
‘Getting Better’—Is It a Feasible Strategy of Broad Pan-Antiherpesviral Drug Targeting by Using the Nuclear Egress-Directed Mechanism?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Multifaceted Aspects of NEC-Directed Antiviral Drug Targeting—An Updating Review
2.1. Investigational Tools That Utilize the Conditional Expression of Viral NEC Proteins
2.1.1. Illustration of the Rate-Limiting Step of Herpesviral Replication by the Generation of Mutant NEC Versions
2.1.2. The ‘Shared-Hook’ Strategy: Addressing Mutant-Specific Cross-Viral NEC Interaction Patterns to Prepare a Platform for Broadly Active NEC Inhibitors
2.2. Antiviral Block of Nuclear Egress Functions by the Intracellular Overexpression of NEC-Inhibitory Hook Fragments
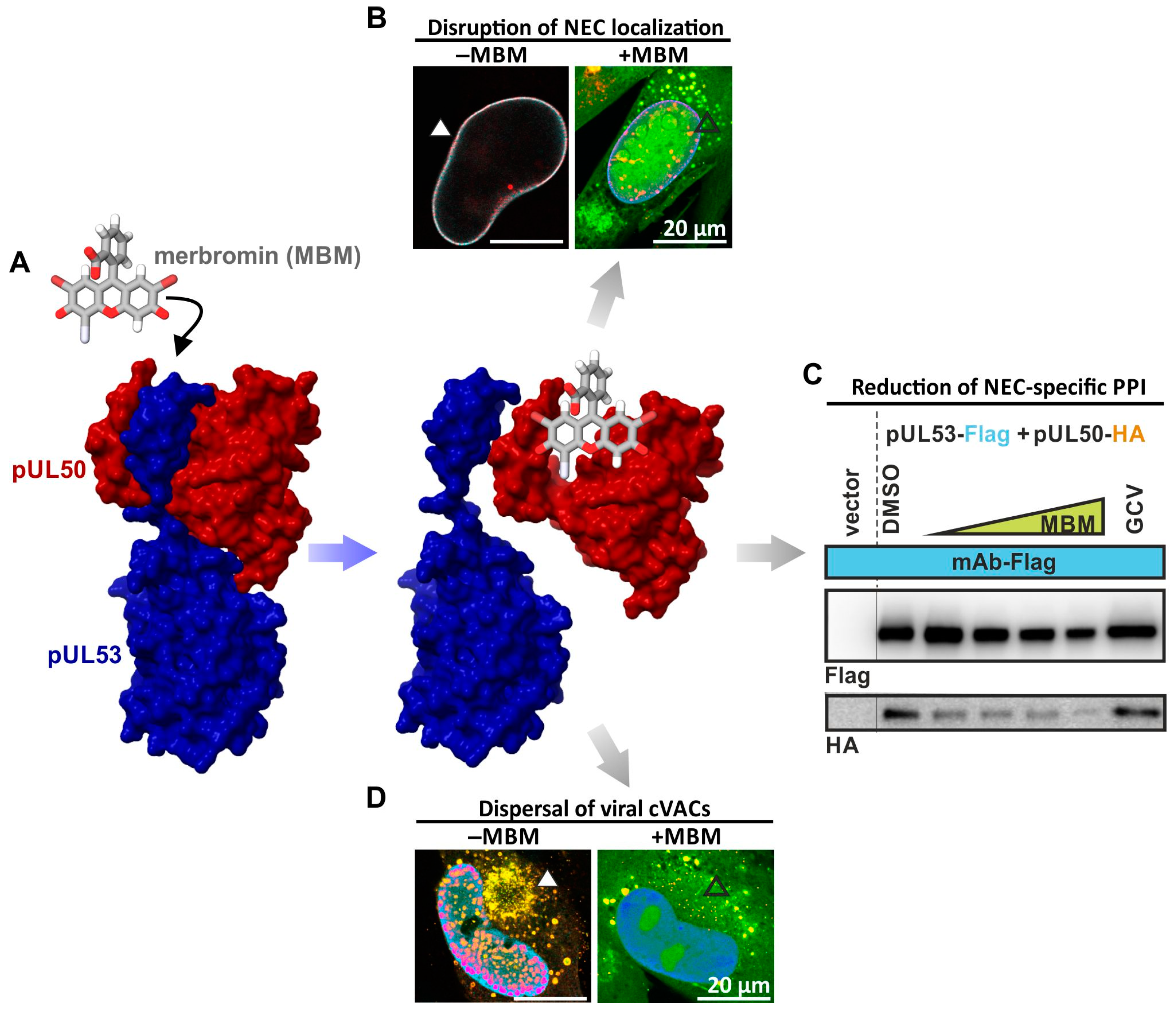
2.3. Identification of First NEC-Directed Small Molecules Comprising a Pronounced Level of Antiviral Activity
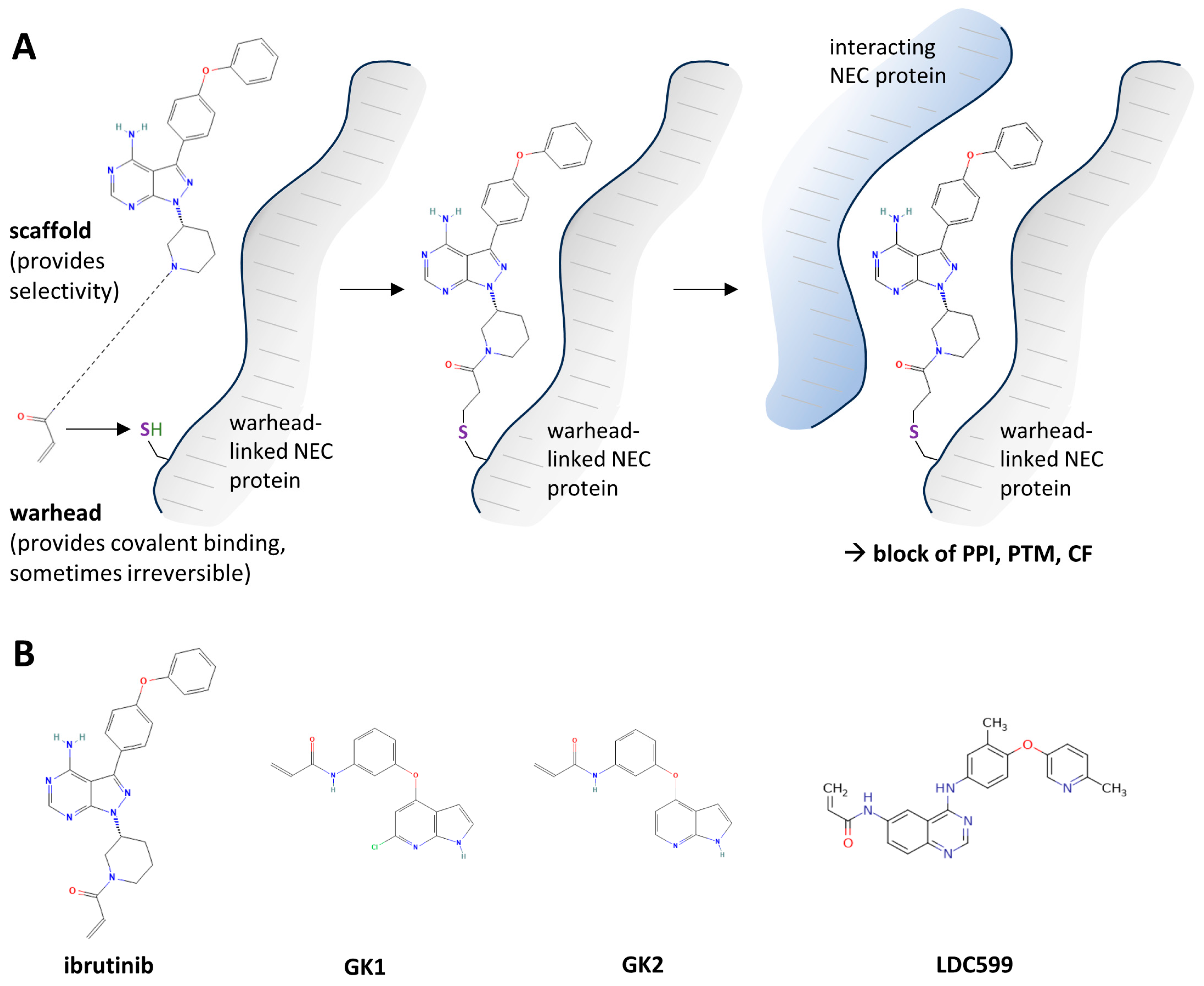
2.4. Covalently Target-Binding Warheads Can Possess an NEC-Directed Antiviral Potency with Amenability for Further Drug Optimization
2.5. Design and Application of Synthetic NEC-Mimetic Cell-Penetrating Peptides (CPPs)
2.6. Kinase Inhibitors Directed to Important NEC-Associated CDK and vCDK Activities
2.7. Further Mechanistic Modes to Interfere with Herpesviral Nuclear Egress
3. Conclusions: Current State of the Art with Various NEC-Directed Investigational Options of Antiviral Interference
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Roizman, B.; Baines, J. The diversity and unity of Herpesviridae. Comp. Immunol. Microbiol. Infect. Dis. 1991, 14, 63–79. [Google Scholar] [CrossRef]
- Boehmer, P.E.; Nimonkar, A.V. Herpes virus replication. IUBMB Life 2003, 55, 13–22. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpesviruses. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston TX, USA, 1996. [Google Scholar]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 2006, 296, 964–973. [Google Scholar] [CrossRef]
- Davison, A.J. Herpesvirus systematics. Vet. Microbiol. 2010, 143, 52–69. [Google Scholar] [CrossRef]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Dai, Y.C.; Liao, Y.T.; Juan, Y.T.; Cheng, Y.Y.; Su, M.T.; Su, Y.Z.; Liu, H.C.; Tsai, C.H.; Lee, C.P.; Chen, M.R. The novel nuclear targeting and BFRF1-interacting domains of BFLF2 are essential for efficient Epstein-Barr virus virion release. J. Virol. 2020, 94, e01498-19. [Google Scholar] [CrossRef]
- Lee, C.P.; Liu, G.T.; Kung, H.N.; Liu, P.T.; Liao, Y.T.; Chow, L.P.; Chang, L.S.; Chang, Y.H.; Chang, C.W.; Shu, W.C.; et al. The Ubiquitin Ligase Itch and Ubiquitination Regulate BFRF1-Mediated Nuclear Envelope Modification for Epstein-Barr Virus Maturation. J. Virol. 2016, 90, 8994–9007. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.T.; Kung, H.N.; Chen, C.K.; Huang, C.; Wang, Y.L.; Yu, C.P.; Lee, C.P. Improving nuclear envelope dynamics by EBV BFRF1 facilitates intranuclear component clearance through autophagy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 3968–3983. [Google Scholar] [CrossRef] [PubMed]
- Luitweiler, E.M.; Henson, B.W.; Pryce, E.N.; Patel, V.; Coombs, G.; McCaffery, J.M.; Desai, P.J. Interactions of the Kaposi’s Sarcoma-associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J. Virol. 2013, 87, 3915–3929. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Microbiol. 2014, 12, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.; Oxman, M.N.; et al. Varicella zoster virus infection. Nat. Rev. Dis. Prim. 2015, 1, 15016. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Shiohara, T. Current understanding of cytomegalovirus infection in immunocompetent individuals. J. Dermatol. Sci. 2000, 22, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Lancini, D.; Faddy, H.M.; Flower, R.; Hogan, C. Cytomegalovirus disease in immunocompetent adults. Med. J. Aust. 2014, 201, 578–580. [Google Scholar] [CrossRef]
- Sissons, J.G.; Carmichael, A.J. Clinical aspects and management of cytomegalovirus infection. J. Infect. 2002, 44, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Fishman, J.A. Infection in solid-organ transplant recipients. N. Engl. J. Med. 2007, 357, 2601–2614. [Google Scholar] [CrossRef] [PubMed]
- Gianella, S.; Letendre, S. Cytomegalovirus and HIV: A Dangerous Pas de Deux. J. Infect. Dis. 2016, 214 (Suppl. S2), S67–S74. [Google Scholar] [CrossRef]
