

Novel and Recurrent Copy Number Variants in ABCA4-Associated Retinopathy

 ,
,  , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Characterization of CNVs in ABCA4-Associated Retinopathies
2.2. Deletions
2.3. Duplications and Complex Rearrangement
2.4. Microhomology and Repetitive Elements
2.5. Literature-Reported CNVs in ABCA4
3. Discussion
4. Materials and Methods
4.1. Sample Origin and Sequencing
4.2. CNV Analysis
4.3. CNV Validation and Breakpoint Analysis
4.4. CNV Literature Selection
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornelis, S.S.; Bauwens, M.; Haer-Wigman, L.; De Bruyne, M.; Pantrangi, M.; De Baere, E.; Hufnagel, R.B.; Dhaenens, C.-M.; Cremers, F.P.M. Compendium of Clinical Variant Classification for 2,246 Unique ABCA4 Variants to Clarify Variant Pathogenicity in Stargardt Disease Using a Modified ACMG/AMP Framework. Hum. Mutat. 2023, 2023, 6815504. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta 2009, 1791, 573–583. [Google Scholar] [CrossRef]
- Quazi, F.; Lenevich, S.; Molday, R.S. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 2012, 3, 925. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef]
- Yatsenko, A.N.; Shroyer, N.F.; Lewis, R.A.; Lupski, J.R. An ABCA4 genomic deletion in patients with Stargardt disease. Hum. Mutat. 2003, 21, 636–644. [Google Scholar] [CrossRef]
- Maugeri, A.; van Driel, M.A.; van de Pol, D.J.; Klevering, B.J.; van Haren, F.J.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; Pinckers, A.J.; et al. The 2588G-->C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am. J. Hum. Genet. 1999, 64, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Xie, Y.A.; Ayuso, C.; Riveiro-Alvarez, R.; Lopez-Martinez, M.A.; Simonelli, F.; Testa, F.; Gorin, M.B.; Strom, S.P.; Bertelsen, M.; et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet. 2014, 23, 6797–6806. [Google Scholar] [CrossRef]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Del Pozo-Valero, M.D.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Iacocca, M.A.; Wang, J.; Sarkar, S.; Dron, J.S.; Lagace, T.; McIntyre, A.D.; Lau, P.; Robinson, J.F.; Yang, P.; Knoll, J.H.; et al. Whole-Gene Duplication of PCSK9 as a Novel Genetic Mechanism for Severe Familial Hypercholesterolemia. Can. J. Cardiol. 2018, 34, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Godoy, V.; Bellucco, F.T.; Colovati, M.; Oliveira-Junior, H.R.; Moyses-Oliveira, M.; Melaragno, M.I. Copy number variation (CNV) identification, interpretation, and database from Brazilian patients. Genet. Mol. Biol. 2020, 43, e20190218. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genom. Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Horn, B.; Campbell, C.; Arno, G.; Barton, S.; Tate, C.; Bhaskar, S.; Sergouniotis, P.I.; Taylor, R.L.; Carss, K.J.; et al. Assessment of the incorporation of CNV surveillance into gene panel next-generation sequencing testing for inherited retinal diseases. J. Med. Genet. 2018, 55, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Falsini, B.; Placidi, G.; De Siena, E.; Chiurazzi, P.; Minnella, A.M.; Savastano, M.C.; Ziccardi, L.; Parisi, V.; Iarossi, G.; Percio, M.; et al. Genetic characteristics of 234 Italian patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2022, 12, 3774. [Google Scholar] [CrossRef]
- Aguirre-Lamban, J.; Riveiro-Alvarez, R.; Maia-Lopes, S.; Cantalapiedra, D.; Vallespin, E.; Avila-Fernandez, A.; Villaverde-Montero, C.; Trujillo-Tiebas, M.J.; Ramos, C.; Ayuso, C. Molecular analysis of the ABCA4 gene for reliable detection of allelic variations in Spanish patients: Identification of 21 novel variants. Br. J. Ophthalmol. 2009, 93, 614–621. [Google Scholar] [CrossRef]
