
Neutrophils and NADPH Oxidases Are Major Contributors to Mild but Not Severe Ischemic Acute Kidney Injury in Mice


 , , and
, , and
Abstract
1. Introduction
2. Results
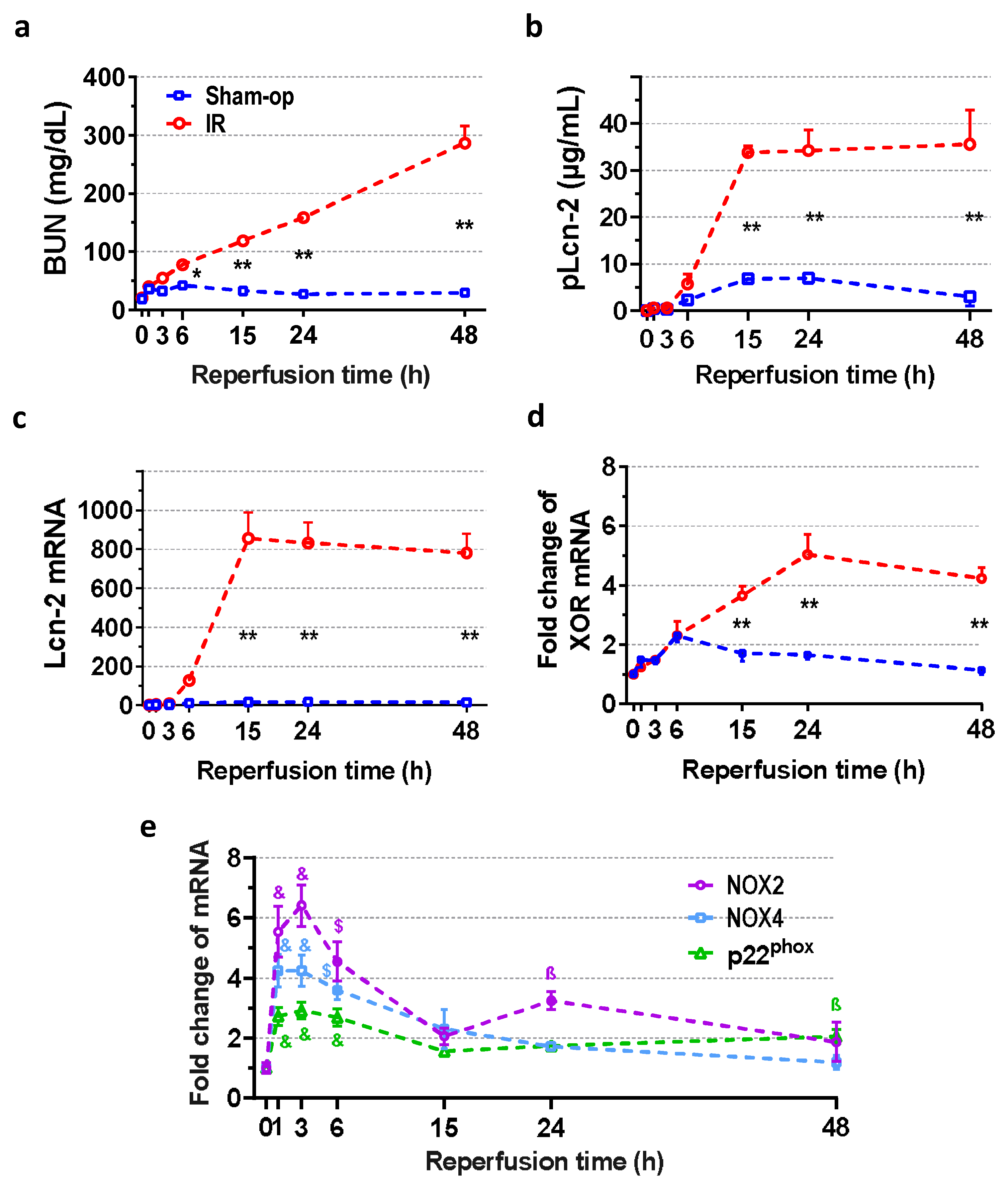
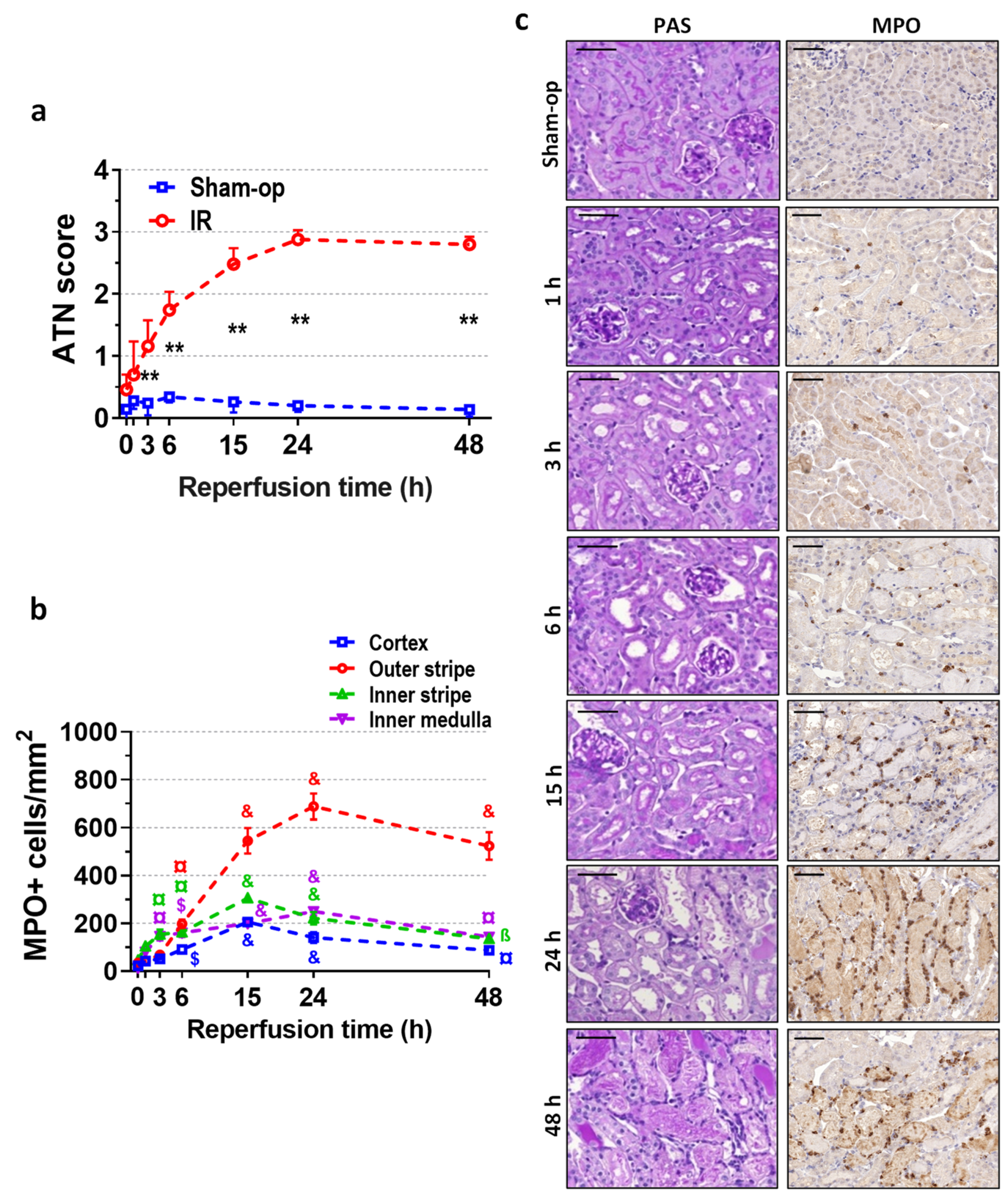
2.1. Severe Ischemia–Reperfusion Impaired Kidney Function and Morphology
2.2. Rapid Renal NOX and Delayed XOR Upregulation after Reperfusion
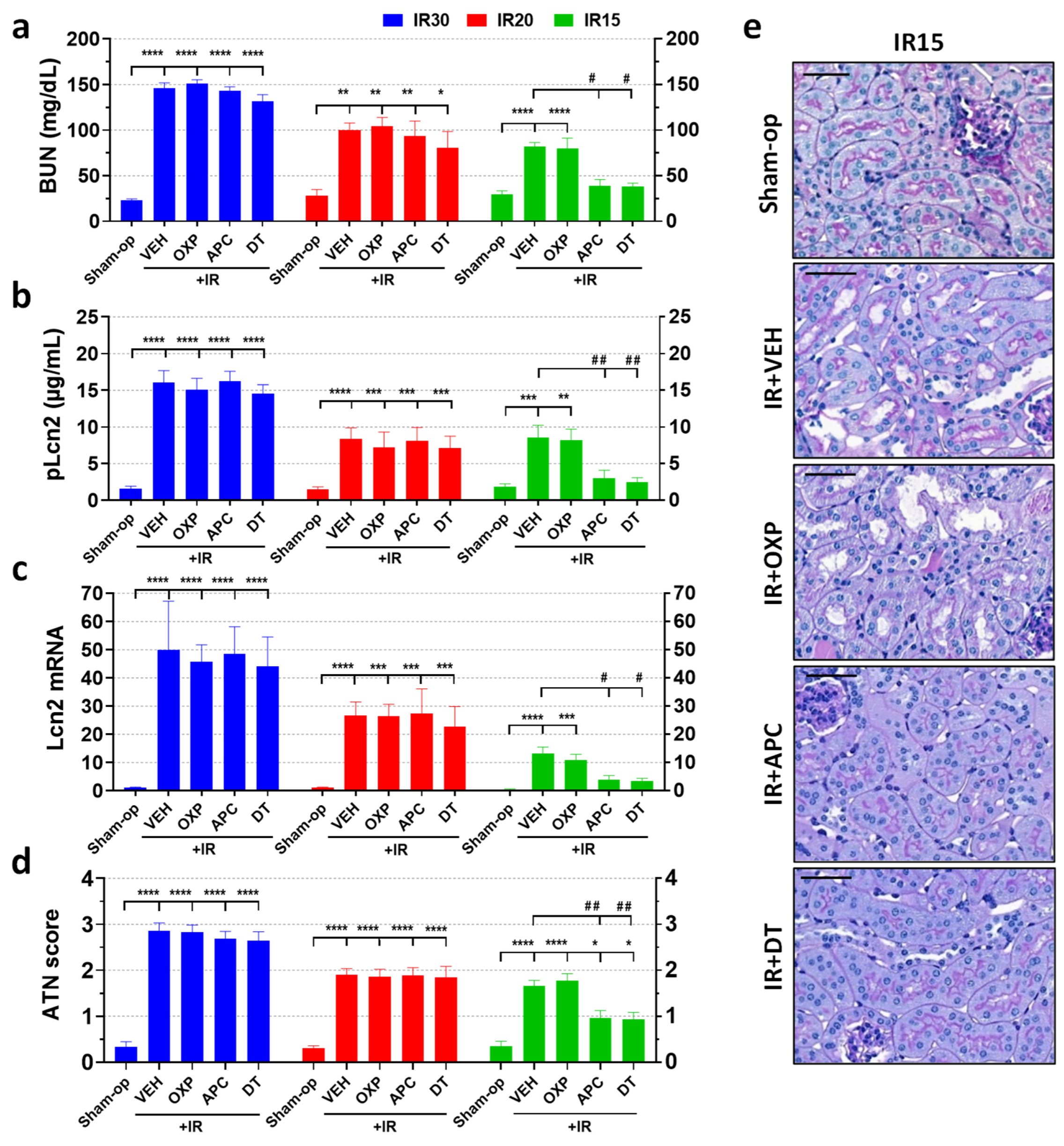
2.3. Pharmacological Inhibition of NOX Enzymes but Not XOR Protected against Mild but Not Moderate or Severe Renal Ischemia
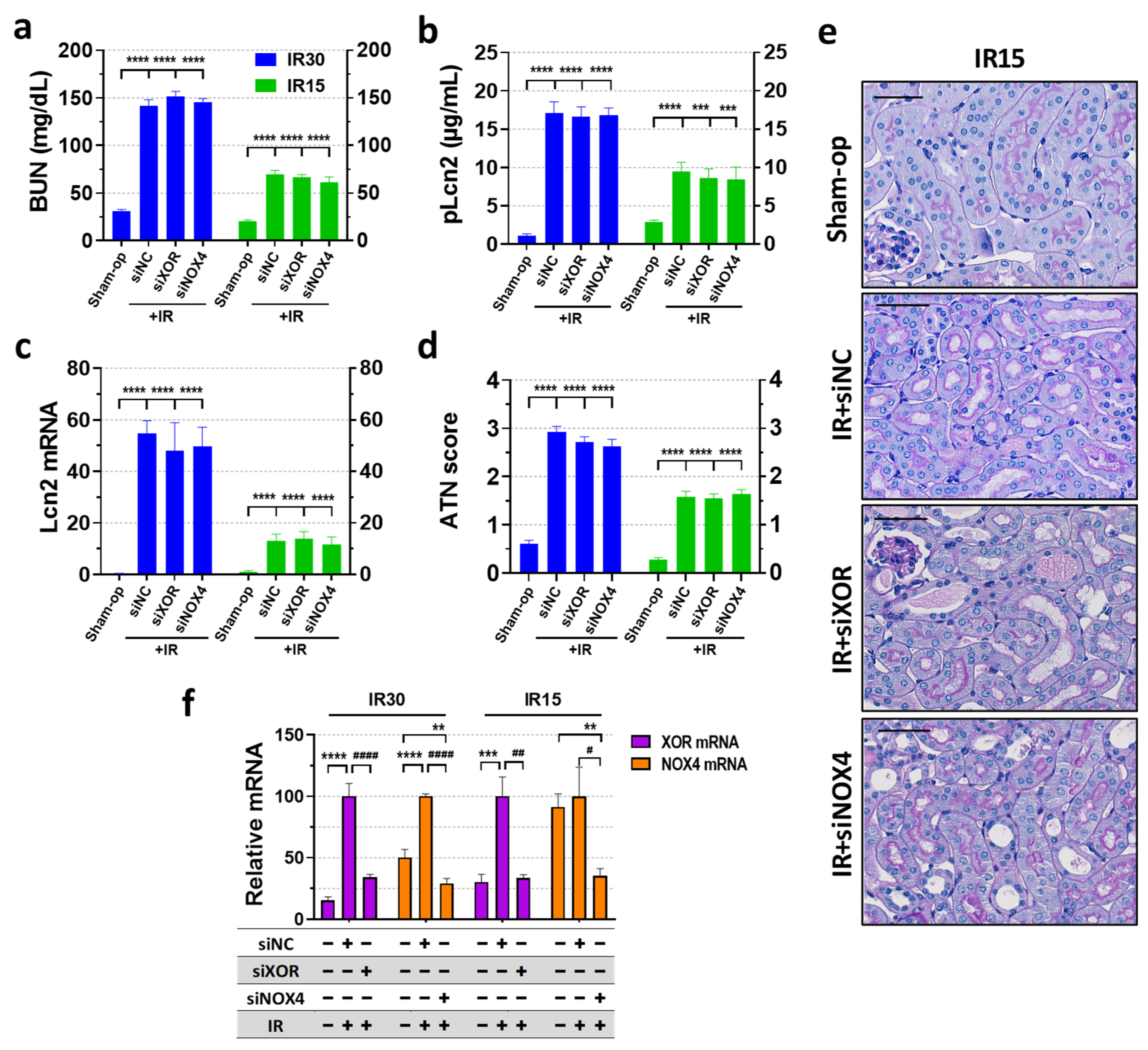
2.4. XOR and NOX4 Silencing Did Not Protect Renal Function Post-Ischemia
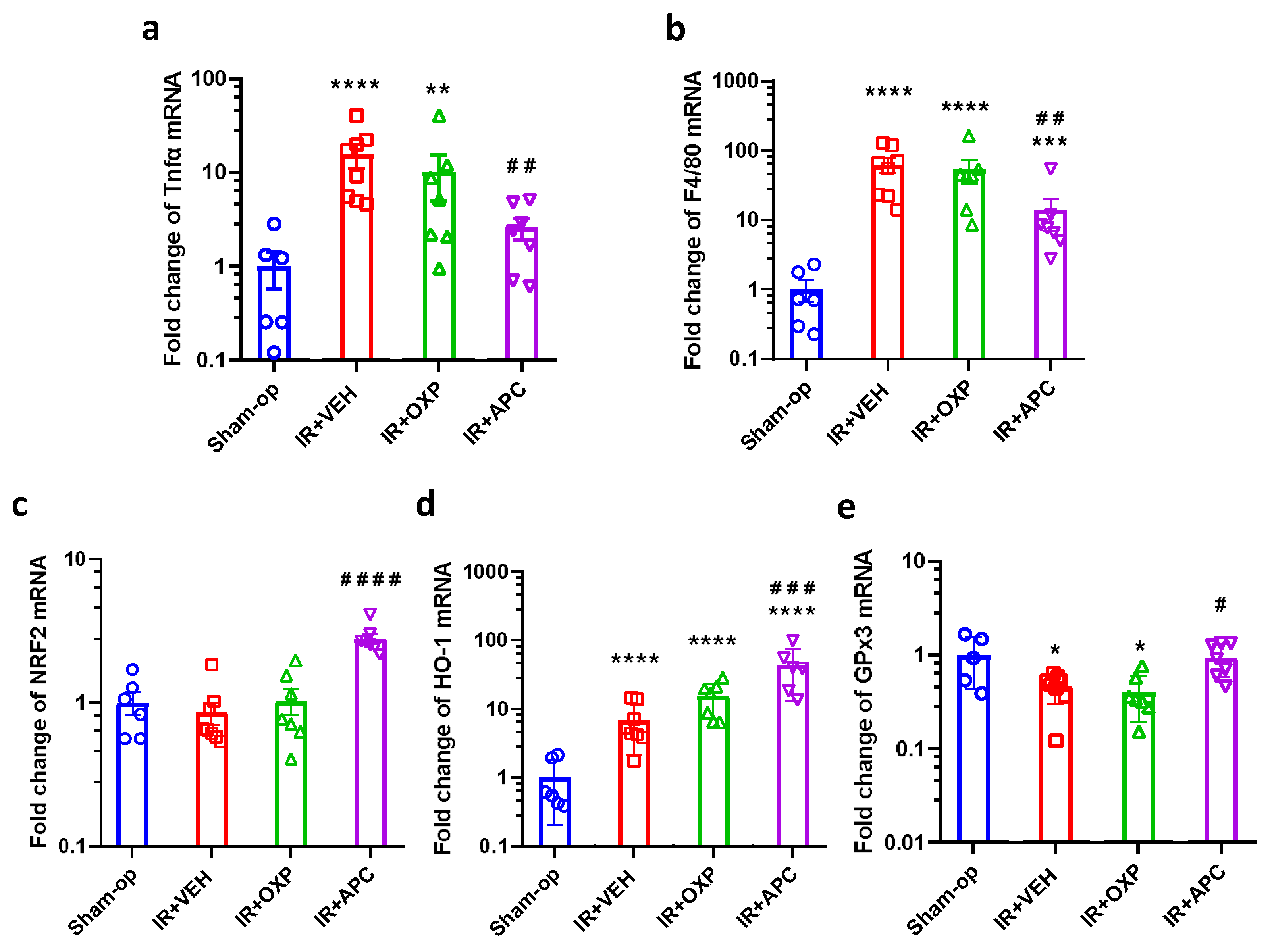
2.5. Pharmacological Inhibition of NOX by Apocynin Increased Antioxidant Defense and Mitigated Inflammation after Mild Renal Ischemia
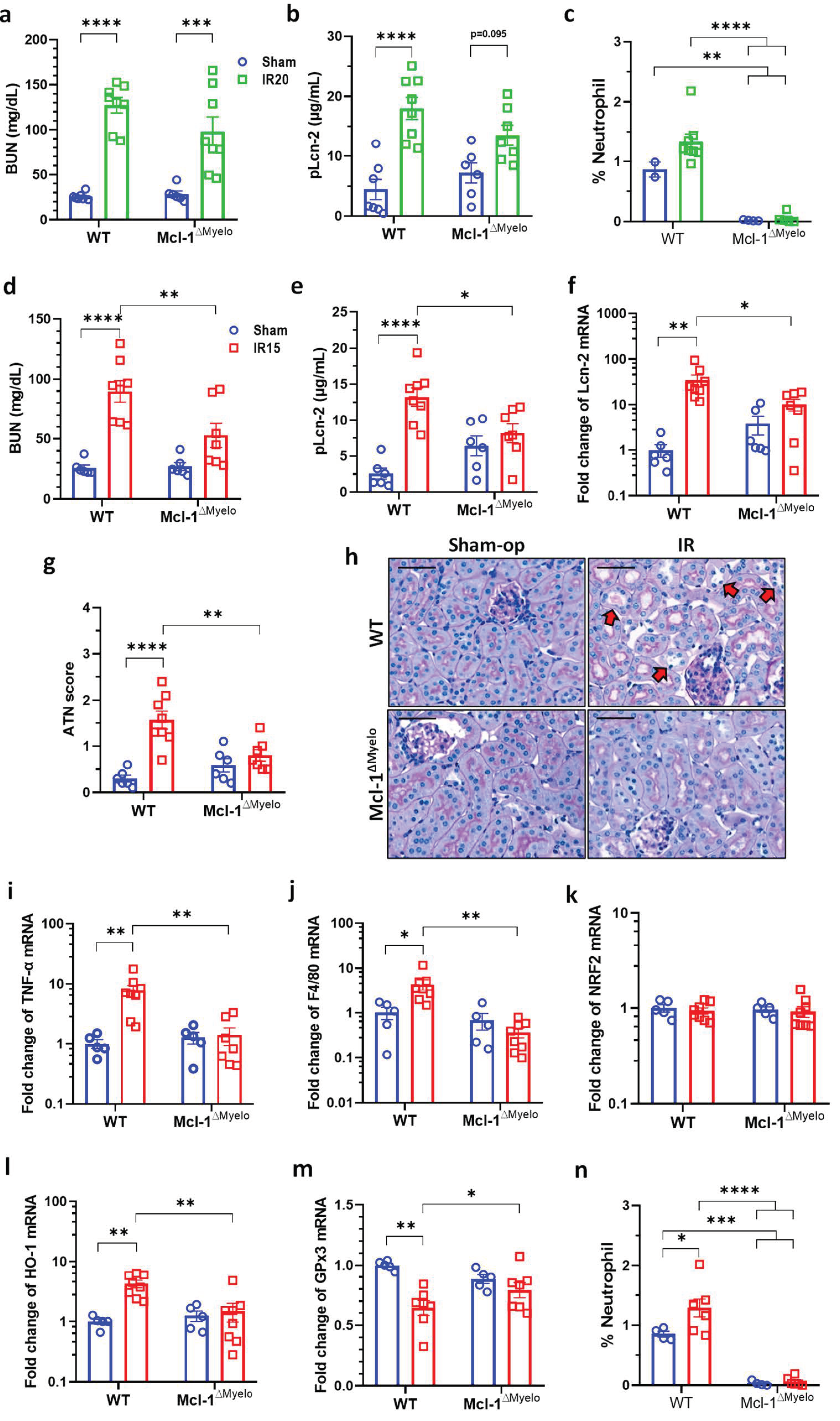
2.6. Neutrophil-Deficient Bone Marrow Chimeric Mice Are Protected from Mild Renal IRI
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. SiRNA Application
4.3. Administration of Pharmacologic Inhibitors
4.4. Kidney Ischemia–Reperfusion
4.5. Animal Sacrifice and Sample Collection
4.6. BUN Assay and Lcn-2 ELISA
4.7. Renal Histology
4.8. RNA Preparation
4.9. Quantitative Real-Time PCR Analysis for mRNA Expression in Renal Tissue
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoste, E.A.J.; Kellum, J.A.; Selby, N.M.; Zarbock, A.; Palevsky, P.M.; Bagshaw, S.M.; Goldstein, S.L.; Cerda, J.; Chawla, L.S. Global epidemiology and outcomes of acute kidney injury. Nat. Rev. Nephrol. 2018, 14, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Tonnus, W.; Meyer, C.; Steinebach, C.; Belavgeni, A.; von Massenhausen, A.; Gonzalez, N.Z.; Maremonti, F.; Gembardt, F.; Himmerkus, N.; Latk, M.; et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 2021, 12, 4402. