
Deletion of Sarcolemmal Membrane-Associated Protein Isoform 3 (SLMAP3) in Cardiac Progenitors Delays Embryonic Growth of Myocardium without Affecting Hippo Pathway


Abstract
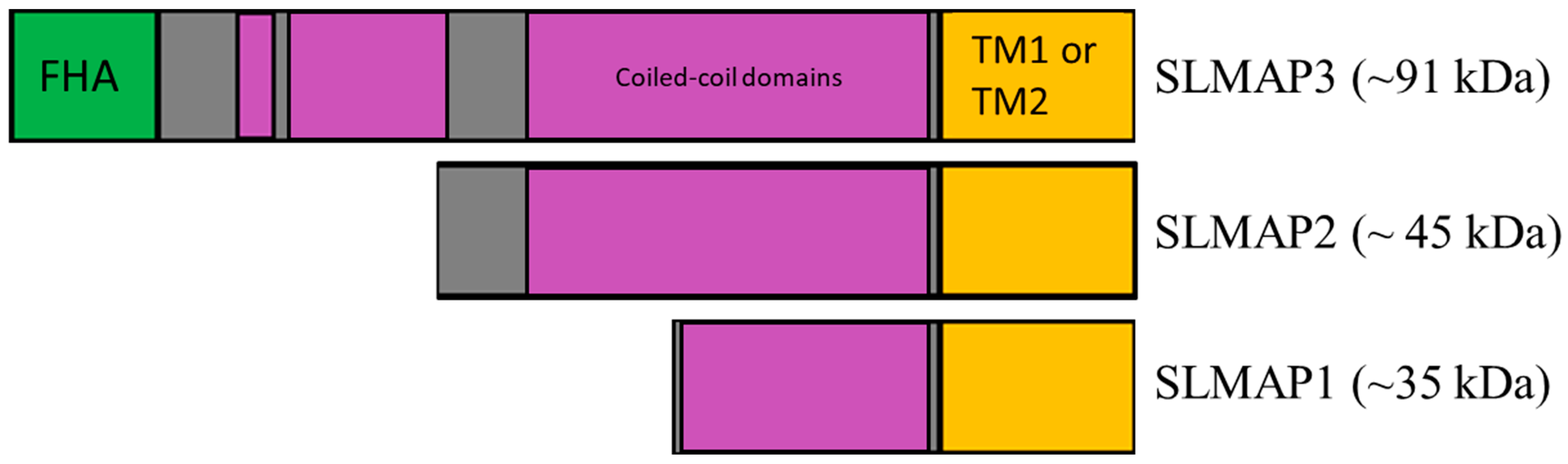
1. Introduction
2. Results
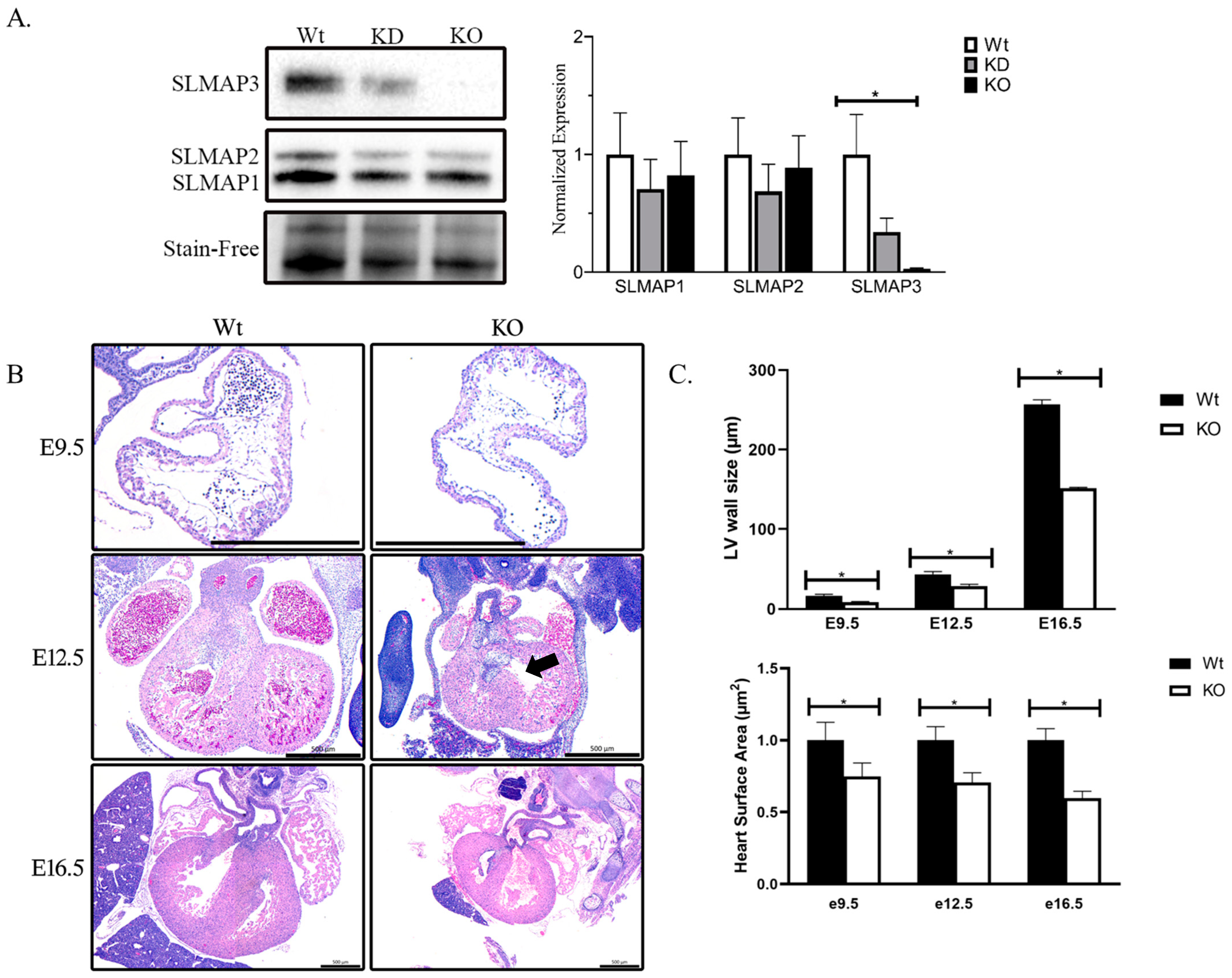
2.1. The Specific Loss of SLMAP3 Isoform in Cardiac Progenitors’ Delays Cardiogenesis
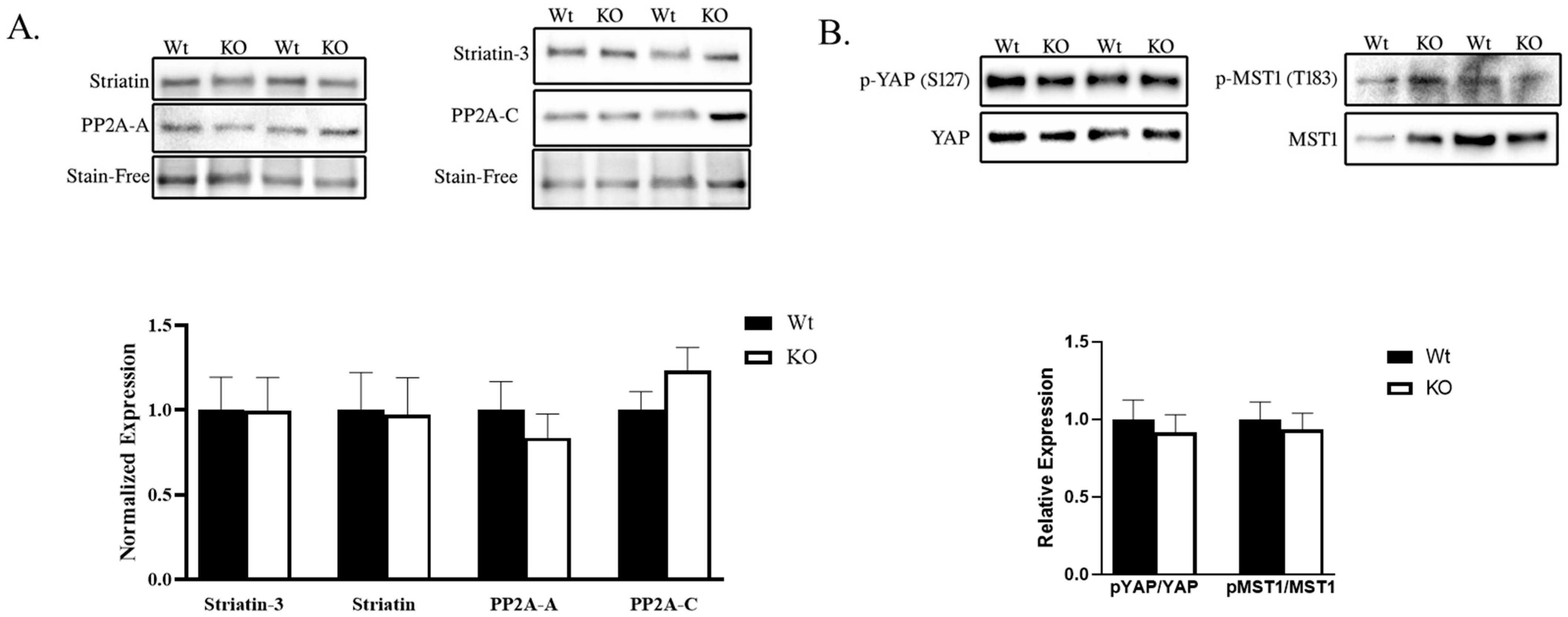
2.2. STRIPAK and Hippo Signaling Was Unaltered in SLMAP3-Deficient Embryonic Myocardium
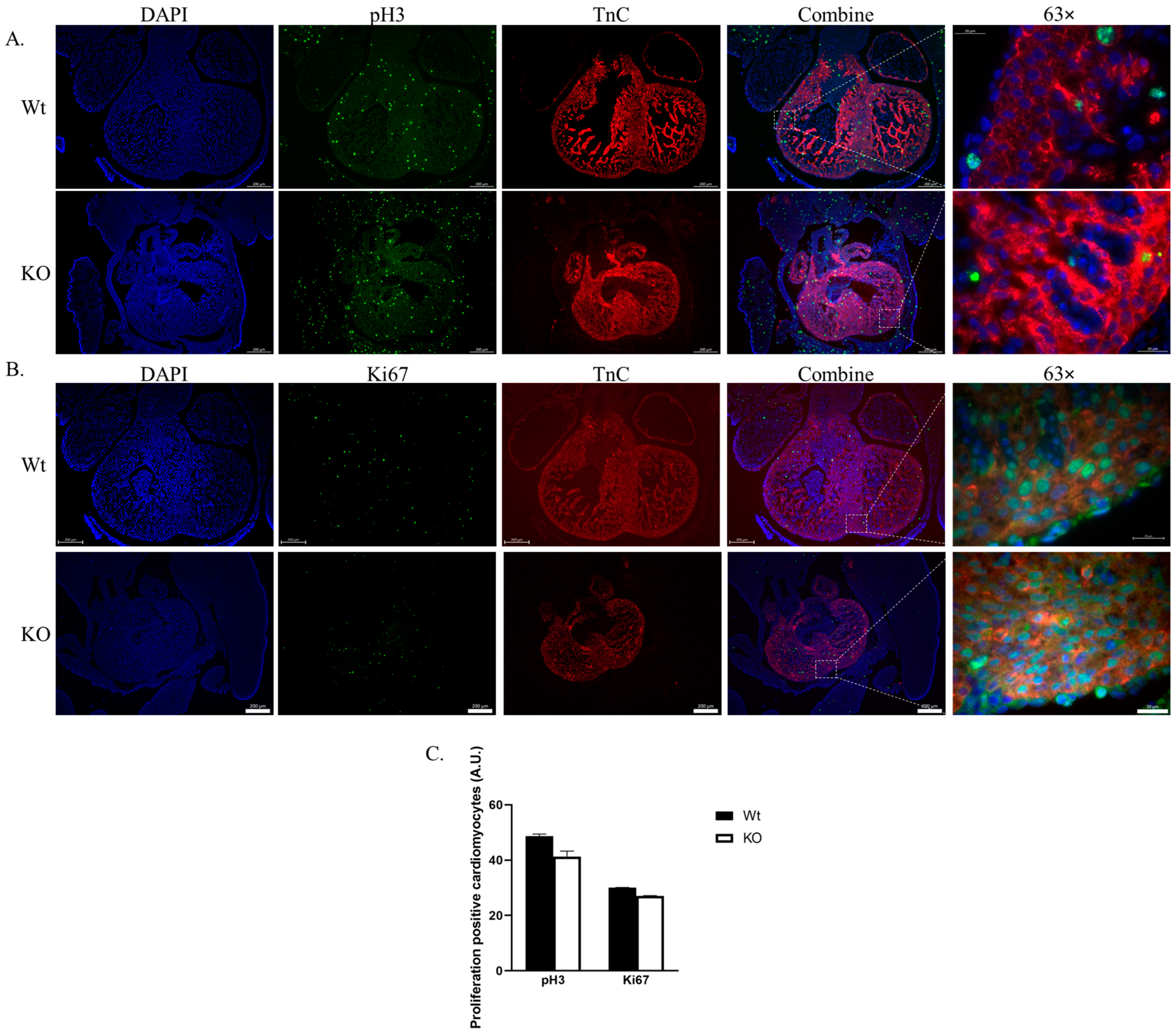
2.3. Cardiomyocyte Proliferation Is Not Affected by SLMAP3-KO during Cardiogenesis
2.4. Cardiomyocyte Size Is Reduced in SLMAP3-KO Embryonic Hearts
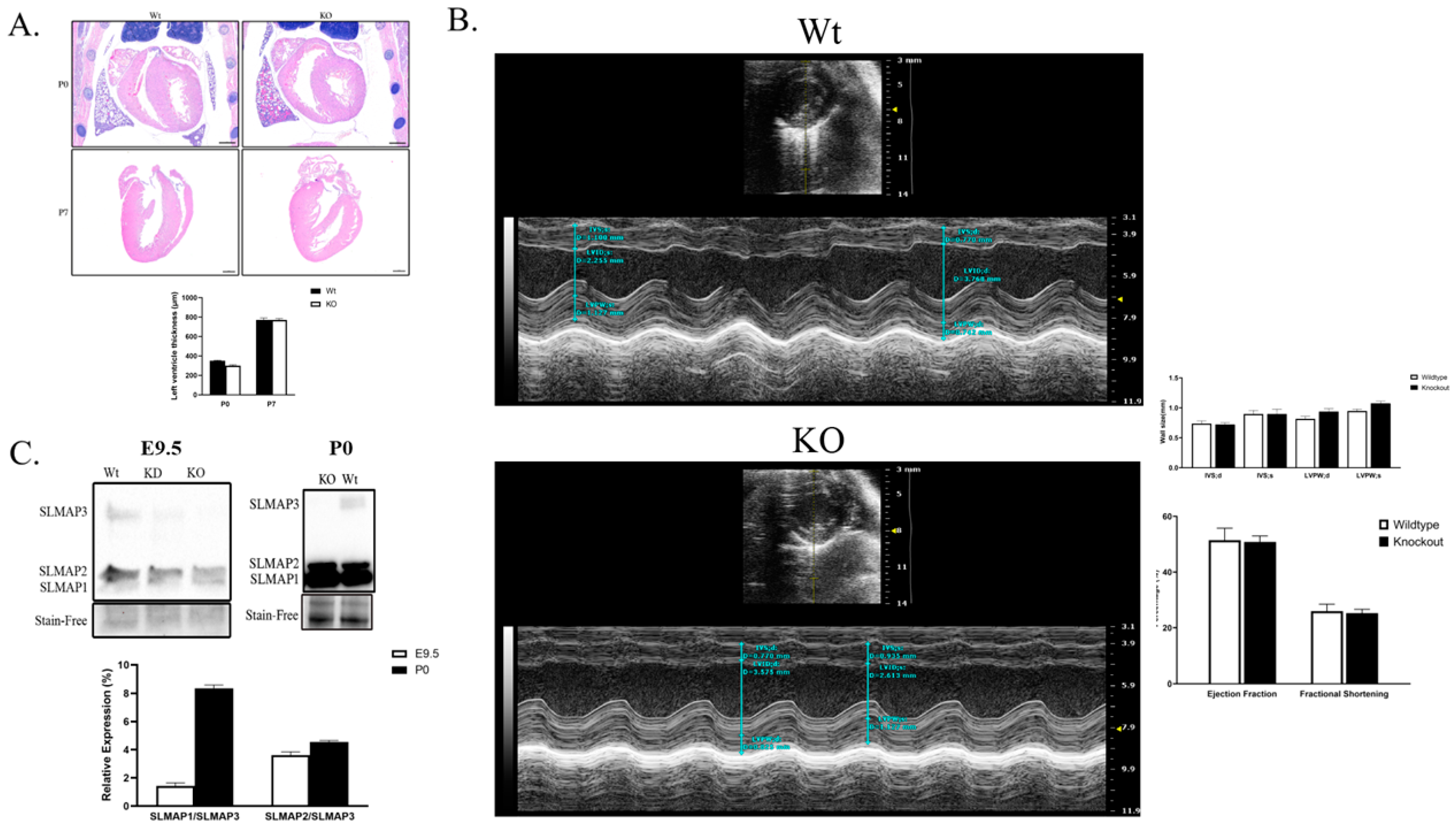
2.5. SLMAP3-KO Hearts Grow to Normal Size and Function in Postnatal Development
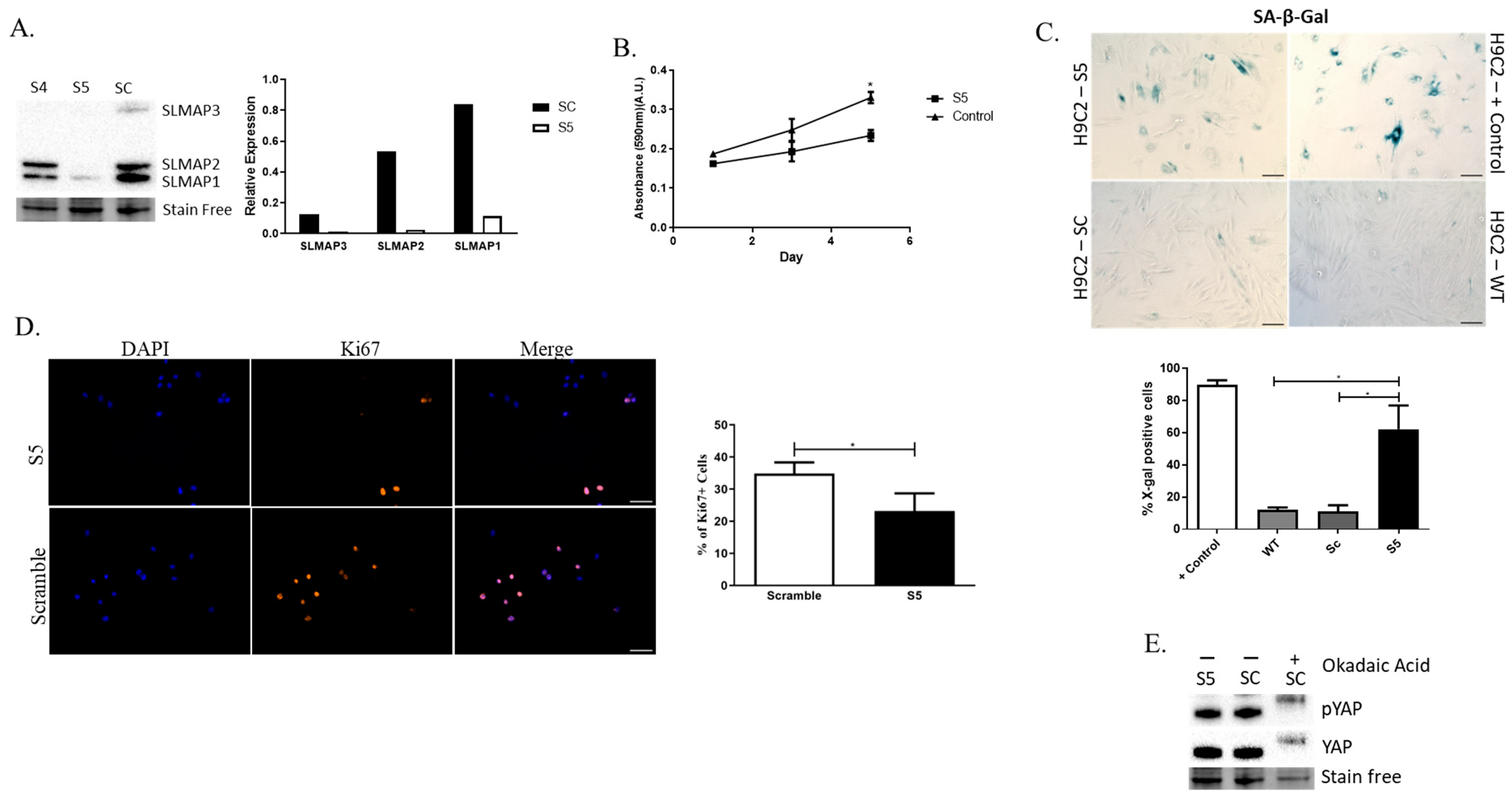
2.6. Depletion of SLMAP Isoforms in Rat Embryonic Cardiomyocytes (H9C2 Cells) Impacts Their Growth
3. Discussion
4. Materials and Methods
4.1. Generating and Genotyping the SLMAP Knockout Model
4.2. Protein Isolation and Western Blotting
4.3. Echocardiography
4.4. Histological and Immunofluorescent Analysis
4.5. Creation of Short-Hairpin RNA (sh-RNA) Lentivirus for Depletion of SLMAP3
4.6. Maintenance and Transduction of H9C2 Rat Cardiomyocyte Cell Line