- Revello, M.G.; Gerna, G. Diagnosis and management of human cytomegalovirus infection in the mother, fetus, and newborn infant. Clin. Microbiol. Rev. 2002, 15, 680–715. [Google Scholar] [CrossRef]
- Tsutsui, Y. Effects of cytomegalovirus infection on embryogenesis and brain development. Congenit. Anom. 2009, 49, 47–55. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- Steingruber, M.; Marschall, M. The cytomegalovirus protein kinase pUL97:host interactions, regulatory mechanisms and antiviral drug targeting. Microorganisms 2020, 8, 515. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, K.; Uchida, A.; Imafuku, H.; Tairaku, S.; Fujioka, K.; Morioka, I.; Yamada, H. The Current Challenges in Developing Biological and Clinical Predictors of Congenital Cytomegalovirus Infection. Int. J. Mol. Sci. 2021, 22, 13487. [Google Scholar] [CrossRef]
- Jha, H.C.; Pei, Y.; Robertson, E.S. Epstein-Barr Virus: Diseases Linked to Infection and Transformation. Front. Microbiol. 2016, 7, 1602. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Curran, M.; Noble, S. Valganciclovir. Drugs 2001, 61, 1145–1150; discussion 1151–1142. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, V.; Talarico, C.L.; Stanat, S.C.; Davis, M.; Coen, D.M.; Biron, K.K. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 1992, 358, 162–164. [Google Scholar] [CrossRef]
- De Clercq, E.; Holý, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef]
- Hakki, M. Moving Past Ganciclovir and Foscarnet: Advances in CMV Therapy. Curr. Hematol. Malig. Rep. 2020, 15, 90–102. [Google Scholar] [CrossRef]
- Biron, K.K. Antiviral drugs for cytomegalovirus diseases. Antivir. Res. 2006, 71, 154–163. [Google Scholar] [CrossRef]
- Harter, G.; Michel, D. Antiviral treatment of cytomegalovirus infection: An update. Expert Opin. Pharmacother. 2012, 13, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Lischka, P.; Zimmermann, H. Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Curr. Opin. Pharmacol. 2008, 8, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Gentry, B.G.; Bogner, E.; Drach, J.C. Targeting the terminase: An important step forward in the treatment and prophylaxis of human cytomegalovirus infections. Antivir. Res. 2019, 161, 116–124. [Google Scholar] [CrossRef]
- Jakharia, N.; Howard, D.; Riedel, D.J. CMV Infection in Hematopoietic Stem Cell Transplantation: Prevention and Treatment Strategies. Curr. Treat. Options Infect. Dis. 2021, 13, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Lischka, P.; Hewlett, G.; Wunberg, T.; Baumeister, J.; Paulsen, D.; Goldner, T.; Ruebsamen-Schaeff, H.; Zimmermann, H. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 2010, 54, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Song, K.; Wu, J.; Bo, T.; Crumpacker, C. Drug Resistance Mutations and Associated Phenotypes Detected in Clinical Trials of Maribavir for Treatment of Cytomegalovirus Infection. J. Infect. Dis. 2022, 226, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.J.; Arthurs, S.K.; Deziel, P.J.; Wilhelm, M.P.; Razonable, R.R. Emergence of drug-resistant cytomegalovirus in the era of valganciclovir prophylaxis: Therapeutic implications and outcomes. Clin. Transplant. 2008, 22, 162–170. [Google Scholar] [CrossRef]
- Razonable, R.R. Drug-resistant cytomegalovirus: Clinical implications of specific mutations. Curr. Opin. Organ Transplant. 2018, 23, 388–394. [Google Scholar] [CrossRef]
- Roller, R.J.; Baines, J.D. Herpesvirus Nuclear Egress. Adv. Anat. Embryol. Cell Biol. 2017, 223, 143–169. [Google Scholar] [CrossRef]
- Avery, R.K.; Alain, S.; Alexander, B.D.; Blumberg, E.A.; Chemaly, R.F.; Cordonnier, C.; Duarte, R.F.; Florescu, D.F.; Kamar, N.; Kumar, D.; et al. Maribavir for Refractory Cytomegalovirus Infections With or Without Resistance Post-Transplant: Results From a Phase 3 Randomized Clinical Trial. Clin. Infect. Dis. 2022, 75, 690–701. [Google Scholar] [CrossRef]
- Wild, M.; Kicuntod, J.; Seyler, L.; Wangen, C.; Bertzbach, L.D.; Conradie, A.M.; Kaufer, B.B.; Wagner, S.; Michel, D.; Eickhoff, J.; et al. Combinatorial Drug Treatments Reveal Promising Anticytomegaloviral Profiles for Clinically Relevant Pharmaceutical Kinase Inhibitors (PKIs). Int. J. Mol. Sci. 2021, 22, 575. [Google Scholar] [CrossRef]