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Kaucsar, T.; Godo, M.; Revesz, C.; Kovacs, M.; Mocsai, A.; Kiss, N.; Albert, M.; Krenacs, T.; Szenasi, G.; Hamar, P. Urine/Plasma Neutrophil Gelatinase Associated Lipocalin Ratio Is a Sensitive and Specific Marker of Subclinical Acute Kidney Injury in Mice. PLoS ONE 2016, 11, e0148043. [Google Scholar] [CrossRef] [PubMed]
- Zelova, H.; Hosek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Wijerathne, C.U.B.; Au-Yeung, K.K.W.; Siow, Y.L.; O, K. 5-Methyltetrahydrofolate Attenuates Oxidative Stress and Improves Kidney Function in Acute Kidney Injury through Activation of Nrf2 and Antioxidant Defense. Antioxidants 2022, 11, 1046. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Altintas, R.; Polat, A.; Vardi, N.; Oguz, F.; Beytur, A.; Sagir, M.; Yildiz, A.; Parlakpinar, H. The protective effects of apocynin on kidney damage caused by renal ischemia/reperfusion. J. Endourol. 2013, 27, 617–624. [Google Scholar] [CrossRef]
- Lima, N.K.S.; Farias, W.R.A.; Cirilo, M.A.S.; Oliveira, A.G.; Farias, J.S.; Aires, R.S.; Muzi-Filho, H.; Paixao, A.D.O.; Vieira, L.D. Renal ischemia-reperfusion leads to hypertension and changes in proximal tubule Na(+) transport and renin-angiotensin-aldosterone system: Role of NADPH oxidase. Life Sci. 2021, 266, 118879. [Google Scholar] [CrossRef]
- Nlandu-Khodo, S.; Dissard, R.; Hasler, U.; Schafer, M.; Pircher, H.; Jansen-Durr, P.; Krause, K.H.; Martin, P.Y.; de Seigneux, S. NADPH oxidase 4 deficiency increases tubular cell death during acute ischemic reperfusion injury. Sci. Rep. 2016, 6, 38598. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Yu, S.L.; Kang, J.; Jeong, B.Y.; Lee, H.Y.; Park, C.G.; Yu, Y.B.; Jin, D.C.; Hwang, W.M.; Yun, S.R.; et al. NADPH oxidase 4 mediates TGF-beta1/Smad signaling pathway induced acute kidney injury in hypoxia. PLoS ONE 2019, 14, e0219483. [Google Scholar] [CrossRef]
