
Aland Island Eye Disease with Retinoschisis in the Clinical Spectrum of CACNA1F-Associated Retinopathy—A Case Report

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
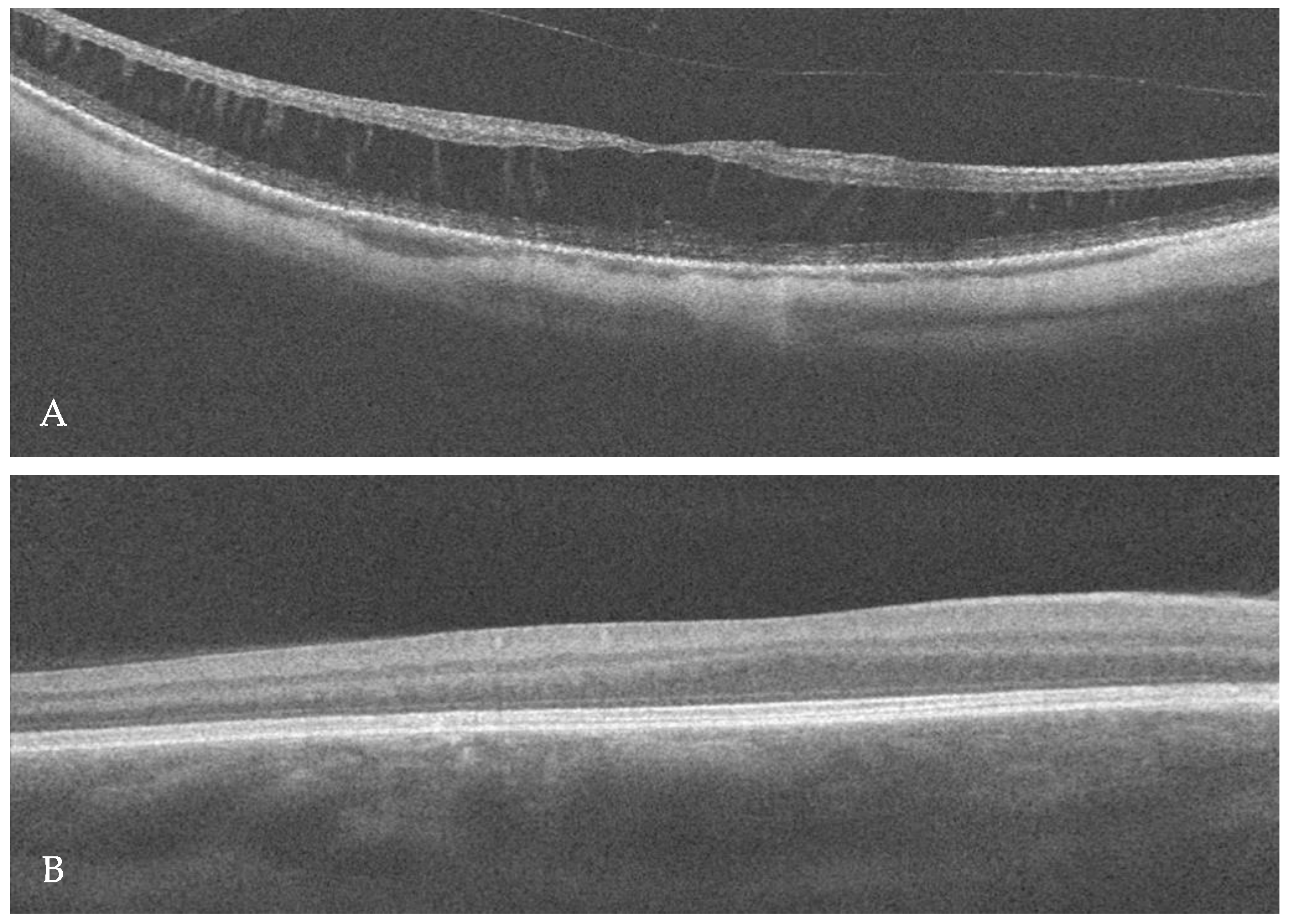
2. Case Report
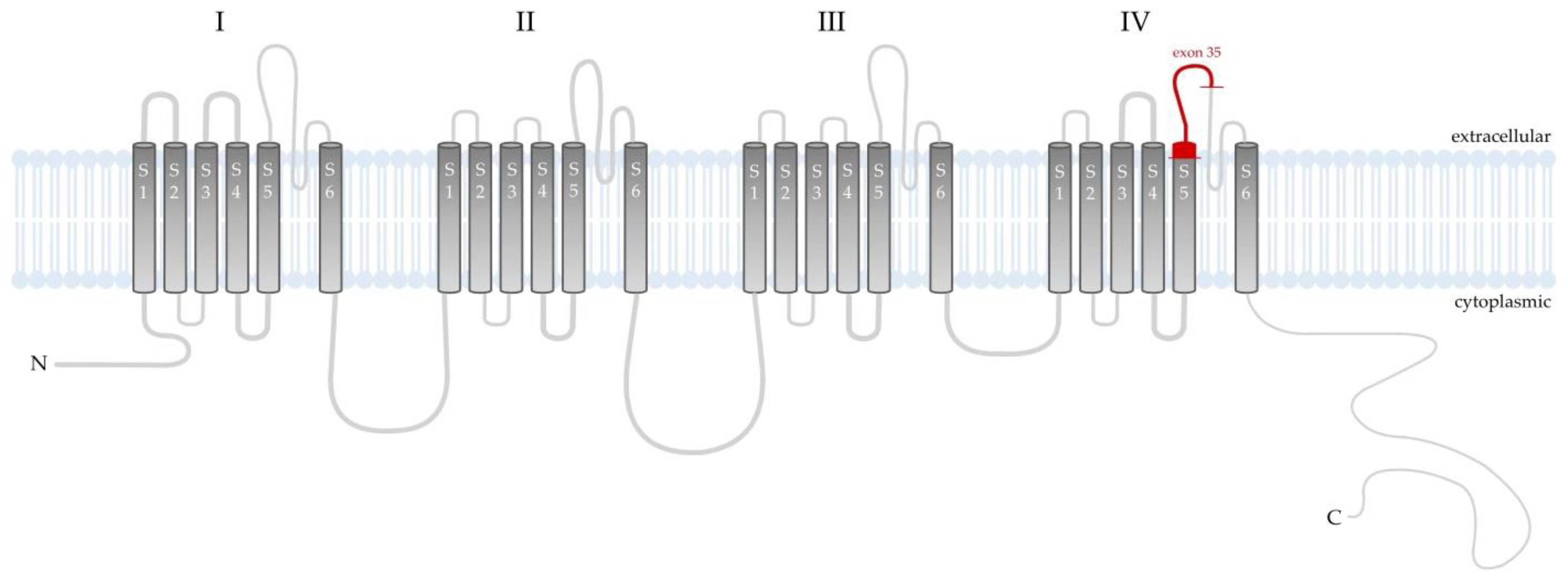
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hove, M.N.; Kilic-Biyik, K.Z.; Trotter, A.; Grønskov, K.; Sander, B.; Larsen, M.; Carroll, J.; Bech-Hansen, T.; Rosenberg, T. Clinical Characteristics, Mutation Spectrum, and Prevalence of Åland Eye Disease/Incomplete Congenital Stationary Night Blindness in Denmark. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6861–6869. [Google Scholar] [CrossRef]
- Jalkanen, R.; Bech-Hansen, N.T.; Tobias, R.; Sankila, E.M.; Mäntyjärvi, M.; Forsius, H.; de la Chapelle, A.; Alitalo, T. A novel CACNA1F gene mutation causes Aland Island eye disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2498–2502. [Google Scholar] [CrossRef]
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, A.G.; Waardenburg, P.J.; Forsius, H.; Eriksson, A.W. Nystagmographical studies in Aland eye disease. Acta Ophthalmol. 1973, 51, 782–790. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, F.E.; Green, W.R.; McKusick, V.A.; Forsius, H.; Eriksson, A.W. Forsius-Eriksson syndrome: Its relation to the Nettleship-Falls X-linked ocular albinism. Clin. Genet. 1980, 17, 403–408. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, D.B.; Eriksson, A.W.; Delleman, J.W.; van Vliet, A.G.; Collewijn, H.; van Balen, A.T.; Forsius, H.R. Aland eye disease: No albino misrouting. Clin. Genet. 1985, 28, 526–531. [Google Scholar] [CrossRef]
- Michalakis, S.; Shaltiel, L.; Sothilingam, V.; Koch, S.; Schludi, V.; Krause, S.; Zeitz, C.; Audo, I.; Lancelot, M.E.; Hamel, C.; et al. Mosaic synaptopathy and functional defects in Cav1.4 heterozygous mice and human carriers of CSNB2. Hum. Mol. Genet. 2014, 23, 1538–1550. [Google Scholar] [CrossRef] [PubMed]
- McRory, J.E.; Hamid, J.; Doering, C.J.; Garcia, E.; Parker, R.; Hamming, K.; Chen, L.; Hildebrand, M.; Beedle, A.M.; Feldcamp, L.; et al. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J. Neurosci. 2004, 24, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Baumann, L.; Gerstner, A.; Zong, X.; Biel, M.; Wahl-Schott, C. Functional characterization of the L-type Ca2+ channel Cav1.4alpha1 from mouse retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 708–713. [Google Scholar] [CrossRef]
