
Transcriptomic Analyses and Experimental Validation Identified Immune-Related lncRNA–mRNA Pair MIR210HG–BPIFC Regulating the Progression of Hypertrophic Cardiomyopathy



Abstract
1. Introduction
2. Results
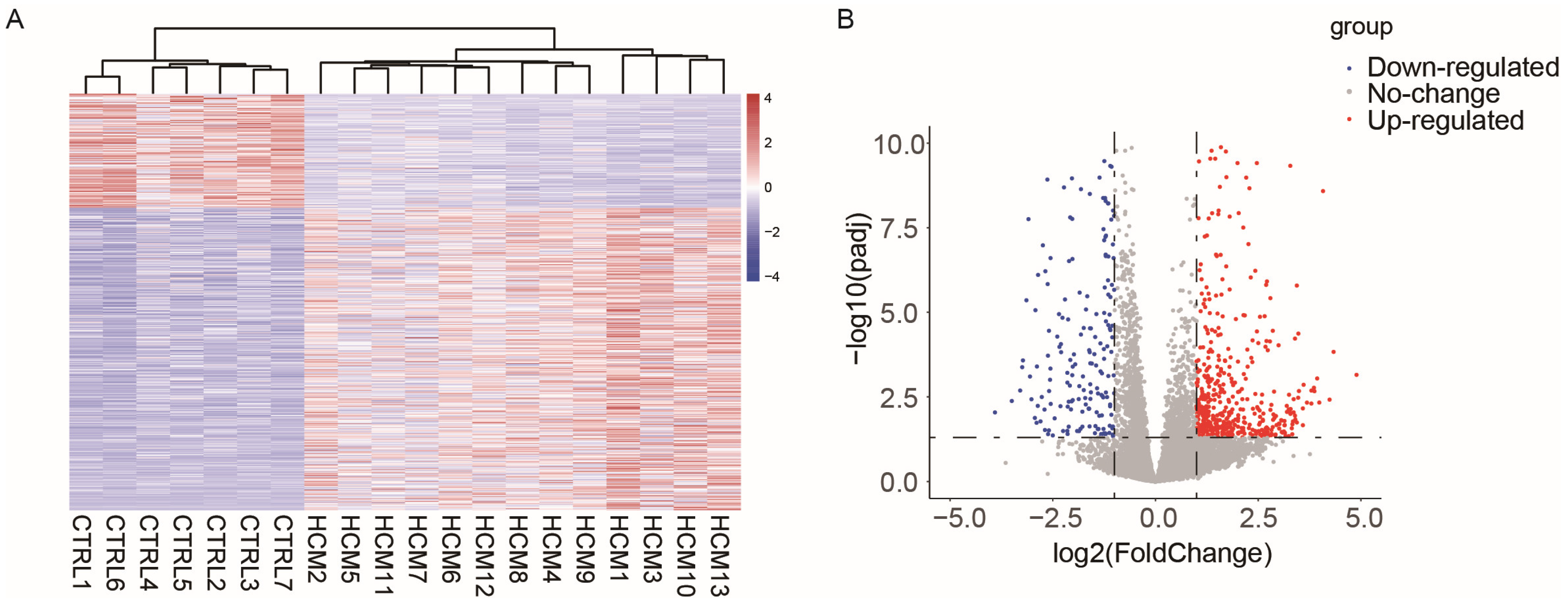
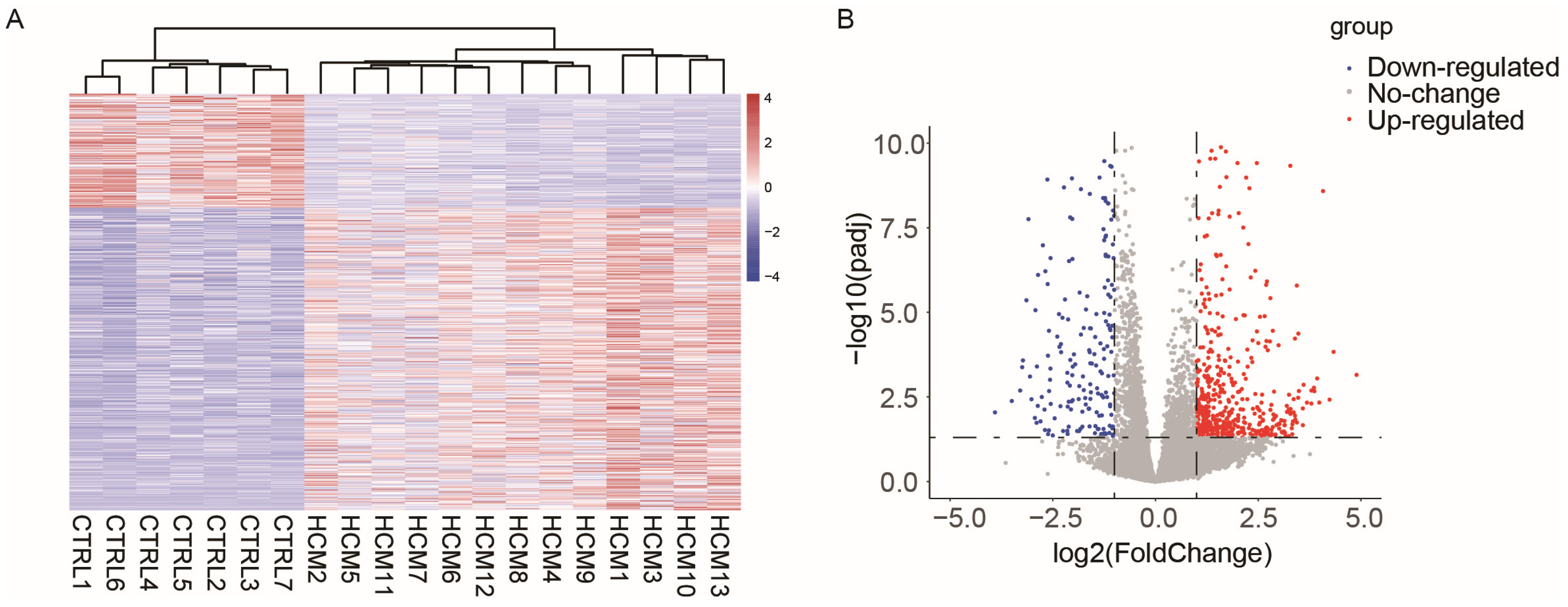
2.1. Data Collection and Analysis of