- Hamilton, S.T.; Marschall, M.; Rawlinson, W.D. Investigational Antiviral Therapy Models for the Prevention and Treatment of Congenital Cytomegalovirus Infection during Pregnancy. Antimicrob. Agents Chemother. 2020, 65, 10–1128. [Google Scholar] [CrossRef]
- Gourin, C.; Alain, S.; Hantz, S. Anti-CMV therapy, what next? A systematic review. Front. Microbiol. 2023, 14, 1321116. [Google Scholar] [CrossRef] [PubMed]
- Häge, S.; Marschall, M. ‘Come together’-The Regulatory Interaction of Herpesviral Nuclear Egress Proteins Comprises Both Essential and Accessory Functions. Cells 2022, 11, 1837. [Google Scholar] [CrossRef] [PubMed]
- Draganova, E.B.; Thorsen, M.K.; Heldwein, E.E. Nuclear Egress. Curr. Issues Mol. Biol. 2021, 41, 125–170. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.T.; Pellet, P.E. The Family Herpesviridae: A Brief Introduction. In Fields Virology, 7th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolter Kluwer: Warsaw, Poland, 2021; Volume 2, pp. 212–234. [Google Scholar]
- Marschall, M.; Häge, S.; Conrad, M.; Alkhashrom, S.; Kicuntod, J.; Schweininger, J.; Kriegel, M.; Lösing, J.; Tillmanns, J.; Neipel, F.; et al. Nuclear egress complexes of HCMV and other herpesviruses: Solving the puzzle of sequence coevolution, conserved structures and subfamily-spanning binding properties. Viruses 2020, 12, 683. [Google Scholar] [CrossRef] [PubMed]
- Panté, N.; Kann, M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol. Biol. Cell 2002, 13, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Muller, Y.A.; Diewald, B.; Sticht, H.; Milbradt, J. The human cytomegalovirus nuclear egress complex unites multiple functions: Recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev. Med. Virol. 2017, 27, e1934. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Hutterer, C.; Bahsi, H.; Wagner, S.; Sonntag, E.; Horn, A.H.; Kaufer, B.B.; Mori, Y.; Sticht, H.; Fossen, T.; et al. The Prolyl Isomerase Pin1 Promotes the Herpesvirus-Induced Phosphorylation-Dependent Disassembly of the Nuclear Lamina Required for Nucleocytoplasmic Egress. PLoS Pathog. 2016, 12, e1005825. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel mode of phosphorylation-triggered reorganization of the nuclear lamina during nuclear egress of human cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, V.; Britt, W. Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions. Viruses 2021, 14, 15. [Google Scholar] [CrossRef]
- Häge, S.; Sonntag, E.; Svrlanska, A.; Borst, E.M.; Stilp, A.C.; Horsch, D.; Müller, R.; Kropff, B.; Milbradt, J.; Stamminger, T.; et al. Phenotypical Characterization of the Nuclear Egress of Recombinant Cytomegaloviruses Reveals Defective Replication upon ORF-UL50 Deletion but Not pUL50 Phosphosite Mutation. Viruses 2021, 13, 165. [Google Scholar] [CrossRef]
- Milbradt, J.; Sonntag, E.; Wagner, S.; Strojan, H.; Wangen, C.; Lenac Rovis, T.; Lisnic, B.; Jonjic, S.; Sticht, H.; Britt, W.J.; et al. Human cytomegalovirus nuclear capsids associate with the core nuclear egress complex and the viral protein kinase pUL97. Viruses 2018, 10, 35. [Google Scholar] [CrossRef]
- Kuny, C.V.; Chinchilla, K.; Culbertson, M.R.; Kalejta, R.F. Cyclin-dependent kinase-like function is shared by the beta- and gamma- subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010, 6, e1001092. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kamil, J.P.; Coughlin, M.; Reim, N.I.; Coen, D.M. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J. Virol. 2014, 88, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Marzi, A.; aus dem Siepen, P.; Jochmann, R.; Kalmer, M.; Auerochs, S.; Lischka, P.; Leis, M.; Stamminger, T. Cellular p32 recruits cytomegalovirus kinase pUL97 to redistribute the nuclear lamina. J. Biol. Chem. 2005, 280, 33357–33367. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Auerochs, S.; Marschall, M. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 2007, 88, 2642–2650. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Auerochs, S.; Sticht, H.; Marschall, M. Cytomegaloviral proteins that associate with the nuclear lamina: Components of a postulated nuclear egress complex. J. Gen. Virol. 2009, 90, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Kraut, A.; Hutterer, C.; Sonntag, E.; Schmeiser, C.; Ferro, M.; Wagner, S.; Lenac, T.; Claus, C.; Pinkert, S.; et al. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol. Cell. Proteom. 2014, 13, 2132–2146. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, J.M.; Heldwein, E.E. Have NEC Coat, Will Travel: Structural Basis of Membrane Budding During Nuclear Egress in Herpesviruses. Adv. Virus. Res. 2017, 97, 107–141. [Google Scholar] [CrossRef]