- Koschak, A.; Fernandez-Quintero, M.L.; Heigl, T.; Ruzza, M.; Seitter, H.; Zanetti, L. Cav1.4 dysfunction and congenital stationary night blindness type 2. Pflugers Arch. 2021, 473, 1437–1454. [Google Scholar] [CrossRef]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, U.; Méjécase, C.; Ali, S.M.A.; Moosajee, M.; Kozak, I. A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness. Genes 2021, 12, 171. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, R.; Mäntyjärvi, M.; Tobias, R.; Isosomppi, J.; Sankila, E.M.; Alitalo, T.; Bech-Hansen, N.T. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J. Med. Genet. 2006, 43, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Odom, J.V.; Bach, M.; Brigell, M.; Holder, G.E.; McCulloch, D.L.; Mizota, A.; Tormene, A.P. International Society for Clinical Electrophysiology of Vision. ISCEV standard for clinical visual evoked potentials: (2016 update). Doc. Ophthalmol. 2016, 133, 77. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Seagar, M.J.; Jones, J.F.; Reber, B.F.; Catterall, W.A. Subunit structure of dihydropyridine-sensitive calcium channels from skeletal muscle. Proc. Natl. Acad. Sci. USA 1987, 84, 5478–5482. [Google Scholar] [CrossRef]
- Koschak, A.; Reimer, D.; Walter, D.; Hoda, J.C.; Heinzle, T.; Grabner, M.; Striessnig, J. Cav1.4alpha1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J. Neurosci. 2003, 23, 6041–6049. [Google Scholar] [CrossRef]
- Singh, A.; Hamedinger, D.; Hoda, J.C.; Gebhart, M.; Koschak, A.; Romanin, C.; Striessnig, J. C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat. Neurosci. 2006, 9, 1108–1116. [Google Scholar] [CrossRef]
- Liu, X.; Kerov, V.; Haeseleer, F.; Majumder, A.; Artemyev, N.; Baker, S.A.; Lee, A. Dysregulation of Ca(v)1.4 channels disrupts the maturation of photoreceptor synaptic ribbons in congenital stationary night blindness type 2. Channels 2013, 7, 514–523. [Google Scholar] [CrossRef]
- Maddox, J.W.; Randall, K.L.; Yadav, R.P.; Williams, B.; Hagen, J.; Derr, P.J.; Kerov, V.; Della Santina, L.; Baker, S.A.; Artemyev, N.; et al. A dual role for Cav1.4 Ca2+ channels in the molecular and structural organization of the rod photoreceptor synapse. eLife 2020, 9, e62184. [Google Scholar] [CrossRef]
- Gupta, S.K.; Chakraborty, R.; Verkicharla, P.K. Electroretinogram responses in myopia: A review. Doc. Ophthalmol. 2022, 2, 77–95. [Google Scholar] [CrossRef]