Differentially Expressed Genes
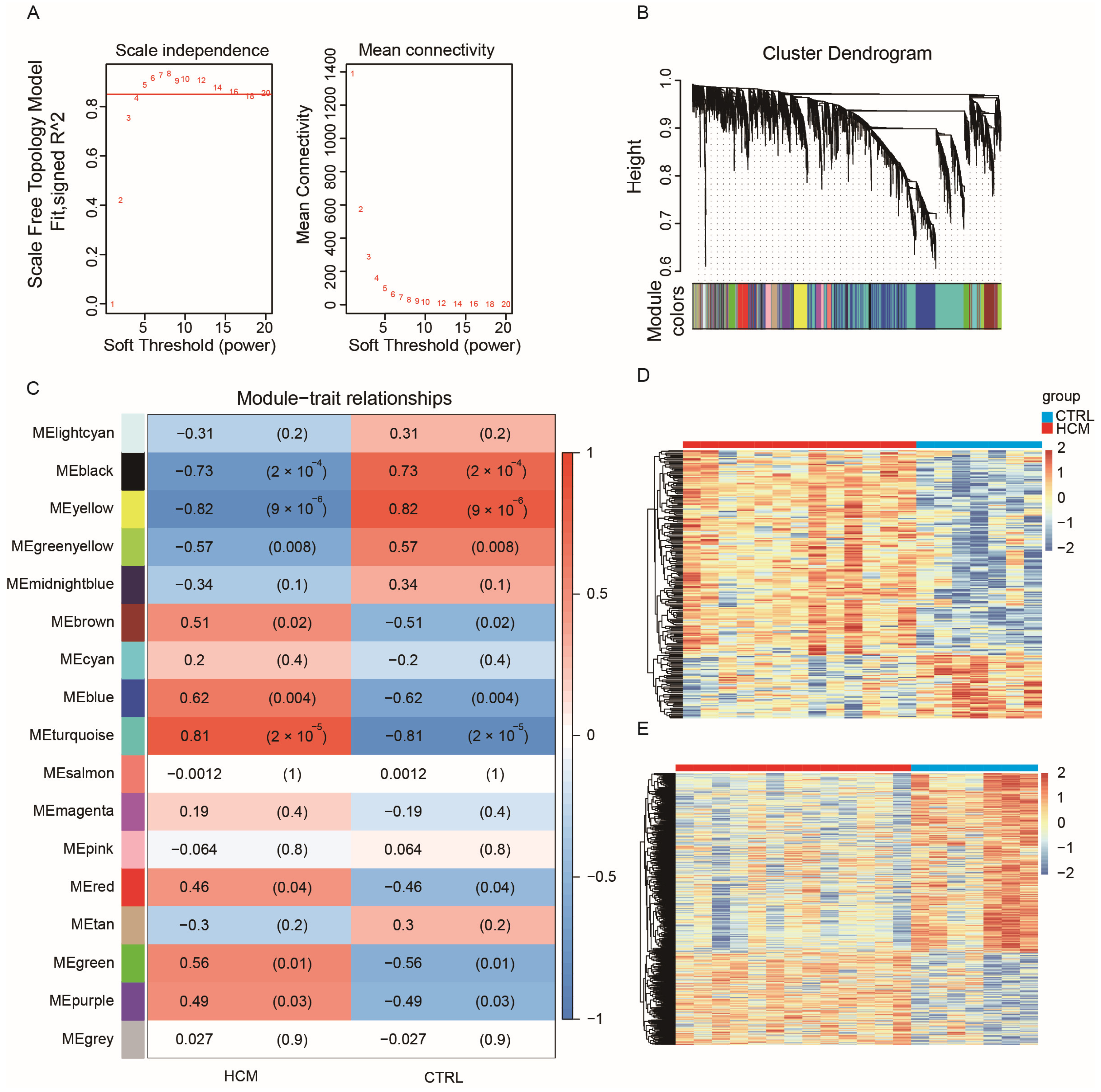
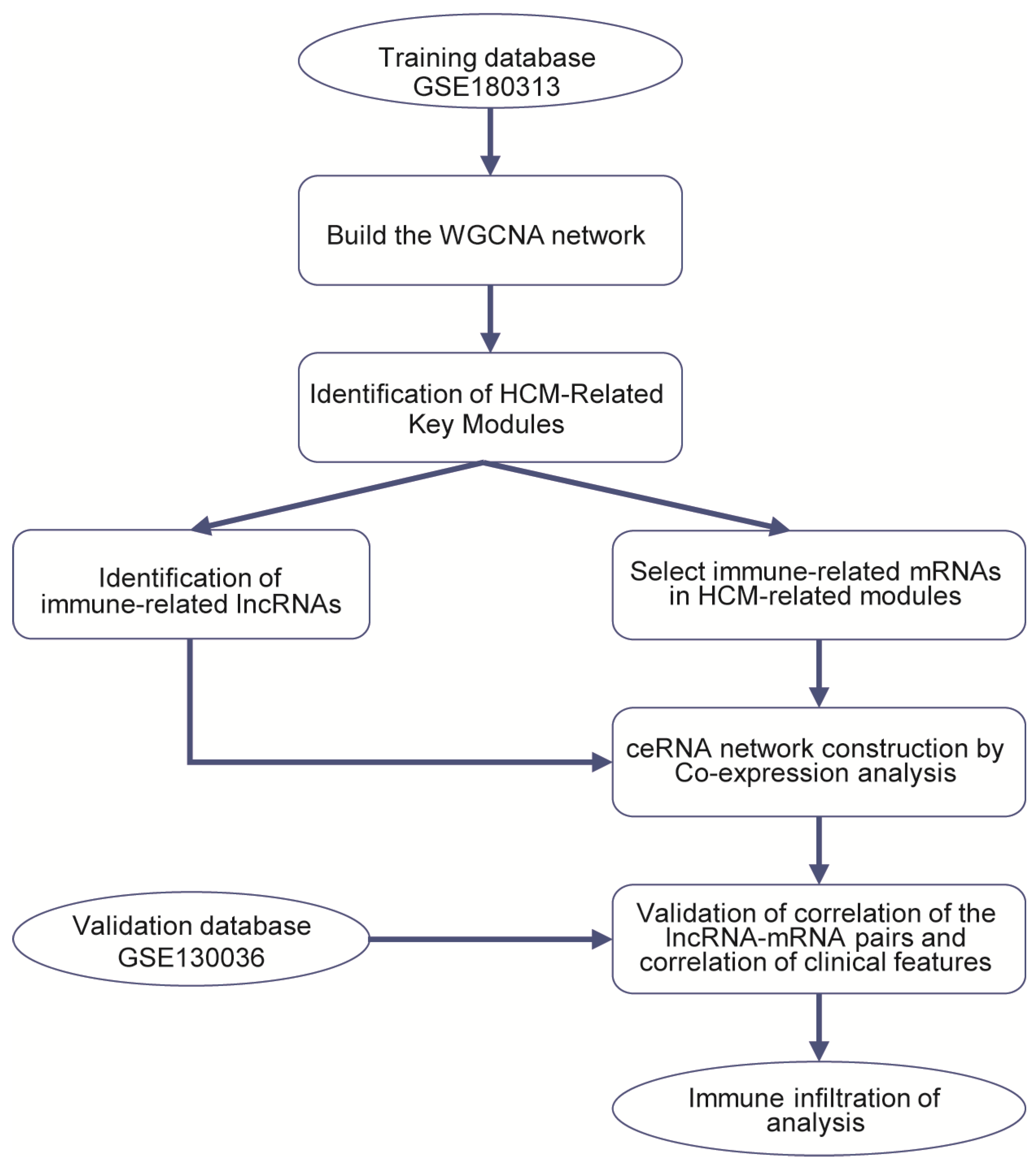
2.2. Construction of Weighted Co-Expression Network