- Lv, Y.; Shen, S.; Xiang, L.; Jia, X.; Hou, Y.; Wang, D.; Deng, H. Functional Identification and Characterization of the Nuclear Egress Complex of a Gammaherpesvirus. J. Virol. 2019, 93, e01422-19. [Google Scholar] [CrossRef]
- Lye, M.F.; Wilkie, A.R.; Filman, D.J.; Hogle, J.M.; Coen, D.M. Getting to and through the inner nuclear membrane during herpesvirus nuclear egress. Curr. Opin. Cell Biol. 2017, 46, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.A.; Hage, S.; Alkhashrom, S.; Hollriegl, T.; Weigert, S.; Dolles, S.; Hof, K.; Walzer, S.A.; Egerer-Sieber, C.; Conrad, M.; et al. High-resolution crystal structures of two prototypical beta- and gamma-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J. Biol. Chem. 2020, 295, 3189–3201. [Google Scholar] [CrossRef]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef]
- Lösing, J.; Häge, S.; Schütz, M.; Wagner, S.; Wardin, J.; Sticht, H.; Marschall, M. ‘Shared-Hook’ and ‘Changed-Hook’ Binding Activities of Herpesviral Core Nuclear Egress Complexes Identified by Random Mutagenesis. Cells 2022, 11, 4030. [Google Scholar] [CrossRef]
- Schweininger, J.; Kriegel, M.; Häge, S.; Conrad, M.; Alkhashrom, S.; Lösing, J.; Weiler, S.; Tillmanns, J.; Egerer-Sieber, C.; Decker, A.; et al. The crystal structure of the varicella-zoster Orf24-Orf27 nuclear egress complex spotlights multiple determinants of herpesvirus subfamily specificity. J. Biol. Chem. 2022, 298, 101625. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [PubMed]
- Häge, S.; Sonntag, E.; Borst, E.M.; Tannig, P.; Seyler, L.; Bauerle, T.; Bailer, S.M.; Lee, C.P.; Muller, R.; Wangen, C.; et al. Patterns of autologous and nonautologous interactions between core nuclear egress complex (NEC) proteins of alpha-, beta- and gamma-herpesviruses. Viruses 2020, 12, 303. [Google Scholar] [CrossRef]
- Zeev-Ben-Mordehai, T.; Weberruß, M.; Lorenz, M.; Cheleski, J.; Hellberg, T.; Whittle, C.; El Omari, K.; Vasishtan, D.; Dent, K.C.; Harlos, K.; et al. Crystal Structure of the Herpesvirus Nuclear Egress Complex Provides Insights into Inner Nuclear Membrane Remodeling. Cell Rep. 2015, 13, 2645–2652. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, M.K.; Draganova, E.B.; Heldwein, E.E. The nuclear egress complex of Epstein-Barr virus buds membranes through an oligomerization-driven mechanism. PLoS Pathog. 2022, 18, e1010623. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat. Commun. 2014, 5, 4131. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.A.; Vasishtan, D.; Bauerlein, F.J.; et al. Structural basis of vesicle formation at the inner nuclear membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed]
- Leigh, K.E.; Sharma, M.; Mansueto, M.S.; Boeszoermenyi, A.; Filman, D.J.; Hogle, J.M.; Wagner, G.; Coen, D.M.; Arthanari, H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9010–9015. [Google Scholar] [CrossRef] [PubMed]
- Milbradt, J.; Auerochs, S.; Sevvana, M.; Muller, Y.A.; Sticht, H.; Marschall, M. Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J. Biol. Chem. 2012, 287, 24004–24016. [Google Scholar] [CrossRef]
- Chang, Y.E.; Sant, C.V.; Krug, P.W.; Sears, A.E.; Roizman, B. The null mutant of the U(L)31 gene of herpes simplex virus 1: Construction and phenotype in infected cells. J. Virol. 1997, 71, 8307–8315. [Google Scholar] [CrossRef]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Osterrieder, N.; Mettenleiter, T.C. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 2002, 76, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Tanaka, M.; Kawaguchi, Y.; Baines, J.D. Cell lines that support replication of a novel herpes simplex virus 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 2004, 329, 68–76. [Google Scholar] [CrossRef]
- Klupp, B.G.; Nixdorf, R.; Mettenleiter, T.C. Pseudorabies virus glycoprotein M inhibits membrane fusion. J. Virol. 2000, 74, 6760–6768. [Google Scholar] [CrossRef]
- Roller, R.J.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; DeSalvo, D. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef]