- McNally, J.S.; Saxena, A.; Cai, H.; Dikalov, S.; Harrison, D.G. Regulation of xanthine oxidoreductase protein expression by hydrogen peroxide and calcium. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef]
- Kang, H.B.; Lim, C.K.; Kim, J.; Han, S.J. Oxypurinol protects renal ischemia/reperfusion injury via heme oxygenase-1 induction. Front. Med. 2023, 10, 1030577. [Google Scholar] [CrossRef] [PubMed]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Franzin, R.; Stasi, A.; Fiorentino, M.; Simone, S.; Oberbauer, R.; Castellano, G.; Gesualdo, L. Renal Delivery of Pharmacologic Agents During Machine Perfusion to Prevent Ischaemia-Reperfusion Injury: From Murine Model to Clinical Trials. Front. Immunol. 2021, 12, 673562. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Votrico, V.; Spadaccino, F.; Catalano, V.; Netti, G.S.; Ranieri, E.; Stallone, G.; Zaza, G. Oxidative Stress and Ischemia/Reperfusion Injury in Kidney Transplantation: Focus on Ferroptosis, Mitophagy and New Antioxidants. Antioxidants 2022, 11, 769. [Google Scholar] [CrossRef] [PubMed]
- Chazelas, P.; Steichen, C.; Favreau, F.; Trouillas, P.; Hannaert, P.; Thuillier, R.; Giraud, S.; Hauet, T.; Guillard, J. Oxidative Stress Evaluation in Ischemia Reperfusion Models: Characteristics, Limits and Perspectives. Int. J. Mol. Sci. 2021, 22, 2366. [Google Scholar] [CrossRef] [PubMed]
- Al-Khafaji, A.B.; Tohme, S.; Yazdani, H.O.; Miller, D.; Huang, H.; Tsung, A. Superoxide induces Neutrophil Extracellular Trap Formation in a TLR-4 and NOX-dependent mechanism. Mol. Med. 2016, 22, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Moure, B.; Caraben-Redano, A.; Aliena-Valero, A.; Cejalvo, D.; Toledo, A.H.; Flores-Bellver, M.; Martinez-Gil, N.; Toledo-Pereyra, L.H.; Lloris Carsi, J.M. Allopurinol in renal ischemia. J. Investig. Surg. 2014, 27, 304–316. [Google Scholar] [CrossRef]