4.7. Senescence Analysis on H9C2
4.8. H9C2 Growth Curve Analysis
4.9. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schleich, J.-M.; Abdulla, T.; Summers, R.; Houyel, L. An overview of cardiac morphogenesis. Arch. Cardiovasc. Dis. 2013, 106, 612–623. [Google Scholar] [CrossRef]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart Fields and Cardiac Morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4, a015750. [Google Scholar] [CrossRef]
- van den Berg, G.; Abu-Issa, R.; de Boer, B.A.; Hutson, M.R.; de Boer, P.A.; Soufan, A.T.; Ruijter, J.M.; Kirby, M.L.; van den Hoff, M.J.; Moorman, A.F. A caudal proliferating growth center contributes to both poles of the forming heart tube. Circ. Res. 2009, 104, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.; de Sena-Tomás, C.; George, V.; Werdich, A.A.; Kapur, S.; MacRae, C.A.; Targoff, K.L. Nkx genes establish second heart field cardiomyocyte progenitors at the arterial pole and pattern the venous pole through Isl1 repression. Development 2018, 145, dev161497. [Google Scholar]
- Hoff, M.J.v.D.; Kruithof, B.P.; Moorman, A.F.; Markwald, R.R.; Wessels, A. Formation of Myocardium after the Initial Development of the Linear Heart Tube. Dev. Biol. 2001, 240, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Yotti, R.; Seidman, C.E.; Seidman, J.G. Advances in the Genetic Basis and Pathogenesis of Sarcomere Cardiomyopathies. Annu. Rev. Genom. Hum. Genet. 