and Identification of HCM-Related Key Modules
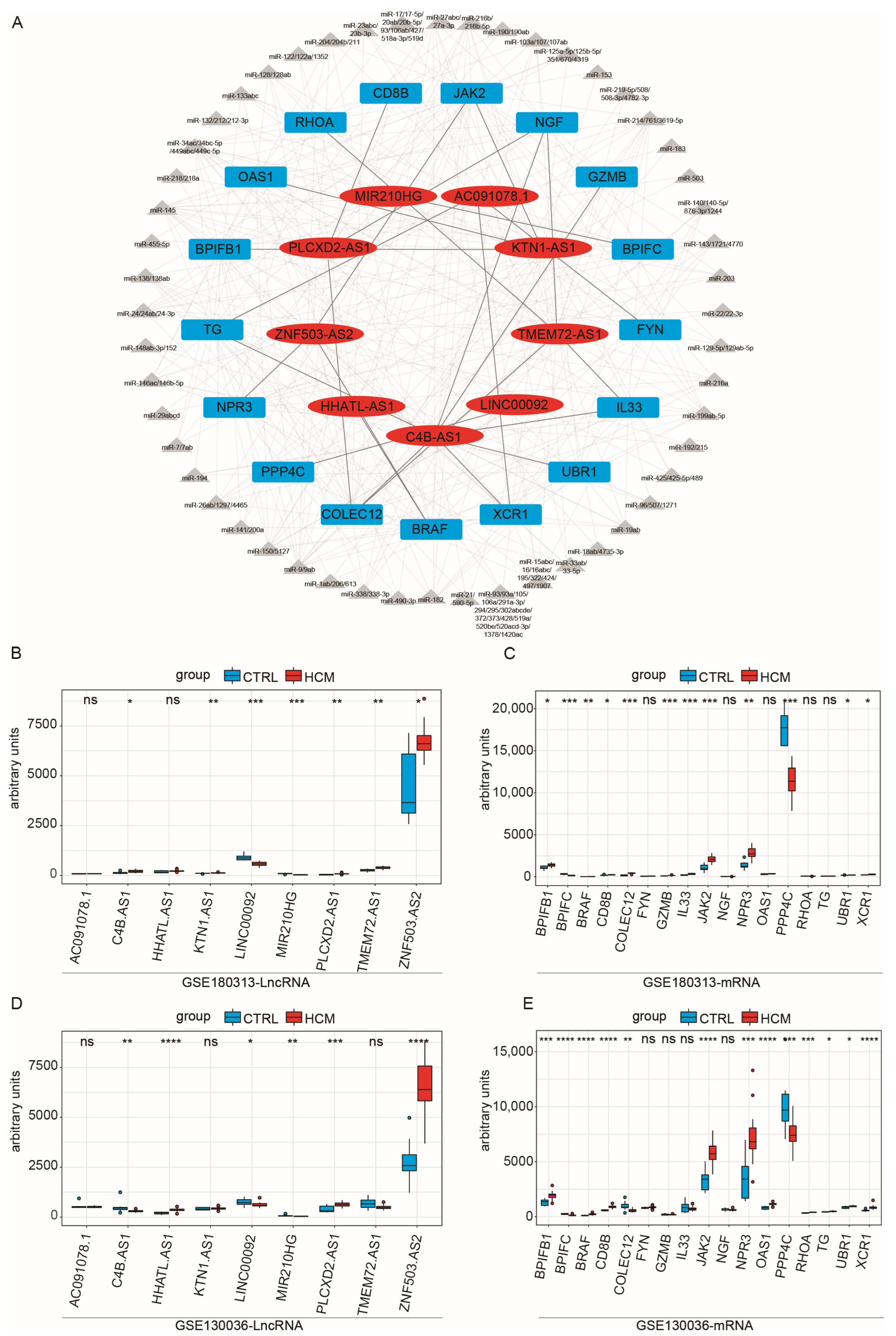
2.3. Construction of Immune-Related ceRNA Network
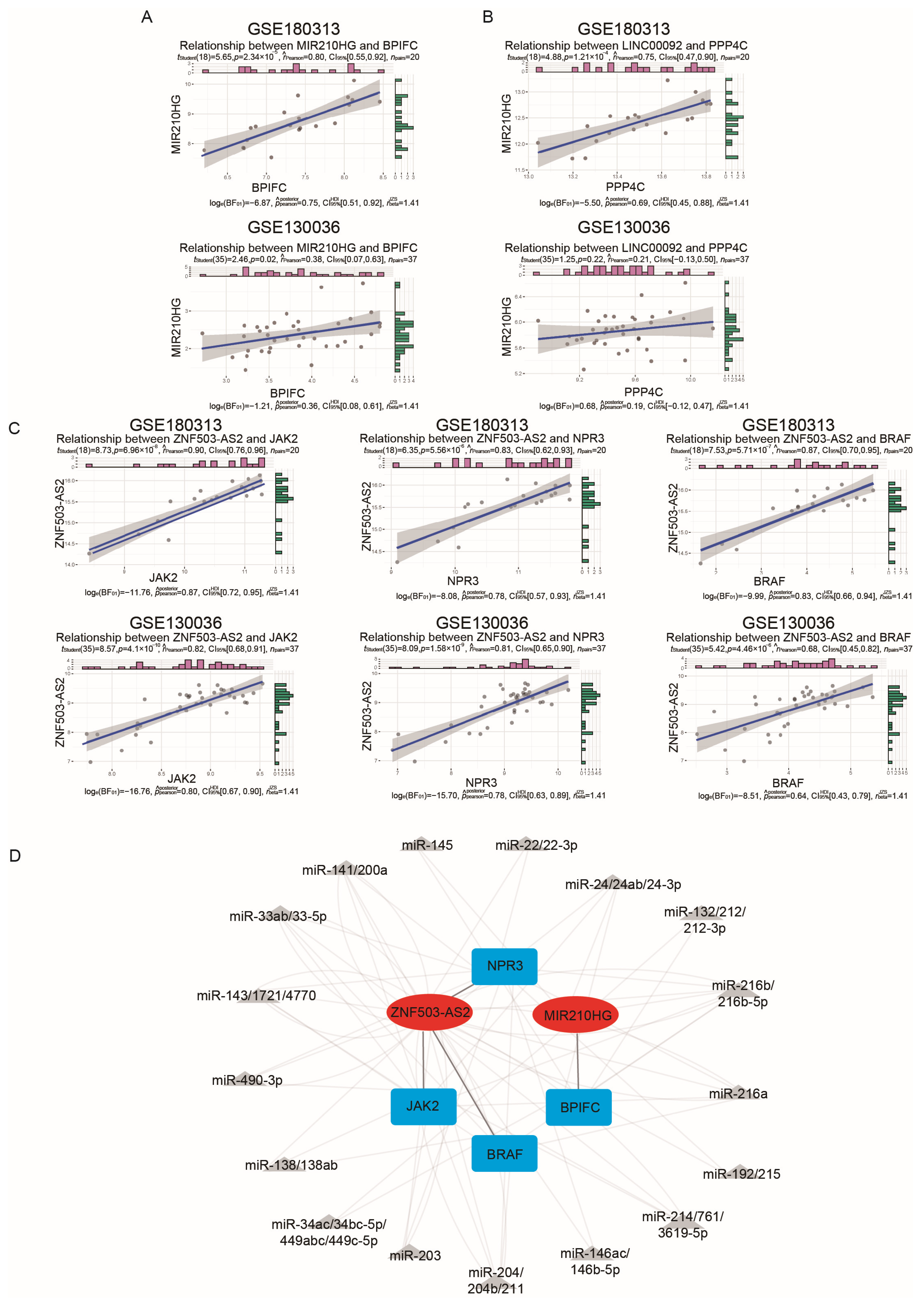