- Zager, R.A.; Fuerstenberg, S.M.; Baehr, P.H.; Myerson, D.; Torok-Storb, B. An evaluation of antioxidant effects on recovery from postischemic acute renal failure. J. Am. Soc. Nephrol. 1994, 4, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Radovic, M.; Miloradovic, Z.; Popovic, T.; Mihailovic-Stanojevic, N.; Jovovic, D.; Tomovic, M.; Colak, E.; Simic-Ogrizovic, S.; Djukanovic, L. Allopurinol and enalapril failed to conserve urinary NOx and sodium in ischemic acute renal failure in spontaneously hypertensive rats. Am. J. Nephrol. 2006, 26, 388–399. [Google Scholar] [CrossRef]
- Munoz, M.; Lopez-Oliva, M.E.; Rodriguez, C.; Martinez, M.P.; Saenz-Medina, J.; Sanchez, A.; Climent, B.; Benedito, S.; Garcia-Sacristan, A.; Rivera, L.; et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020, 28, 101330. [Google Scholar] [CrossRef]
- Turan, N.N.; Akar, F.; Budak, B.; Seren, M.; Parlar, A.I.; Surucu, S.; Ulus, A.T. How DMSO, a widely used solvent, affects spinal cord injury. Ann. Vasc. Surg. 2008, 22, 98–105. [Google Scholar] [CrossRef]
- Pascher, A.; Klupp, J. Biologics in the treatment of transplant rejection and ischemia/reperfusion injury: New applications for TNFα inhibitors? BioDrugs 2005, 19, 211–231. [Google Scholar] [CrossRef]
- Huen, S.C.; Cantley, L.G. Macrophages in Renal Injury and Repair. Annu. Rev. Physiol. 2017, 79, 449–469. [Google Scholar] [CrossRef]
- Park, Y.R.; Kong, M.J.; Noh, M.R.; Park, K.M. Rac1 inhibition protects the kidney against kidney ischemia/reperfusion through the inhibition of macrophage migration. Korean J. Physiol. Pharmacol. 2023, 27, 257–265. [Google Scholar] [CrossRef]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef] [PubMed]