2019, 20, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pu, W. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Wigle, J.; Demchyshyn, L.; Pratt, M.; Staines, W.; Salih, M.; Tuana, B. Molecular cloning, expression, and chromosomal assignment of sarcolemmal-associated proteins. A family of acidic amphipathic alpha-helical proteins associated with the membrane. J. Biol. Chem. 1997, 272, 32384–32394. [Google Scholar] [CrossRef]
- Guzzo, R.M.; Salih, M.; Moore, E.D.; Tuana, B.S. Molecular properties of cardiac tail-anchored membrane protein SLMAP are consistent with structural role in arrangement of excitation-contraction coupling apparatus. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1810–H1819. [Google Scholar] [CrossRef]
- Byers, J.T.; Guzzo, R.M.; Salih, M.; Tuana, B.S. Hydrophobic profiles of the tail anchors in SLMAP dictate subcellular targeting. BMC Cell Biol. 2009, 10, 48. [Google Scholar] [CrossRef]
- Guzzo, R.M.; Sevinc, S.; Salih, M.; Tuana, B.S. A novel isoform of sarcolemmal membrane-associated protein (SLMAP) is a component of the microtubule organizing centre. J. Cell Sci. 2004, 117, 2271–2281. [Google Scholar] [CrossRef]
- Ishikawa, T.; Sato, A.; Marcou, C.; Tester, D.; Ackerman, M.; Crotti, L.; Schwartz, P.; On, Y.; Park, J.; Nakamura, K.; et al. A novel disease gene for Brugada syndrome: Sarcolemmal membrane-associated protein gene mutations impair intracellular trafficking of hNav1.5. Circ. Arrhythm. Electrophysiol. 2012, 5, 1098–1107. [Google Scholar] [CrossRef]
- Nader, M.; Westendorp, B.; Hawari, O.; Salih, M.; Stewart, A.F.R.; Leenen, F.H.H.; Tuana, B.S. Tail-anchored membrane protein SLMAP is a novel regulator of cardiac function at the sarcoplasmic reticulum. Am. J. Physiol. Circ. Physiol. 2012, 302, H1138–H1145. [Google Scholar] [CrossRef] [PubMed]
- Mlynarova, J.; Trentin-Sonoda, M.; da Silva, F.G.; Major, J.L.; Salih, M.; Carneiro-Ramos, M.S.; Tuana, B.S. SLMAP3 isoform modulates cardiac gene expression and function. PLoS ONE 2019, 14, e0214669. [Google Scholar] [CrossRef] [PubMed]
- Wielowieyski, P.; Sevinc, S.; Guzzo, R.; Salih, M.; Wigle, J.; Tuana, B. Alternative splicing, expression, and genomic structure of the 3’ region of the gene encoding the sarcolemmal-associated proteins (SLAPs) defines a novel class of coiled-coil tail-anchored membrane proteins. J. Biol. Chem. 2000, 275, 38474–38481. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Guzzo, R.M.; Wigle, J.; Salih, M.; Moore, E.D.; Tuana, B.S. Regulated expression and temporal induction of the tail-anchored sarcolemmal-membrane-associated protein is critical for myoblast fusion. Biochem. J. 2004, 381, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Yuan, C.; Lee, H.; Chen, E.S.-W.; Wu, P.-Y.; Tsai, M.-D. Structure and Function of the Phosphothreonine-Specific FHA Domain. Sci. Signal. 2008, 1, re12. [Google Scholar] [CrossRef]
- Yu, F.; Zhao, B.; Guan, K. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Goudreault, M.; D’Ambrosio, L.M.; Kean, M.J.; Mullin, M.J.; Larsen, B.G.; Sanchez, A.; Chaudhry, S.; Chen, G.I.; Sicheri, F.; Nesvizhskii, A.I.; et al. A PP2A Phosphatase High Density Interaction Network Identifies a Novel Striatin-interacting Phosphatase and Kinase Complex Linked to the Cerebral Cavernous Malformation 3 (CCM3) Protein. Mol. Cell. Proteom. 2009, 8, 157–171. [Google Scholar] [CrossRef]
- Jeong, B.-C.; Bae, S.J.; Ni, L.; Zhang, X.; Bai, X.-C.; Luo, X. Cryo-EM structure of the Hippo signaling integrator human STRIPAK. Nat. Struct. Mol. Biol. 2021, 28, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Couzens, A.L.; Knight, J.D.R.; Kean, M.J.; Teo, G.; Weiss, A.; Dunham, W.H.; Lin, Z.