- Ye, G.J.; Roizman, B. The essential protein encoded by the UL31 gene of herpes simplex virus 1 depends for its stability on the presence of UL34 protein. Proc. Natl. Acad. Sci. USA 2000, 97, 11002–11007. [Google Scholar] [CrossRef] [PubMed]
- Grimm, K.S.; Klupp, B.G.; Granzow, H.; Müller, F.M.; Fuchs, W.; Mettenleiter, T.C. Analysis of viral and cellular factors influencing herpesvirus-induced nuclear envelope breakdown. J. Virol. 2012, 86, 6512–6521. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J. Virol. 2011, 85, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Etingov, I.; Pante, N. Effect of viral infection on the nuclear envelope and nuclear pore complex. Int. Rev. Cell Mol. Biol. 2012, 299, 117–159. [Google Scholar] [CrossRef]
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. ESCRT-III controls nuclear envelope reformation. Nature 2015, 522, 236–239. [Google Scholar] [CrossRef]
- Tillmanns, J.; Häge, S.; Borst, E.M.; Wardin, J.; Eickhoff, J.; Klebl, B.; Wagner, S.; Wangen, C.; Hahn, F.; Socher, E.; et al. Assessment of Covalently Binding Warhead Compounds in the Validation of the Cytomegalovirus Nuclear Egress Complex as an Antiviral Target. Cells 2023, 12, 1162. [Google Scholar] [CrossRef]
- Häge, S.; Horsch, D.; Stilp, A.C.; Kicuntod, J.; Müller, R.; Hamilton, S.T.; Egilmezer, E.; Rawlinson, W.D.; Stamminger, T.; Sonntag, E.; et al. A quantitative nuclear egress assay to investigate the nucleocytoplasmic capsid release of human cytomegalovirus. J. Virol. Methods 2020, 283, 113909. [Google Scholar] [CrossRef]
- Fossum, E.; Friedel, C.C.; Rajagopala, S.V.; Titz, B.; Baiker, A.; Schmidt, T.; Kraus, T.; Stellberger, T.; Rutenberg, C.; Suthram, S.; et al. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 2009, 5, e1000570. [Google Scholar] [CrossRef]
- Santarelli, R.; Farina, A.; Granato, M.; Gonnella, R.; Raffa, S.; Leone, L.; Bei, R.; Modesti, A.; Frati, L.; Torrisi, M.R.; et al. Identification and characterization of the product encoded by ORF69 of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 4562–4572. [Google Scholar] [CrossRef]
- Schnee, M.; Ruzsics, Z.; Bubeck, A.; Koszinowski, U.H. Common and specific properties of herpesvirus UL34/UL31 protein family members revealed by protein complementation assay. J. Virol. 2006, 80, 11658–11666. [Google Scholar] [CrossRef] [PubMed]
- Alkhashrom, S.; Kicuntod, J.; Häge, S.; Schweininger, J.; Muller, Y.A.; Lischka, P.; Marschall, M.; Eichler, J. Exploring the Human Cytomegalovirus Core Nuclear Egress Complex as a Novel Antiviral Target: A New Type of Small Molecule Inhibitors. Viruses 2021, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Alkhashrom, S.; Kicuntod, J.; Stillger, K.; Lützenburg, T.; Anzenhofer, C.; Neundorf, I.; Marschall, M.; Eichler, J. A Peptide Inhibitor of the Human Cytomegalovirus Core Nuclear Egress Complex. Pharmaceuticals 2022, 15, 1040. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lye, M.F.; Gorgulla, C.; Ficarro, S.B.; Cuny, G.D.; Scott, D.A.; Wu, F.; Rothlauf, P.W.; Wang, X.; Fernandez, R.; et al. A small molecule exerts selective antiviral activity by targeting the human cytomegalovirus nuclear egress complex. PLoS Pathog. 2023, 19, e1011781. [Google Scholar] [CrossRef]
- Draganova, E.B.; Heldwein, E.E. Virus-derived peptide inhibitors of the herpes simplex virus type 1 nuclear egress complex. Sci. Rep. 2021, 11, 4206. [Google Scholar] [CrossRef]
- Kicuntod, J.; Alkhashrom, S.; Häge, S.; Diewald, B.; Müller, R.; Hahn, F.; Lischka, P.; Sticht, H.; Eichler, J.; Marschall, M. Properties of Oligomeric Interaction of the Cytomegalovirus Core Nuclear Egress Complex (NEC) and Its Sensitivity to an NEC Inhibitory Small Molecule. Viruses 2021, 13, 462. [Google Scholar] [CrossRef]
- Kicuntod, J.; Häge, S.; Lösing, J.; Kopar, S.; Muller, Y.A.; Marschall, M. An antiviral targeting strategy based on the inducible interference with cytomegalovirus nuclear egress complex. Antivir. Res. 2023, 212, 105557. [Google Scholar] [CrossRef]
- Kicuntod, J.; Häge, S.; Hahn, F.; Sticht, H.; Marschall, M. The Oligomeric Assemblies of Cytomegalovirus Core Nuclear Egress Proteins Are Associated with Host Kinases and Show Sensitivity to Antiviral Kinase Inhibitors. Viruses 2022, 14, 1021. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Burley, S.K.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. The archiving and dissemination of biological structure data. Curr. Opin. Struct. Biol. 2016, 40, 17–22. [Google Scholar] [CrossRef]