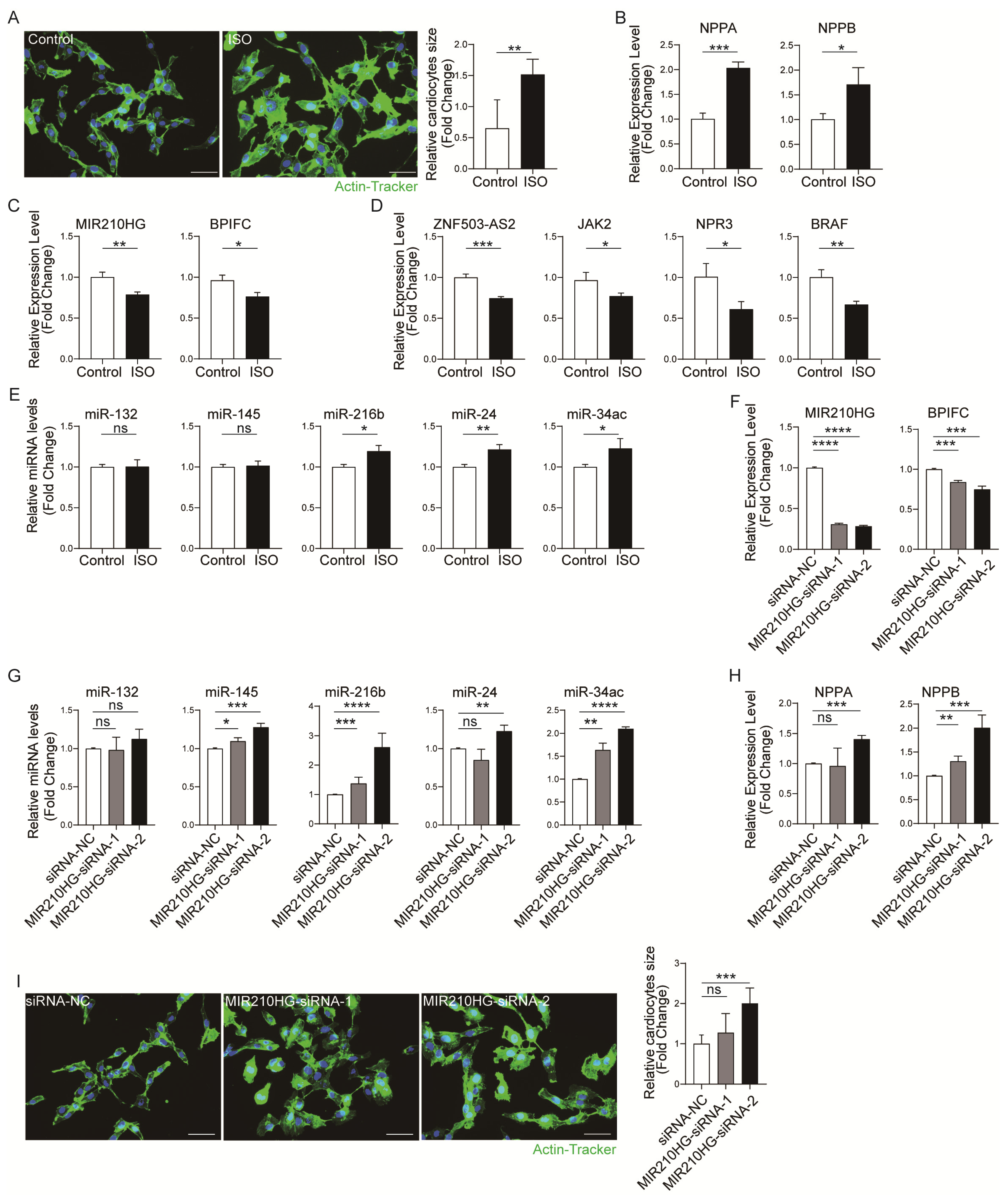
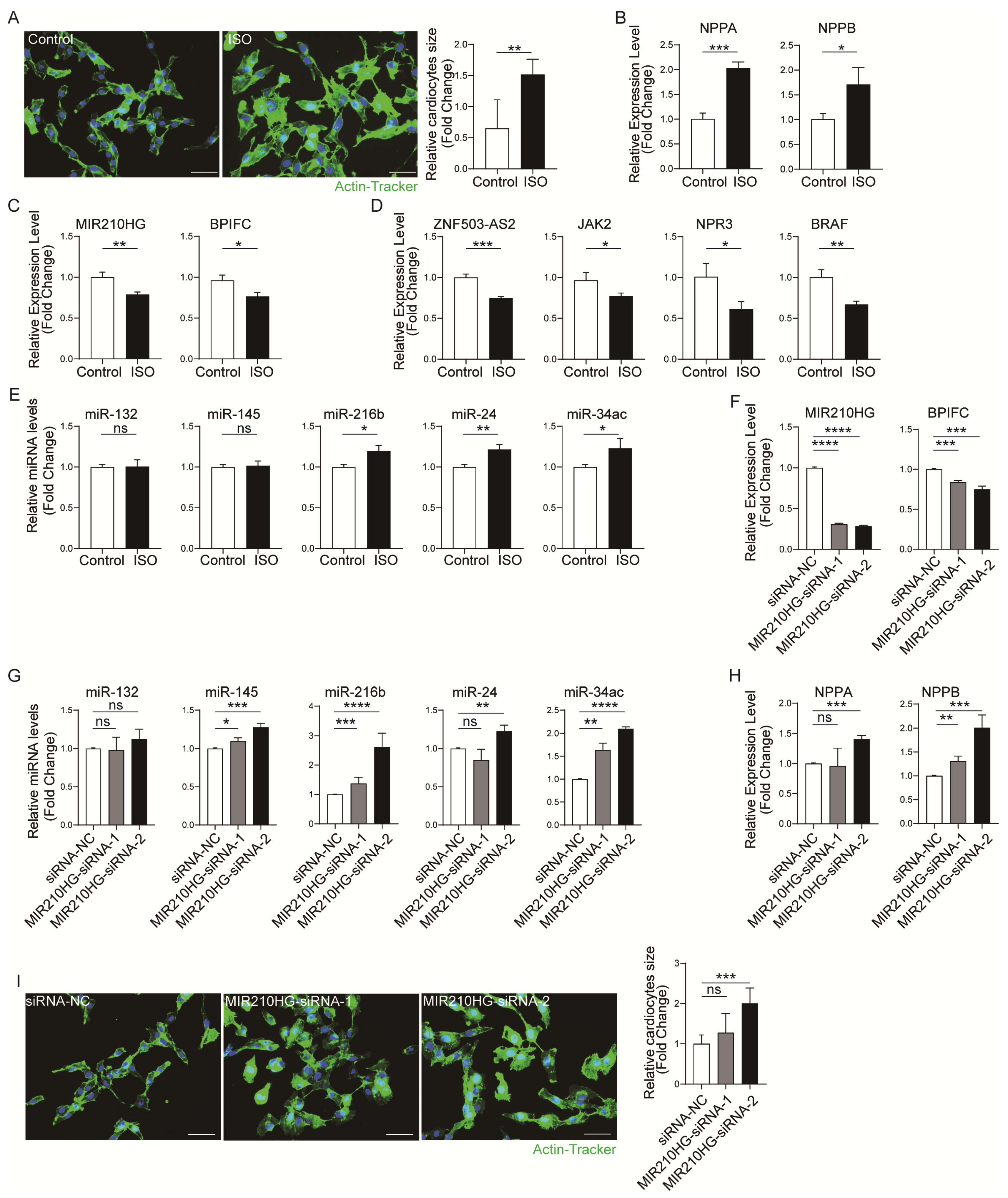
2.4. Validation of the Immune-Related lncRNA–mRNA Pairs