-Y.; Bagshaw, R.D.; Sicheri, F.; Pawson, T.; et al. Protein Interaction Network of the Mammalian Hippo Pathway Reveals Mechanisms of Kinase-Phosphatase Interactions. Sci. Signal. 2013, 6, rs15. [Google Scholar] [CrossRef] [PubMed]
- von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N.; Ma, Q.; Ishiwata, T.; Zhou, B.; Camargo, F.D.; et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2394–2399. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of Insulin-Like Growth Factor Signaling by Yap Governs Cardiomyocyte Proliferation and Embryonic Heart Size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, B.; Wang, L.; Lei, H.; Prieto, K.D.P.; Pan, D. Homeostatic Control of Hpo/MST Kinase Activity through Autophosphorylation-Dependent Recruitment of the STRIPAK PP2A Phosphatase Complex. Cell Rep. 2017, 21, 3612–3623. [Google Scholar] [CrossRef]
- Stanley, E.G.; Biben, C.; Elefanty, A.; Barnett, L.; Koentgen, F.; Robb, L.; Harvey, R.P. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3’UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int. J. Dev. Biol. 2002, 46, 431–439. [Google Scholar]
- Bae, S.; Ni, L.; Osinski, A.; Tomchick, D.; Brautigam, C.; Luo, X. SAV1 promotes Hippo kinase activation through antagonizing the PP2A phosphatase STRIPAK. eLife 2017, 6, 30278. [Google Scholar] [CrossRef]