- Ormö, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef]
- Acosta, E.; Bowlin, T.; Brooks, J.; Chiang, L.; Hussein, I.; Kimberlin, D.; Kauvar, L.M.; Leavitt, R.; Prichard, M.; Whitley, R. Advances in the Development of Therapeutics for Cytomegalovirus Infections. J. Infect. Dis. 2020, 221, S32–S44. [Google Scholar] [CrossRef]
- Kotton, C.N. Updates on antiviral drugs for cytomegalovirus prevention and treatment. Curr. Opin. Organ Transplant. 2019, 24, 469–475. [Google Scholar] [CrossRef]
- Li, M.K.; Liu, Y.Y.; Wei, F.; Shen, M.X.; Zhong, Y.; Li, S.; Chen, L.J.; Ma, N.; Liu, B.Y.; Mao, Y.D.; et al. Antiviral activity of arbidol hydrochloride against herpes simplex virus I in vitro and in vivo. Int. J. Antimicrob. Agents 2018, 51, 98–106. [Google Scholar] [CrossRef]
- Sharma, M.; Coen, D.M. Comparison of effects of inhibitors of viral and cellular protein kinases on human cytomegalovirus disruption of nuclear lamina and nuclear egress. J. Virol. 2014, 88, 10982–10985. [Google Scholar] [CrossRef]
- Péczka, N.; Orgován, Z.; Ábrányi-Balogh, P.; Keserű, G.M. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin. Drug Discov. 2022, 17, 413–422. [Google Scholar] [CrossRef]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef]
- Ahangarpour, M.; Kavianinia, I.; Brimble, M.A. Thia-Michael addition: The route to promising opportunities for fast and cysteine-specific modification. Org. Biomol. Chem. 2023, 21, 3057–3072. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Orally effective FDA-approved protein kinase targeted covalent inhibitors (TCIs). Pharmacol. Res. 2021, 165, 105422. [Google Scholar] [CrossRef] [PubMed]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. Engl. 2016, 55, 13408–13421. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Burgess, J.; Colclough, N.; Davies, N.L.; Lenz, E.M.; Orton, A.L.; Ward, R.A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57, 3124–3137. [Google Scholar] [CrossRef]
- Sam, M.D.; Evans, B.T.; Coen, D.M.; Hogle, J.M. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J. Virol. 2009, 83, 2996–3006. [Google Scholar] [CrossRef]
- Zhang, T.; Hatcher, J.M.; Teng, M.; Gray, N.S.; Kostic, M. Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation. Cell Chem. Biol. 2019, 26, 1486–1500. [Google Scholar] [CrossRef]
- Feni, L.; Neundorf, I. The Current Role of Cell-Penetrating Peptides in Cancer Therapy. Adv. Exp. Med. Biol. 2017, 1030, 279–295. [Google Scholar] [CrossRef]
- Habault, J.; Poyet, J.L. Recent Advances in Cell Penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Gräslund, A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef]
- Nigatu, A.S.; Vupputuri, S.; Flynn, N.; Ramsey, J.D. Effects of cell-penetrating peptides on transduction efficiency of PEGylated adenovirus. Biomed. Pharmacother. 2015, 71, 153–160. [Google Scholar] [CrossRef]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Vale, N.; Duarte, D.; Silva, S.; Correia, A.S.; Costa, B.; Gouveia, M.J.; Ferreira, A. Cell-penetrating peptides in oncologic pharmacotherapy: A review. Pharmacol. Res. 2020, 162, 105231. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Cao, X.W.; Fu, L.Y.; Zhang, T.Z.; Wang, F.J.; Zhao, J. Screening and characterization of a novel high-efficiency tumor-homing cell-penetrating peptide from the buffalo cathelicidin family. J. Pept. Sci. 2019, 25, e3201. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, C.; Borst, E.; Sticht, H.; Marschall, M.; Milbradt, J. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J. Gen. Virol. 2013, 94, 2056–2069. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Haas, J.; Wagner, M.; Krohne, G.; Koszinowski, U.H. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 2002, 297, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, E.; Hamilton, S.T.; Bahsi, H.; Wagner, S.; Jonjic, S.; Rawlinson, W.D.; Marschall, M.; Milbradt, J. Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J. Gen. Virol. 2016, 97, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Hage, S.; Kraut, A.; Hesse, A.M.; Amin, B.; Sonnewald, U.; Coute, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Arii, J. Host and Viral Factors Involved in Nuclear Egress of Herpes Simplex Virus 1. Viruses 2021, 13, 754. [Google Scholar] [CrossRef] [PubMed]