- Bax, N.M.; Sangermano, R.; Roosing, S.; Thiadens, A.A.; Hoefsloot, L.H.; van den Born, L.I.; Phan, M.; Klevering, B.J.; Westeneng-van Haaften, C.; Braun, T.A.; et al. Heterozygous deep-intronic variants and deletions in ABCA4 in persons with retinal dystrophies and one exonic ABCA4 variant. Hum. Mutat. 2015, 36, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Corradi, Z.; Khan, M.; Hitti-Malin, R.; Mishra, K.; Whelan, L.; Cornelis, S.S.; Group, A.B.-S.; Hoyng, C.B.; Kampjarvi, K.; Klaver, C.C.W.; et al. Targeted sequencing and in vitro splice assays shed light on ABCA4-associated retinopathies missing heritability. Hum. Genet. Genom. Adv. 2023, 4, 100237. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo-Valero, M.; Riveiro-Alvarez, R.; Blanco-Kelly, F.; Aguirre-Lamban, J.; Martin-Merida, I.; Iancu, I.F.; Swafiri, S.; Lorda-Sanchez, I.; Rodriguez-Pinilla, E.; Trujillo-Tiebas, M.J.; et al. Genotype-Phenotype Correlations in a Spanish Cohort of 506 Families With Biallelic ABCA4 Pathogenic Variants. Am. J. Ophthalmol. 2020, 219, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hidalgo, M.; de Bruijn, S.E.; Corradi, Z.; Rodenburg, K.; Lara-López, A.; Valverde-Megías, A.; Ávila-Fernández, A.; Fernandez-Caballero, L.; Del Pozo-Valero, M.; Corominas, J.; et al. ABCA4 c.6480-35A>G, a novel branchpoint variant associated with Stargardt disease. Front. Genet. 2023, 14, 1234032. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, D.; Runhart, E.H.; Bax, N.M.; Liefers, B.; Lambertus, S.L.; Sanchez, C.I.; Cremers, F.P.M.; Hoyng, C.B. Highly Variable Disease Courses in Siblings with Stargardt Disease. Ophthalmology 2019, 126, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Muller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Xie, Y.; Zernant, J.; Yuan, B.; Bearelly, S.; Tsang, S.H.; Lupski, J.R.; Allikmets, R. Complex inheritance of ABCA4 disease: Four mutations in a family with multiple macular phenotypes. Hum. Genet. 2016, 135, 9–19. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Hanany, M.; Khalaileh, A.; Beryozkin, A.; Meyer, S.; Abu-Diab, A.; Abu Turky, F.; Mizrahi-Meissonnier, L.; Lieberman, S.; Ben-Yosef, T.; et al. Identification of genomic deletions causing inherited retinal degenerations by coverage analysis of whole exome sequencing data. J. Med. Genet. 2016, 53, 600–607. [Google Scholar] [CrossRef]
- Tian, L.; Chen, C.; Song, Y.; Zhang, X.; Xu, K.; Xie, Y.; Jin, Z.B.; Li, Y. Phenotype-Based Genetic Analysis Reveals Missing Heritability of ABCA4-Related Retinopathy: Deep Intronic Variants and Copy Number Variations. Investig. Ophthalmol. Vis. Sci. 2022, 63, 5. [Google Scholar] [CrossRef]
- Scortecci, J.F.; Molday, L.L.; Curtis, S.B.; Garces, F.A.; Panwar, P.; Van Petegem, F.; Molday, R.S. Cryo-EM structures of the ABCA4 importer reveal mechanisms underlying substrate binding and Stargardt disease. Nat. Commun. 2021, 12, 5902. [Google Scholar] [CrossRef] [PubMed]
- Kaltak, M.; Blanco-Garavito, R.; Molday, L.L.; Dhaenens, C.M.; Souied, E.E.; Platenburg, G.; Swildens, J.; Molday, R.S.; Cremers, F.P.M. Stargardt disease-associated in-frame ABCA4 exon 17 skipping results in significant ABCA4 function. J. Transl. Med. 2023, 21, 546. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; Bhatt, S.S.; Janssen, I.M.; Xia, Z.; Lalani, S.R.; Pfundt, R.; Derwinska, K.; de Vries, B.B.; Gilissen, C.; Hoischen, A.; et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009, 18, 3579–3593. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Beck, C.R.; Du, R.; Campbell, I.M.; Coban-Akdemir, Z.; Gu, S.; Breman, A.M.; Stankiewicz, P.; Ira, G.; Shaw, C.A.; et al. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018, 28, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.J.; Lupski, J.R. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum. Genet. 2005, 116, 1–7. [Google Scholar] [CrossRef]