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.-L. Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Matsui, T. Phosphoinositide-3 Kinase Signaling in Cardiac Hypertrophy and Heart Failure. Curr. Pharm. Des. 2011, 17, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Rehmani, T.; Mlynarova, J.; Byers, J.; Salih, M.; Tuana, B.S. Specific Deletion of the FHA Domain Containing SLMAP3 Isoform in Postnatal Myocardium Has No Impact on Structure or Function. Cardiogenetics 2021, 11, 164–184. [Google Scholar] [CrossRef]
- George, V.; Colombo, S.; Targoff, K.L. An early requirement for nkx2.5 ensures the first and second heart field ventricular identity and cardiac function into adulthood. Dev. Biol. 2015, 400, 10–22. [Google Scholar] [CrossRef]
- Cambier, L.; Plate, M.; Sucov, H.M.; Pashmforoush, M. Nkx2-5 regulates cardiac growth through modulation of Wnt signaling by R-spondin3. Development 2014, 141, 2959–2971. [Google Scholar] [CrossRef]
- Becker, R.; Leone, M.; Engel, F.B. Microtubule Organization in Striated Muscle Cells. Cells 2020, 9, 1395. [Google Scholar] [CrossRef]
- Available online: https://www.novusbio.com/support-by-application/Fluorescent-IHC-Staining-of-FFPE-Tissue (accessed on 1 November 2019).