- Potteti, H.R.; Noone, P.M.; Tamatam, C.R.; Ankireddy, A.; Noel, S.; Rabb, H.; Reddy, S.P. Nrf2 mediates hypoxia-inducible HIF1alpha activation in kidney tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2021, 320, F464–F474. [Google Scholar] [CrossRef] [PubMed]
- Grunenwald, A.; Roumenina, L.T.; Frimat, M. Heme Oxygenase 1: A Defensive Mediator in Kidney Diseases. Int. J. Mol. Sci. 2021, 22, 2009. [Google Scholar] [CrossRef]
- Mahmoud, N.A.; Hassanein, E.H.M.; Bakhite, E.A.; Shaltout, E.S.; Sayed, A.M. Apocynin and its chitosan nanoparticles attenuated cisplatin-induced multiorgan failure: Synthesis, characterization, and biological evaluation. Life Sci. 2023, 314, 121313. [Google Scholar] [CrossRef]
- Bhatt, N.P.; Park, J.Y.; Lee, H.J.; Kim, S.S.; Kwon, Y.S.; Chun, W. Apocynin protects mesangial cells from lipopolysaccharide-induced inflammation by exerting heme oxygenase 1-mediated monocyte chemoattractant protein-1 suppression. Int. J. Mol. Med. 2017, 40, 1294–1301. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Hue, M.; Rayego-Mateos, S.; Vazquez-Carballo, C.; Palomino-Antolin, A.; Garcia-Caballero, C.; Opazo-Rios, L.; Morgado-Pascual, J.L.; Herencia, C.; Mas, S.; Ortiz, A.; et al. Protective Role of Nrf2 in Renal Disease. Antioxidants 2020, 10, 39. [Google Scholar] [CrossRef]
- Pei, J.; Tian, X.; Yu, C.; Luo, J.; Zhang, J.; Hua, Y.; Wei, G. GPX3 and GSTT1 as biomarkers related to oxidative stress during renal ischemia reperfusion injuries and their relationship with immune infiltration. Front. Immunol. 2023, 14, 1136146. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, H.; Xu, Y.; Wen, R.; Gong, M.; Hong, G.; Xu, S. Selenoprotein Gene mRNA Expression Evaluation During Renal Ischemia-Reperfusion Injury in Rats and Ebselen Intervention Effects. Biol. Trace Elem. Res. 2023, 201, 1792–1805. [Google Scholar] [CrossRef]