- de Smith, A.J.; Walters, R.G.; Coin, L.J.; Steinfeld, I.; Yakhini, Z.; Sladek, R.; Froguel, P.; Blakemore, A.I. Small deletion variants have stable breakpoints commonly associated with alu elements. PLoS ONE 2008, 3, e3104. [Google Scholar] [CrossRef] [PubMed]
- Falfoul, Y.; Habibi, I.; Turki, A.; Chebil, A.; Hassairi, A.; Schorderet, D.F.; El Matri, L. Phenotypic Progression of Stargardt Disease in a Large Consanguineous Tunisian Family Harboring New ABCA4 Mutations. J. Ophthalmol. 2018, 2018, 1030184. [Google Scholar] [CrossRef] [PubMed]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675.e19. [Google Scholar] [CrossRef]
- Travers, K.J.; Chin, C.S.; Rank, D.R.; Eid, J.S.; Turner, S.W. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010, 38, e159. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Ritz, A.; Bashir, A.; Sindi, S.; Hsu, D.; Hajirasouliha, I.; Raphael, B.J. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics 2014, 30, 3458–3466. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhang, Y.; Ying, C.; Wang, D.; Du, C. Nanopore-based fourth-generation DNA sequencing technology. Genom. Proteom. Bioinform. 2015, 13, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Cretu Stancu, M.; van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; de Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Espejo Valle-Inclan, J.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.T.; Hastie, A.; Lin, C.; Ehrlich, D.; Das, S.K.; Austin, M.D.; Deshpande, P.; Cao, H.; Nagarajan, N.; Xiao, M.; et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012, 30, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Levy-Sakin, M.; Pastor, S.; Mostovoy, Y.; Li, L.; Leung, A.K.Y.; McCaffrey, J.; Young, E.; Lam, E.T.; Hastie, A.R.; Wong, K.H.Y.; et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun. 2019, 10, 1025. [Google Scholar] [CrossRef] [PubMed]
- Hitti-Malin, R.J.; Dhaenens, C.-M.; Panneman, D.M.; Corradi, Z.; Khan, M.; den Hollander, A.I.; Farrar, G.J.; Gilissen, C.; Hoischen, A.; van de Vorst, M.; et al. Using single molecule Molecular Inversion Probes as a cost-effective, high-throughput sequencing approach to target all genes and loci associated with macular diseases. Hum. Mutat. 2022, 43, 2234–2250. [Google Scholar] [CrossRef] [PubMed]
- Panneman, D.M.; Hitti-Malin, R.J.; Holtes, L.K.; de Bruijn, S.E.; Reurink, J.; Boonen, E.G.M.; Khan, M.I.; Ali, M.; Andreasson, S.; De Baere, E.; et al. Cost-effective sequence analysis of 113 genes in 1,192 probands with retinitis pigmentosa and Leber congenital amaurosis. Front. Cell Dev. Biol. 2023, 11, 1112270. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Khan, M.I.; Elmelik, D.; Manders, E.; Bakker, S.; Derks, R.; Neveling, K.; van DeVorst, M.; Gilissen, C.; et al. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum. Mutat. 2019, 40, 1749–1759. [Google Scholar] [CrossRef]
- Ebert, P.; Audano, P.A.; Zhu, Q.; Rodriguez-Martin, B.; Porubsky, D.; Bonder, M.J.; Sulovari, A.; Ebler, J.; Zhou, W.; Serra Mari, R.; et al. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 2021, 372, eabf7117. [Google Scholar] [CrossRef]
- Babadi, M.; Fu, J.M.; Lee, S.K.; Smirnov, A.N.; Gauthier, L.D.; Walker, M.; Benjamin, D.I.; Zhao, X.; Karczewski, K.J.; Wong, I.; et al. GATK-gCNV enables the discovery of rare copy number variants from exome sequencing data. Nat. Genet. 2023, 55, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0. 1996–2010. Available online: https://www.repeatmasker.org/ (accessed on 27 March 2024).