- Sin, T.N.; Kim, S.; Li, Y.; Wang, J.; Chen, R.; Chung, S.H.; Kim, S.; Casanova, M.I.; Park, S.; Smit-McBride, Z.; et al. A Spontaneous Nonhuman Primate Model of Myopic Foveoschisis. Investig. Ophthalmol. Vis. Sci. 2023, 64, 18. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Ko, M.L.; Ko, G.Y. Retinoschisin Facilitates the Function of L-Type Voltage-Gated Calcium Channels. Front. Cell. Neurosci. 2017, 11, 232. [Google Scholar] [CrossRef]
- Weber, B.H.; Schrewe, H.; Molday, L.L.; Gehrig, A.; White, K.L.; Seeliger, M.W.; Jaissle, G.B.; Friedburg, C.; Tamm, E.; Molday, R.S. Inactivation of the murine X-linked juvenile retinoschisis gene, Rs1h, suggests a role of retinoschisin in retinal cell layer organization and synaptic structure. Proc. Natl. Acad. Sci. USA 2002, 99, 6222–6227. [Google Scholar] [CrossRef]
- Ko, M.L.; Liu, Y.; Shi, L.; Trump, D.; Ko, G.Y. Circadian regulation of retinoschisin in the chick retina. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1615–1621. [Google Scholar] [CrossRef]
- Shi, L.; Jian, K.; Ko, M.L.; Trump, D.; Ko, G.Y. Retinoschisin, a new binding partner for L-type voltage-gated calcium channels in the retina. J. Biol. Chem. 2009, 284, 3966–3975. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Pearce, W.G.; Bech-Hansen, N.T. Clinical variability among patients with incomplete X-linked congenital stationary night blindness and a founder mutation in CACNA1F. Can. J. Ophthalmol. 2000, 35, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Mihalich, A.; Cammarata, G.; Tremolada, G.; Pollazzon, M.; Di Blasio, A.M.; Marzoli, S.B. Two novel CACNA1F gene mutations cause two different phenotypes: Aland Eye Disease and incomplete Congenital Stationary Night Blindness. Exp. Eye Res. 2022, 221, 109143. [Google Scholar] [CrossRef]
- Burtscher, V.; Schicker, K.; Novikova, E.; Pöhn, B.; Stockner, T.; Kugler, C.; Singh, A.; Zeitz, C.; Lancelot, M.E.; Audo, I.; et al. Spectrum of Cav1.4 dysfunction in congenital stationary night blindness type 2. Biochim. Biophys. Acta 2014, 8, 2053–2065. [Google Scholar] [CrossRef]
- Vincent, A.; Wright, T.; Day, M.A.; Westall, C.A.; Héon, E. A novel p.Gly603Arg mutation in CACNA1F causes Åland island eye disease and incomplete congenital stationary night blindness phenotypes in a family. Mol. Vis. 2011, 17, 3262–3270. [Google Scholar]