- Li, L.; He, M.; Tang, X.; Huang, J.; Li, J.; Hong, X.; Fu, H.; Liu, Y. Proteomic landscape of the extracellular matrix in the fibrotic kidney. Kidney Int. 2023, 103, 1063–1076. [Google Scholar] [CrossRef]
- Awad, A.S.; Rouse, M.; Huang, L.; Vergis, A.L.; Reutershan, J.; Cathro, H.P.; Linden, J.; Okusa, M.D. Compartmentalization of neutrophils in the kidney and lung following acute ischemic kidney injury. Kidney Int. 2009, 75, 689–698. [Google Scholar] [CrossRef]
- Simone, S.; Rascio, F.; Castellano, G.; Divella, C.; Chieti, A.; Ditonno, P.; Battaglia, M.; Crovace, A.; Staffieri, F.; Oortwijn, B.; et al. Complement-dependent NADPH oxidase enzyme activation in renal ischemia/reperfusion injury. Free Radic. Biol. Med. 2014, 74, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Horvath, B.; Zsengeller, Z.; Batkai, S.; Cao, Z.; Kechrid, M.; Holovac, E.; Erdelyi, K.; Tanchian, G.; Liaudet, L.; et al. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: Therapeutic potential of mitochondrially targeted antioxidants. Free Radic. Biol. Med. 2012, 53, 1123–1138. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Zhu, P.; Carman, C.V.; Lieberman, J.; Shimaoka, M. Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc. Natl. Acad. Sci. USA 2007, 104, 4095–4100. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, T.; Sperandio, M.; Mocsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.C.; Nemeth, T.; Csepregi, J.Z.; Dudeck, A.; Roers, A.; Ozsvari, B.; Oswald, E.; Puskas, L.G.; Jakob, T.; Mocsai, A.; et al. Neutrophils are required for both the sensitization and elicitation phase of contact hypersensitivity. J. Exp. Med. 2015, 212, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Csepregi, J.Z.; Orosz, A.; Zajta, E.; Kasa, O.; Nemeth, T.; Simon, E.; Fodor, S.; Csonka, K.; Baratki, B.L.; Kovesdi, D.; et al. Myeloid-Specific Deletion of Mcl-1 Yields Severely Neutropenic Mice That Survive and Breed in Homozygous Form. J. Immunol. 