- Jurka, J.; Klonowski, P.; Dagman, V.; Pelton, P. Censor—a program for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 1996, 20, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Bruce, K.H.; Mudunuri, U.S.; Yi, M.; Volfovsky, N.; Luke, B.T.; Bacolla, A.; Collins, J.R.; Stephens, R.M. Non-B DB: A database of predicted non-B DNA-forming motifs in mammalian genomes. Nucleic Acids Res. 2010, 39, D383–D391. [Google Scholar] [CrossRef] [PubMed]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef] [PubMed]
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| CNV | Type | Region | IN/OUT | # Alleles | cDNA Variant | Protein Variant | Genomic Variant (hg19) | Size (nt) |
|---|---|---|---|---|---|---|---|---|
| 1 | del | ex6_int6 | OUT | 2 | c.699_768+341del | p.(?) | g.94564009_94564419del | 411 |
| 2 | del | int9_int11 | IN | 1 | c.1239+303_1555-5571del | p.(Ala414_Glu518del) | g.94534444_94544575del | 10,132 |
| 3 | del | int14_ex16 | OUT | 1 | c.2160+531_2569del | p.(?) | g.94520685_94525562del | 4878 |
| 4a | del | int19_int22 | OUT | 1 | c.(2918+757_2918+981)_ | p.(Ser974Glnfs*64) | g.(94507378_94507602)_ | n/a |
| (3329-420_3329-644)del | (94511494_94511718)del | |||||||
| 4b | del | int19_int22 | OUT | 1 | c.2918+532_3329-622del | p.(Ser974Glnfs*64) | g.94507580_94511943del | 4363 |
| 5 | del | int23_ex50 | n/a | 1 | n/a | p.(?) | g.(?_94458389)_(94505684_94506764)del | n/a |
| 6 | del | int26_int30 | OUT | 1 | c.3863-553_4539+578del | p.(Gly1288Glufs*41) | g.94494423_94498152del | 3728 |
| 7 | del | int28_ex33 | OUT | 1 | c.4254-197_4672delinsGCTTTTT | p.(?) | g.94487503_94496279delinsAAAAAGC | 8779 |
| 8 | del | int29_int30 | OUT | 1 | c.4352+123_4540-585del | p.(Tyr1453Hisfs*11) | g.94491189_94495861del | 4396 |
| 9 | del | int30_int31 | OUT | 1 | c.4540-1000_4635-389delinsTGCCCG | p.(Arg1514Leufs*9) | g.94489363_94491604delisCGGGCA | 4591 |
| 10 | del | int30_int31 | OUT | 1 | c.4539+872_4635-565delins28 | p.(Arg1514Leufs*9) | g.94489539_94494129delins28 | 2242 |