- Available online: https://www.genscript.com/tools/create-scrambled-sequence-reference (accessed on 31 October 2022).
- Available online: https://en.vectorbuilder.com/design/pLV_shRNA_U6.html-reference (accessed on 1 November 2022).
- Available online: https://www.addgene.org/12260/ (accessed on 1 November 2022).
- Available online: https://www.addgene.org/12259/ (accessed on 1 November 2022).
- Available online: https://www.addgene.org/protocols/lentivirus-production/ (accessed on 1 November 2022).
- Available online: https://www.addgene.org/protocols/generating-stable-cell-lines/-reference (accessed on 1 November 2022).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Manufacturer | Dilution |
|---|---|---|
| SLMAP | Novus Biologicals (NBP1-81397) | 1:1000 (WB) |
| Striatin-1 | BD Transduction Laboratories (610838) | 1:1000 (WB) |
| Striatin-3 | Novus Biological (NBP-74572) | 1:1000 (WB) |
| STRIP1 | Bethyl Laboratories (A304-644A) | 1:1000 (WB) |
| PP2A-C α/β subunit | Santa Cruz Biotechnology (sc-80665) | 1:200 (WB) |
| PP2A-A α subunit | Millipore Sigma (07-250) | 1:600 (WB) |
| Phospho-YAP (S127) | Cell Signaling Technology (13008S) | 1:1000 (WB) |
| YAP | Cell Signaling Technology (14074S) | 1:1000 (WB) |
| pH3 | Abcam (ab5176) | 1:200 (IF) |
| Ki67 | Abcam (ab15580) | 1:200 (IF) |
| Troponin C (TnC) | Santa Cruz (sc-52265) | 1:200 (IF) |
| pAKT1 | Cell Signaling Technology (9018S) | 1:1000 (WB) |
| AKT1 | Cell Signaling Technology (9272) | 1:1000 (WB) |
| pMTOR1 | Cell Signaling Technology (5536) | 1:1000 (WB) |
| MTOR1 | Cell Signaling Technology (2972) | 1:1000 (WB) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehmani, T.; Dias, A.P.; Kamal, M.; Salih, M.; Tuana, B.S. Deletion of Sarcolemmal Membrane-Associated Protein Isoform 3 (SLMAP3) in Cardiac Progenitors Delays Embryonic Growth of Myocardium without Affecting Hippo Pathway. Int. J. Mol. Sci. 2024, 25, 2888. https://doi.org/10.3390/ijms25052888
Rehmani T, Dias AP, Kamal M, Salih M, Tuana BS. Deletion of Sarcolemmal Membrane-Associated Protein Isoform 3 (SLMAP3) in Cardiac Progenitors Delays Embryonic Growth of Myocardium without Affecting Hippo Pathway. International Journal of Molecular Sciences. 2024; 25(5):2888. https://doi.org/10.3390/ijms25052888
Chicago/Turabian StyleRehmani, Taha, Ana Paula Dias, Marsel Kamal, Maysoon Salih, and Balwant S. Tuana. 2024. "Deletion of Sarcolemmal Membrane-Associated Protein Isoform 3 (SLMAP3) in Cardiac Progenitors Delays Embryonic Growth of Myocardium without Affecting Hippo Pathway" International Journal of Molecular Sciences 25, no. 5: 2888. https://doi.org/10.3390/ijms25052888
APA StyleRehmani, T., Dias, A. P., Kamal, M., Salih, M., & Tuana, B. S. (2024). Deletion of Sarcolemmal Membrane-Associated Protein Isoform 3 (SLMAP3) in Cardiac Progenitors Delays Embryonic Growth of Myocardium without Affecting Hippo Pathway. International Journal of Molecular Sciences, 25(5), 2888. https://doi.org/10.3390/ijms25052888