2018, 201, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Hamar, P.; Song, E.; Kokeny, G.; Chen, A.; Ouyang, N.; Lieberman, J. Small interfering RNA targeting Fas protects mice against renal ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2004, 101, 14883–14888. [Google Scholar] [CrossRef]
- Liu, F.; Fan, L.M.; Michael, N.; Li, J.M. In vivo and in silico characterization of apocynin in reducing organ oxidative stress: A pharmacokinetic and pharmacodynamic study. Pharmacol. Res. Perspect. 2020, 8, e00635. [Google Scholar] [CrossRef]
- Vora, B.; Brackman, D.J.; Zou, L.; Garcia-Cremades, M.; Sirota, M.; Savic, R.M.; Giacomini, K.M. Oxypurinol pharmacokinetics and pharmacodynamics in healthy volunteers: Influence of BCRP Q141K polymorphism and patient characteristics. Clin. Transl. Sci. 2021, 14, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Noiri, E.; Nakao, A.; Uchida, K.; Tsukahara, H.; Ohno, M.; Fujita, T.; Brodsky, S.; Goligorsky, M.S. Oxidative and nitrosative stress in acute renal ischemia. Am. J. Physiol. Renal Physiol. 2001, 281, F948–F957. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Takata, T.; Yamada, K.; Yamamoto, M.; Mae, Y.; Iyama, T.; Ikeda, S.; Kanda, T.; Sugihara, T.; Isomoto, H. Steatosis is involved in the progression of kidney disease in a high-fat-diet-induced non-alcoholic steatohepatitis mouse model. PLoS ONE 2022, 17, e0265461. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Sequence (5′ to 3′) | Reverse Sequence (5′ to 3′) |
|---|---|---|
| 18S | CTCAACACGGGAAACCTCAC | CGCTCCACCAACTAAGAACG |
| GAPDH | CCAGAATGAGGATCCCAGAA | ACCACCTGAAACATGCAACA |
| Lcn-2 | AGGTGGTACGTTGTGGGC | CTGTACCTGAGGATACCTGTG |
