein Oxidation in Rat Brain. Neurochem. Int. 2000, 36, 185–191. [Google Scholar] [CrossRef]
- Zenobia, C.; Hajishengallis, G. Basic Biology and Role of Interleukin-17 in Immunity and Inflammation. Periodontol. 2000 2015, 69, 142–159. [Google Scholar] [CrossRef]
- Yue, F.Y.; Merchant, A.; Kovacs, C.M.; Loutfy, M.; Persad, D.; Ostrowski, M.A. Virus-Specific Interleukin-17-Producing CD4+ T Cells Are Detectable in Early Human Immunodeficiency Virus Type 1 Infection. J. Virol. 2008, 82, 6767–6771. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Paiardini, M.; Knox, K.S.; Asher, A.I.; Cervasi, B.; Asher, T.E.; Scheinberg, P.; Price, D.A.; Hage, C.A.; Kholi, L.M.; et al. Differential Th17 CD4 T-Cell Depletion in Pathogenic and Nonpathogenic Lentiviral Infections. Blood 2008, 112, 2826–2835. [Google Scholar] [CrossRef]
- Christensen-Quick, A.; Lafferty, M.; Sun, L.; Marchionni, L.; DeVico, A.; Garzino-Demo, A. Human Th17 Cells Lack HIV-Inhibitory RNases and Are Highly Permissive to Productive HIV Infection. J. Virol. 2016, 90, 7833–7847. [Google Scholar] [CrossRef] [PubMed]
- Iannello, A.; Boulassel, M.-R.; Samarani, S.; Debbeche, O.; Tremblay, C.; Toma, E.; Routy, J.-P.; Ahmad, A. Dynamics and Consequences of IL-21 Production in HIV-Infected Individuals: A Longitudinal and Cross-Sectional Study. J. Immunol. 2010, 184, 114–126. [Google Scholar] [CrossRef]
- McGary, C.S.; Alvarez, X.; Harrington, S.; Cervasi, B.; Ryan, E.S.; Iriele, R.I.; Paganini, S.; Harper, J.L.; Easley, K.; Silvestri, G.; et al. The Loss of CCR6+ and CD161+ CD4+ T-Cell Homeostasis Contributes to Disease Progression in SIV-Infected Rhesus Macaques. Mucosal Immunol. 2017, 10, 1082–1096. [Google Scholar] [CrossRef]
- Murray, L.W.; Satti, I.; Meyerowitz, J.; Jones, M.; Willberg, C.B.; Ussher, J.E.; Goedhals, D.; Hurst, J.; Phillips, R.E.; McShane, H.; et al. HIV Infection Impairs Th1 and Th17 Mycobacterium Tuberculosis-Specific T Cell Responses. J. Infect. Dis. 2018, 217, 1782–1792. [Google Scholar] [CrossRef]
- Hernández-Santos, N.; Gaffen, S.L. Th17 Cells in Immunity to Candida Albicans. Cell Host Microbe 2012, 11, 425–435. [Google Scholar] [CrossRef]
- Shacklett, B.L.; Anton, P.A. HIV Infection and Gut Mucosal Immune Function: Updates on Pathogenesis with Implications for Management and Intervention. Curr. Infect. Dis. Rep. 2010, 12, 19–27. [Google Scholar] [CrossRef]
- Bonetti, L.; Horkova, V.; Longworth, J.; Guerra, L.; Kurniawan, H.; Franchina, D.G.; Soriano-Baguet, L.; Grusdat, M.; Spath, S.; Koncina, E.; et al. A Th17 Cell-Intrinsic Glutathione/Mitochondrial-IL-22 Axis Protects against Intestinal Inflammation. bioRxiv 2023. [Google Scholar] [CrossRef]
- Müller, M.; Wandel, S.; Colebunders, R.; Attia, S.; Furrer, H.; Egger, M. IeDEA Southern and Central Africa Immune Reconstitution Inflammatory Syndrome in Patients Starting Antiretroviral Therapy for HIV Infection: A Systematic Review and Meta-Analysis. Lancet Infect. Dis. 2010, 10, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Lanzafame, M.; Vento, S. Tuberculosis-Immune Reconstitution Inflammatory Syndrome. J. Clin. Tuberc. Other Mycobact. Dis. 2016, 3, 6–9. [Google Scholar] [CrossRef]
- Guerra, C.; Morris, D.; Sipin, A.; Kung, S.; Franklin, M.; Gray, D.; Tanzil, M.; Guilford, F.; Khasawneh, F.T.; Venketaraman, V. Glutathione and Adaptive Immune Responses against Mycobacterium Tuberculosis Infection in Healthy and HIV Infected Individuals. PLoS ONE 2011, 6, e28378. [Google Scholar] [CrossRef]
- Wong, K.; Nguyen, J.; Blair, L.; Banjanin, M.; Grewal, B.; Bowman, S.; Boyd, H.; Gerstner, G.; Cho, H.J.; Panfilov, D.; et al. Pathogenesis of Human Immunodeficiency Virus-Mycobacterium Tuberculosis Co-Infection. J. Clin. Med. 2020, 9, 3575. [Google Scholar] [CrossRef]
- Wen, L.; Shi, L.; Wan, S.-S.; Xu, T.; Zhang, L.; Zhou, Z.-G. Changes in the Balance of Th17/Treg Cells and Oxidative Stress Markers in Patients with HIV-associated Pulmonary Tuberculosis Who Develop IRIS. Exp. Ther. Med. 2023, 25, 271. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, N.; Erdos, T.; Louie, C.; Desai, R.; Lin, N.; Ayzenberg, G.; Venketaraman, V. The Role of Glutathione in the Management of Cell-Mediated Immune Responses in Individuals with HIV. Int. J. Mol. Sci. 2024, 25, 2952. https://doi.org/10.3390/ijms25052952
Lin N, Erdos T, Louie C, Desai R, Lin N, Ayzenberg G, Venketaraman V. The Role of Glutathione in the Management of Cell-Mediated Immune Responses in Individuals with HIV. International Journal of Molecular Sciences. 2024; 25(5):2952. https://doi.org/10.3390/ijms25052952
Chicago/Turabian StyleLin, Nicole, Thomas Erdos, Carson Louie, Raina Desai, Naomi Lin, Gregory Ayzenberg, and Vishwanath Venketaraman. 2024. "The Role of Glutathione in the Management of Cell-Mediated Immune Responses in Individuals with HIV" International Journal of Molecular Sciences 25, no. 5: 2952. https://doi.org/10.3390/ijms25052952
APA StyleLin, N., Erdos, T., Louie, C., Desai, R., Lin, N., Ayzenberg, G., & Venketaraman, V. (2024). The Role of Glutathione in the Management of Cell-Mediated Immune Responses in Individuals with HIV. International Journal of Molecular Sciences, 25(5), 2952. https://doi.org/10.3390/ijms25052952