| 11 | del | ex42_ex44 | OUT | 1 | c.5864_6085del | p.(?) | g.94471059_94473825del | 2469 |
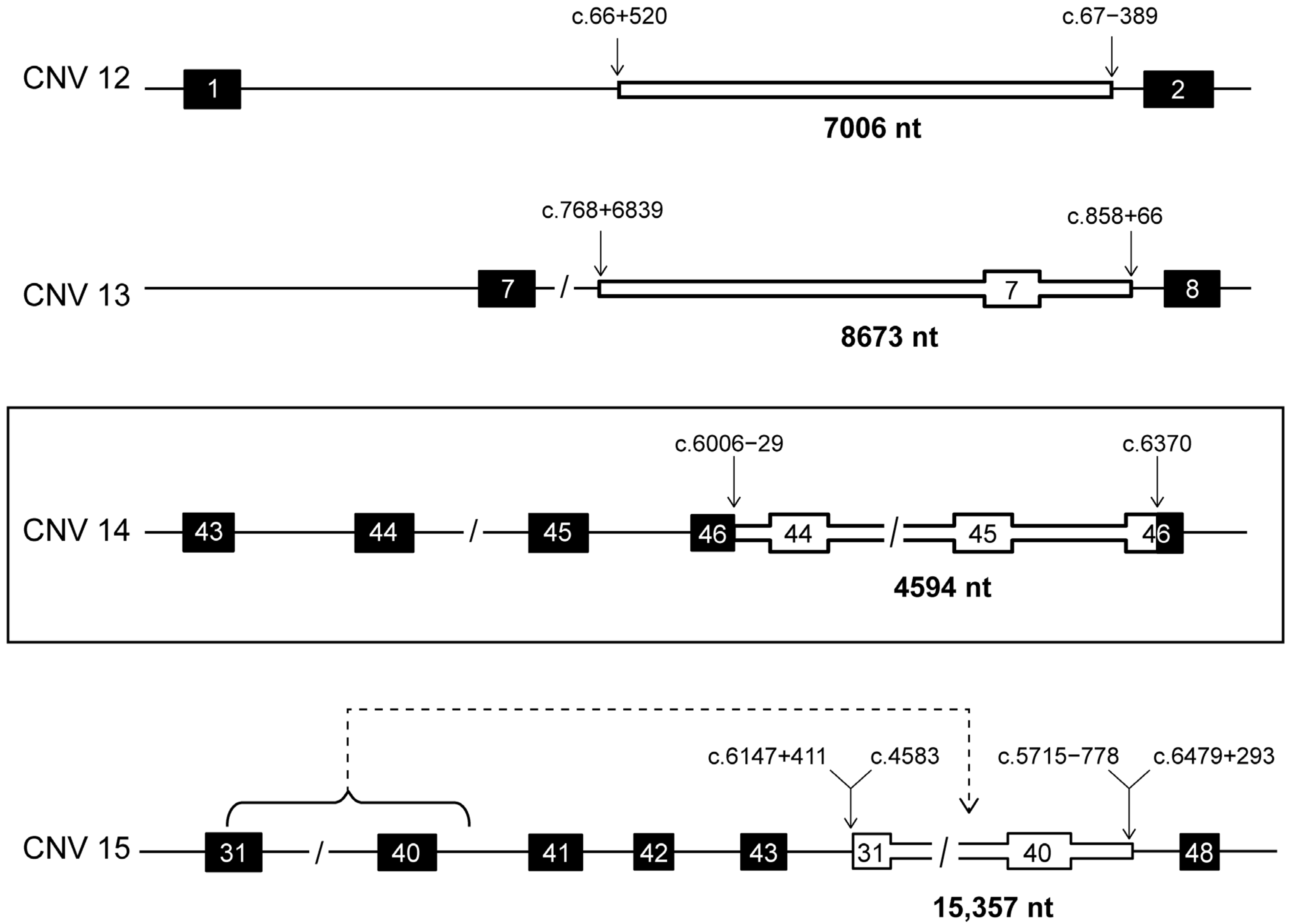
| 12 | dup | int1_int1 | IN | 3 | c.66+520_67-389dup | p.(?) | g.94579011_94586016dup | 7006 |
| 13 | dup | int6_int7 | IN | 1 | c.768+6839_858+66dup | p.(Leu257_Glu286dup) | g.94548842_94557511dup | 8670 |
| 14 | dup | int43_ex46 | OUT | 1 | c.6006-29_6370dup | p.(?) | g.94466574_94471167dup | 4594 |
| 15 | ins | ex31_int40/ | OUT | 2 | c.6147+411_6479+293delins4583_5715-778 | p.(?) | g.94466098_94470586delins | 15,357 |
| int44_int47 | 94475205_94490561 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corradi, Z.; Dhaenens, C.-M.; Grunewald, O.; Kocabaş, I.S.; Meunier, I.; Banfi, S.; Karali, M.; Cremers, F.P.M.; Hitti-Malin, R.J. Novel and Recurrent Copy Number Variants in ABCA4-Associated Retinopathy. Int. J. Mol. Sci. 2024, 25, 5940. https://doi.org/10.3390/ijms25115940
Corradi Z, Dhaenens C-M, Grunewald O, Kocabaş IS, Meunier I, Banfi S, Karali M, Cremers FPM, Hitti-Malin RJ. Novel and Recurrent Copy Number Variants in ABCA4-Associated Retinopathy. International Journal of Molecular Sciences. 2024; 25(11):5940. https://doi.org/10.3390/ijms25115940
Chicago/Turabian StyleCorradi, Zelia, Claire-Marie Dhaenens, Olivier Grunewald, Ipek Selen Kocabaş, Isabelle Meunier, Sandro Banfi, Marianthi Karali, Frans P. M. Cremers, and Rebekkah J. Hitti-Malin. 2024. "Novel and Recurrent Copy Number Variants in ABCA4-Associated Retinopathy" International Journal of Molecular Sciences 25, no. 11: 5940. https://doi.org/10.3390/ijms25115940
APA StyleCorradi, Z., Dhaenens, C.-M., Grunewald, O., Kocabaş, I. S., Meunier, I., Banfi, S., Karali, M., Cremers, F. P. M., & Hitti-Malin, R. J. (2024). Novel and Recurrent Copy Number Variants in ABCA4-Associated Retinopathy. International Journal of Molecular Sciences, 25(11), 5940. https://doi.org/10.3390/ijms25115940