| NOX2 | TGCCACCAGTCTGAAACTCAA | AGCAAAGTGATTGGCCTGAGA |
| NOX4 | CCAGAATGAGGATCCCAGAA | ACCACCTGAACCATGCAACA |
| p22phox | CGATCAGTGAGGACTTGCGA | CACACCTGCAGCGATAGAGT |
| XOR | GGCCATTTATGAAGCCTGTCA | GAAGTAGTGGAAGGGGTTCC |
| NRF2 | CCTCACCTCTGCTGCAAGTA | GCTCATAGTCCTTCTGTCGCT |
| HO-1 | ACAGAAGAGGCTAAGACCGC | GGCAGTATCTTGCACCAGG |
| TNF-α | AAATGGCCTCCCTCTCATCA | AGATAGCAAATCGGCTGACG |
| F4/80 | TTTCCTCGCCTGCTTCTTC | CCCCGTCTCTGTATTCAACC |
| GPx3 | CATCCTGCCTTCTGTCCCTG | CGATGGTGAGGGCTCCATAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Révész, C.; Kaucsár, T.; Godó, M.; Bocskai, K.; Krenács, T.; Mócsai, A.; Szénási, G.; Hamar, P. Neutrophils and NADPH Oxidases Are Major Contributors to Mild but Not Severe Ischemic Acute Kidney Injury in Mice. Int. J. Mol. Sci. 2024, 25, 2948. https://doi.org/10.3390/ijms25052948
Révész C, Kaucsár T, Godó M, Bocskai K, Krenács T, Mócsai A, Szénási G, Hamar P. Neutrophils and NADPH Oxidases Are Major Contributors to Mild but Not Severe Ischemic Acute Kidney Injury in Mice. International Journal of Molecular Sciences. 2024; 25(5):2948. https://doi.org/10.3390/ijms25052948
Chicago/Turabian StyleRévész, Csaba, Tamás Kaucsár, Mária Godó, Krisztián Bocskai, Tibor Krenács, Attila Mócsai, Gábor Szénási, and Péter Hamar. 2024. "Neutrophils and NADPH Oxidases Are Major Contributors to Mild but Not Severe Ischemic Acute Kidney Injury in Mice" International Journal of Molecular Sciences 25, no. 5: 2948. https://doi.org/10.3390/ijms25052948
APA StyleRévész, C., Kaucsár, T., Godó, M., Bocskai, K., Krenács, T., Mócsai, A., Szénási, G., & Hamar, P. (2024). Neutrophils and NADPH Oxidases Are Major Contributors to Mild but Not Severe Ischemic Acute Kidney Injury in Mice. International Journal of Molecular Sciences, 25(5), 2948. https://doi.org/10.3390/ijms25052948