- Bailer, S.M. Venture from the Interior-Herpesvirus pUL31 Escorts Capsids from Nucleoplasmic Replication Compartments to Sites of Primary Envelopment at the Inner Nuclear Membrane. Cells 2017, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, J.M.; Heldwein, E.E. Nuclear Exodus: Herpesviruses Lead the Way. Annu. Rev. Virol. 2016, 3, 387–409. [Google Scholar] [CrossRef]
- Bosse, J.B.; Enquist, L.W. The diffusive way out: Herpesviruses remodel the host nucleus, enabling capsids to access the inner nuclear membrane. Nucleus 2016, 7, 13–19. [Google Scholar] [CrossRef]
- Hellberg, T.; Passvogel, L.; Schulz, K.S.; Klupp, B.G.; Mettenleiter, T.C. Nuclear Egress of Herpesviruses: The Prototypic Vesicular Nucleocytoplasmic Transport. Adv. Virus Res. 2016, 94, 81–140. [Google Scholar] [CrossRef]
- Lee, C.P.; Chen, M.R. Conquering the Nuclear Envelope Barriers by EBV Lytic Replication. Viruses 2021, 13, 702. [Google Scholar] [CrossRef] [PubMed]
- Roller, R.J.; Johnson, D.C. Herpesvirus Nuclear Egress across the Outer Nuclear Membrane. Viruses 2021, 13, 2356. [Google Scholar] [CrossRef] [PubMed]
- Schütz, M.; Cordsmeier, A.; Wangen, C.; Horn, A.H.C.; Wyler, E.; Ensser, A.; Sticht, H.; Marschall, M. The Interactive Complex between Cytomegalovirus Kinase vCDK/pUL97 and Host Factors CDK7-Cyclin H Determines Individual Patterns of Transcription in Infected Cells. Int. J. Mol. Sci. 2023, 24, 17421. [Google Scholar] [CrossRef] [PubMed]
- Schütz, M.; Wangen, C.; Sommerer, M.; Kögler, M.; Eickhoff, J.; Degenhart, C.; Klebl, B.; Naing, Z.; Egilmezer, E.; Hamilton, S.T.; et al. Cytomegalovirus cyclin-dependent kinase ortholog vCDK/pUL97 undergoes regulatory interaction with human cyclin H and CDK7 to codetermine viral replication efficiency. Virus Res. 2023, 335, 199200. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Bender, B.J.; Kamil, J.P.; Lye, M.F.; Pesola, J.M.; Reim, N.I.; Hogle, J.M.; Coen, D.M. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J. Virol. 2015, 89, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Bahnamiri, M.M.; Roller, R.J. Distinct roles of viral US3 and UL13 protein kinases in herpes virus simplex type 1 (HSV-1) nuclear egress. bioRxiv 2023. [Google Scholar] [CrossRef]
- Cano-Monreal, G.L.; Wylie, K.M.; Cao, F.; Tavis, J.E.; Morrison, L.A. Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology 2009, 392, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Huang, Y.H.; Lin, S.F.; Chang, Y.; Chang, Y.H.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 2008, 82, 11913–11926. [Google Scholar] [CrossRef] [PubMed]
- Krosky, P.M.; Baek, M.C.; Coen, D.M. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 2003, 77, 905–914. [Google Scholar] [CrossRef]
- Prichard, M.N.; Gao, N.; Jairath, S.; Mulamba, G.; Krosky, P.; Coen, D.M.; Parker, B.O.; Pari, G.S. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 1999, 73, 5663–5670. [Google Scholar] [CrossRef]
- Wolf, D.G.; Courcelle, C.T.; Prichard, M.N.; Mocarski, E.S. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc. Natl. Acad. Sci. USA 2001, 98, 1895–1900. [Google Scholar] [CrossRef]
- Kang, C. Maribavir: First Approval. Drugs 2022, 82, 335–340. [Google Scholar] [CrossRef]
- Herget, T.; Freitag, M.; Morbitzer, M.; Kupfer, R.; Stamminger, T.; Marschall, M. Novel chemical class of pUL97 protein kinase-specific inhibitors with strong anticytomegaloviral activity. Antimicrob. Agents Chemother. 2004, 48, 4154–4162. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, C.; Hamilton, S.; Steingruber, M.; Zeitträger, I.; Bahsi, H.; Thuma, N.; Naing, Z.; Örfi, Z.; Örfi, L.; Socher, E.; et al. The chemical class of quinazoline compounds provides a core structure for the design of anticytomegaloviral kinase inhibitors. Antivir. Res. 2016, 134, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Freitag, M.; Suchy, P.; Romaker, D.; Kupfer, R.; Hanke, M.; Stamminger, T. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 2003, 311, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Schleiss, M.R. Cytomegalovirus vaccine development. Curr. Top Microbiol. Immunol. 2008, 325, 361–382. [Google Scholar] [CrossRef]
- Wild, M.; Hahn, F.; Brückner, N.; Schütz, M.; Wangen, C.; Wagner, S.; Sommerer, M.; Strobl, S.; Marschall, M. Cyclin-Dependent Kinases (CDKs) and the Human Cytomegalovirus-Encoded CDK Ortholog pUL97 Represent Highly Attractive Targets for Synergistic Drug Combinations. Int. J. Mol. Sci. 2022, 23, 2493. [Google Scholar] [CrossRef]