- Hope, C.I.; Sharp, D.M.; Hemara-Wahanui, A.; Sissingh, J.I.; Lundon, P.; Mitchell, E.A.; Maw, M.A.; Clover, G.M. Clinical manifestations of a unique X-linked retinal disorder in a large New Zealand family with a novel mutation in CACNA1F, the gene responsible for CSNB2. Clin. Exp. Ophthalmol. 2005, 33, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Ito, S.; Piao, C.H.; Terasaki, H.; Miyake, Y. Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family. Arch. Ophthalmol. 2003, 121, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 9, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.A.; Li, T.; Yang, B.; Spalding, M.H. TALEN-mediated genome editing: Prospects and perspectives. Biochem. J. 2014, 462, 15–24. [Google Scholar] [CrossRef]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40, BSR20200127. [Google Scholar] [CrossRef]
- Waldner, D.M.; Ito, K.; Chen, L.L.; Nguyen, L.; Chow, R.L.; Lee, A.; Rancourt, D.E.; Tremblay, F.; Stell, W.K.; Bech-Hansen, N.T. Transgenic Expression of Cacna1f Rescues Vision and Retinal Morphology in a Mouse Model of Congenital Stationary Night Blindness 2A (CSNB2A). Transl. Vis. Sci. Technol. 2020, 11, 19. [Google Scholar] [CrossRef]
- Laird, J.G.; Gardner, S.H.; Kopel, A.J.; Kerov, V.; Lee, A.; Baker, S.A. Rescue of Rod Synapses by Induction of Cav Alpha 1F in the Mature Cav1.4 Knock-Out Mouse Retina. Investig. Ophthalmol. Vis. Sci. 2019, 8, 3150–3161. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyględowska-Promieńska, D.; Świerczyńska, M.; Śpiewak, D.; Pojda-Wilczek, D.; Tronina, A.; Dorecka, M.; Smędowski, A. Aland Island Eye Disease with Retinoschisis in the Clinical Spectrum of CACNA1F-Associated Retinopathy—A Case Report. Int. J. Mol. Sci. 2024, 25, 2928. https://doi.org/10.3390/ijms25052928
Wyględowska-Promieńska D, Świerczyńska M, Śpiewak D, Pojda-Wilczek D, Tronina A, Dorecka M, Smędowski A. Aland Island Eye Disease with Retinoschisis in the Clinical Spectrum of CACNA1F-Associated Retinopathy—A Case Report. International Journal of Molecular Sciences. 2024; 25(5):2928. https://doi.org/10.3390/ijms25052928
Chicago/Turabian StyleWyględowska-Promieńska, Dorota, Marta Świerczyńska, Dorota Śpiewak, Dorota Pojda-Wilczek, Agnieszka Tronina, Mariola Dorecka, and Adrian Smędowski. 2024. "Aland Island Eye Disease with Retinoschisis in the Clinical Spectrum of CACNA1F-Associated Retinopathy—A Case Report" International Journal of Molecular Sciences 25, no. 5: 2928. https://doi.org/10.3390/ijms25052928
APA StyleWyględowska-Promieńska, D., Świerczyńska, M., Śpiewak, D., Pojda-Wilczek, D., Tronina, A., Dorecka, M., & Smędowski, A. (2024). Aland Island Eye Disease with Retinoschisis in the Clinical Spectrum of CACNA1F-Associated Retinopathy—A Case Report. International Journal of Molecular Sciences, 25(5), 2928. https://doi.org/10.3390/ijms25052928