- Sharma, M.; Kamil, J.P.; Coen, D.M. Preparation of the Human Cytomegalovirus Nuclear Egress Complex and Associated Proteins. Methods Enzymol. 2016, 569, 517–526. [Google Scholar] [CrossRef]
- Lemnitzer, F.; Raschbichler, V.; Kolodziejczak, D.; Israel, L.; Imhof, A.; Bailer, S.M.; Koszinowski, U.; Ruzsics, Z. Mouse cytomegalovirus egress protein pM50 interacts with cellular endophilin-A2. Cell. Microbiol. 2013, 15, 335–351. [Google Scholar] [CrossRef]
- Liu, Z.; Kato, A.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kawaguchi, Y. Role of Host Cell p32 in Herpes Simplex Virus 1 De-Envelopment during Viral Nuclear Egress. J. Virol. 2015, 89, 8982–8998. [Google Scholar] [CrossRef]
- Yadav, S.; Libotte, F.; Buono, E.; Valia, S.; Farina, G.A.; Faggioni, A.; Farina, A. EBV early lytic protein BFRF1 alters emerin distribution and post-translational modification. Virus Res. 2017, 232, 113–122. [Google Scholar] [CrossRef]
- Hamirally, S.; Kamil, J.P.; Ndassa-Colday, Y.M.; Lin, A.J.; Jahng, W.J.; Baek, M.C.; Noton, S.; Silva, L.A.; Simpson-Holley, M.; Knipe, D.M.; et al. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009, 5, e1000275. [Google Scholar] [CrossRef]
- Heald, R.; McKeon, F. Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 1990, 61, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.A.; DeLassus, G.S. Breach of the nuclear lamina during assembly of herpes simplex viruses. Nucleus 2011, 2, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.L.; Mathias, R.A. The human cytomegalovirus decathlon: Ten critical replication events provide opportunities for restriction. Front. Cell Dev. Biol. 2022, 10, 1053139. [Google Scholar] [CrossRef] [PubMed]
- Schütz, M.; Thomas, M.; Wangen, C.; Wagner, S.; Rauschert, L.; Errerd, T.; Kießling, M.; Sticht, H.; Milbradt, J.; Marschall, M. The peptidyl-prolyl cis/trans isomerase Pin1 interacts with three early regulatory proteins of human cytomegalovirus. Virus Res. 2020, 285, 198023. [Google Scholar] [CrossRef] [PubMed]
- Draganova, E.B.; Zhang, J.; Zhou, Z.H.; Heldwein, E.E. Structural basis for capsid recruitment and coat formation during HSV-1 nuclear egress. eLife 2020, 9, e56627. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.; Ott, M.; Raschbichler, V.; Nagel, C.H.; Binz, A.; Sodeik, B.; Bauerfeind, R.; Bailer, S.M. The herpes simplex virus protein pUL31 escorts nucleocapsids to sites of nuclear egress, a process coordinated by its N-terminal domain. PLoS Pathog. 2015, 11, e1004957. [Google Scholar] [CrossRef]
- Takeshima, K.; Arii, J.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Identification of the Capsid Binding Site in the Herpes Simplex Virus 1 Nuclear Egress Complex and Its Role in Viral Primary Envelopment and Replication. J. Virol. 2019, 93, e01290-19. [Google Scholar] [CrossRef]
- Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M.S. Lenacapavir: A first-in-class HIV-1 capsid inhibitor. Curr. Opin. HIV AIDS 2022, 17, 15–21. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tillmanns, J.; Kicuntod, J.; Lösing, J.; Marschall, M. ‘Getting Better’—Is It a Feasible Strategy of Broad Pan-Antiherpesviral Drug Targeting by Using the Nuclear Egress-Directed Mechanism? Int. J. Mol. Sci. 2024, 25, 2823. https://doi.org/10.3390/ijms25052823
Tillmanns J, Kicuntod J, Lösing J, Marschall M. ‘Getting Better’—Is It a Feasible Strategy of Broad Pan-Antiherpesviral Drug Targeting by Using the Nuclear Egress-Directed Mechanism? International Journal of Molecular Sciences. 2024; 25(5):2823. https://doi.org/10.3390/ijms25052823
Chicago/Turabian StyleTillmanns, Julia, Jintawee Kicuntod, Josephine Lösing, and Manfred Marschall. 2024. "‘Getting Better’—Is It a Feasible Strategy of Broad Pan-Antiherpesviral Drug Targeting by Using the Nuclear Egress-Directed Mechanism?" International Journal of Molecular Sciences 25, no. 5: 2823. https://doi.org/10.3390/ijms25052823
APA StyleTillmanns, J., Kicuntod, J., Lösing, J., & Marschall, M. (2024). ‘Getting Better’—Is It a Feasible Strategy of Broad Pan-Antiherpesviral Drug Targeting by Using the Nuclear Egress-Directed Mechanism? International Journal of Molecular Sciences, 25(5), 2823. https://doi.org/10.3390/ijms25052823