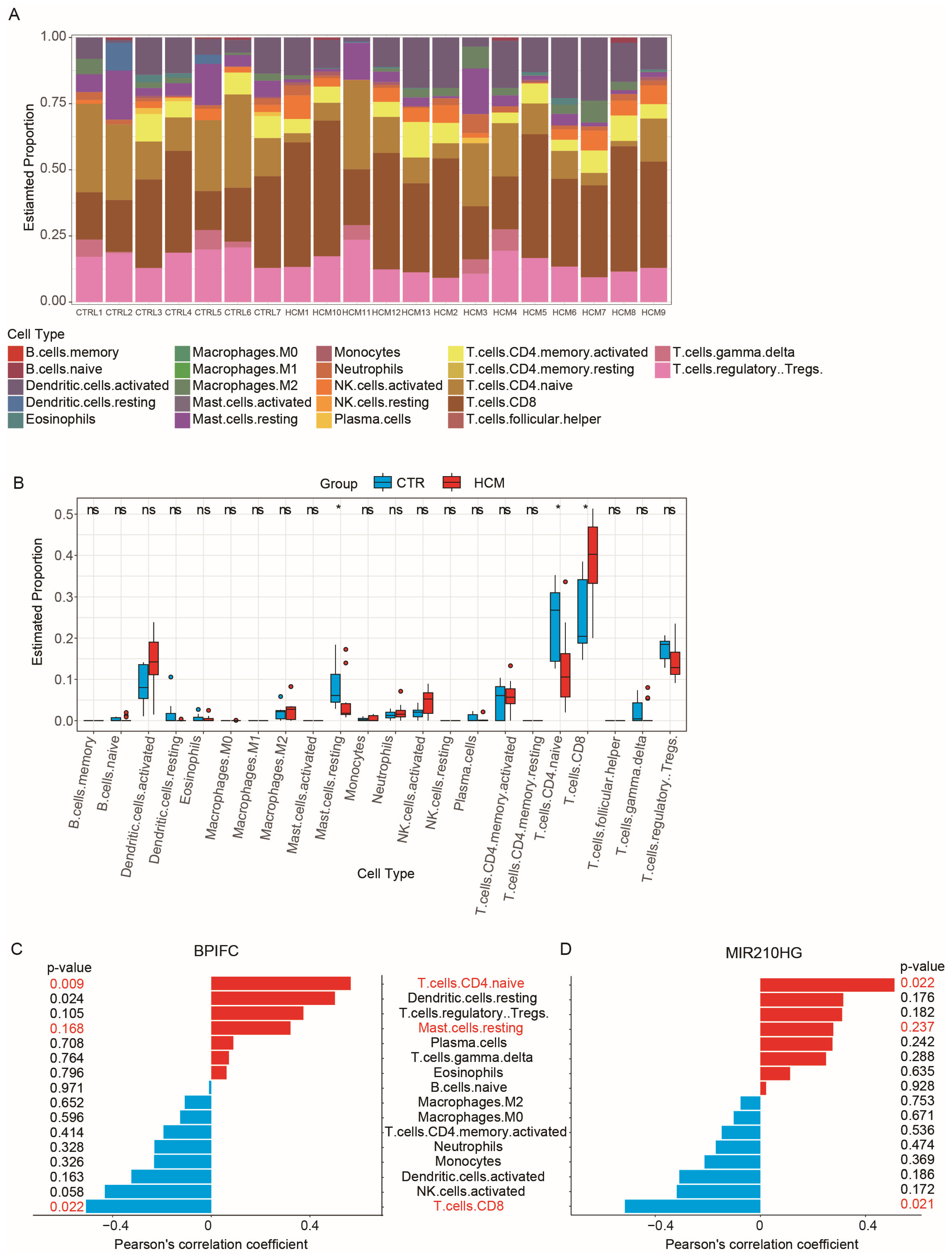
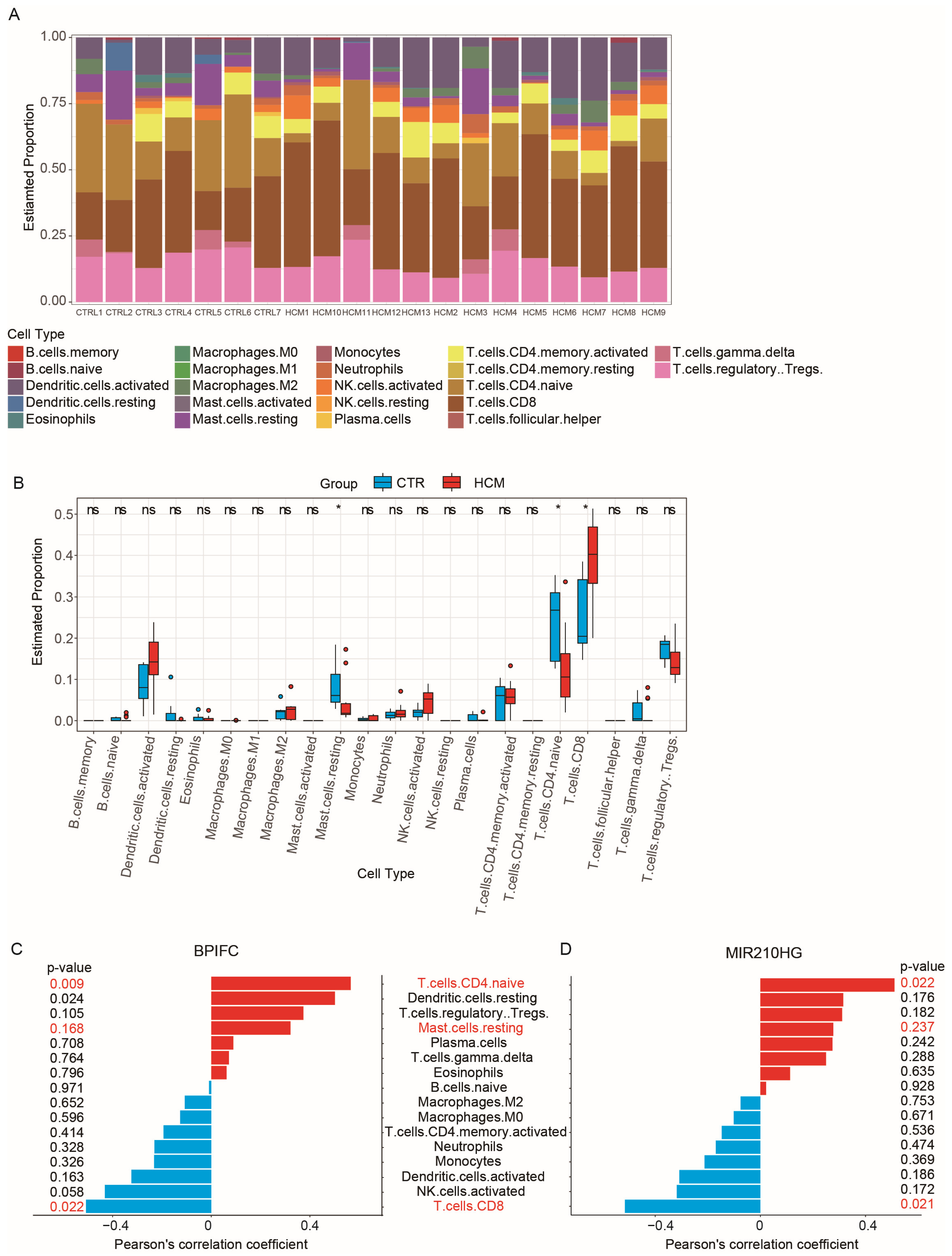
2.5. Analysis of Immune Cell Infiltration
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Data Preprocessing and DEG Screening
4.3. Weighted Gene Co-Expression Network Analysis
4.4. Identification of Immune-Related RNAs and ceRNA Network Construction
4.5. Cell Culture and HCM Cell Model
4.6. Measurement of Cell Surface Area
4.7. Quantitative Real-Time PCR (qRT-PCR)
4.8. CIBERSORTx and Statistical Analysis
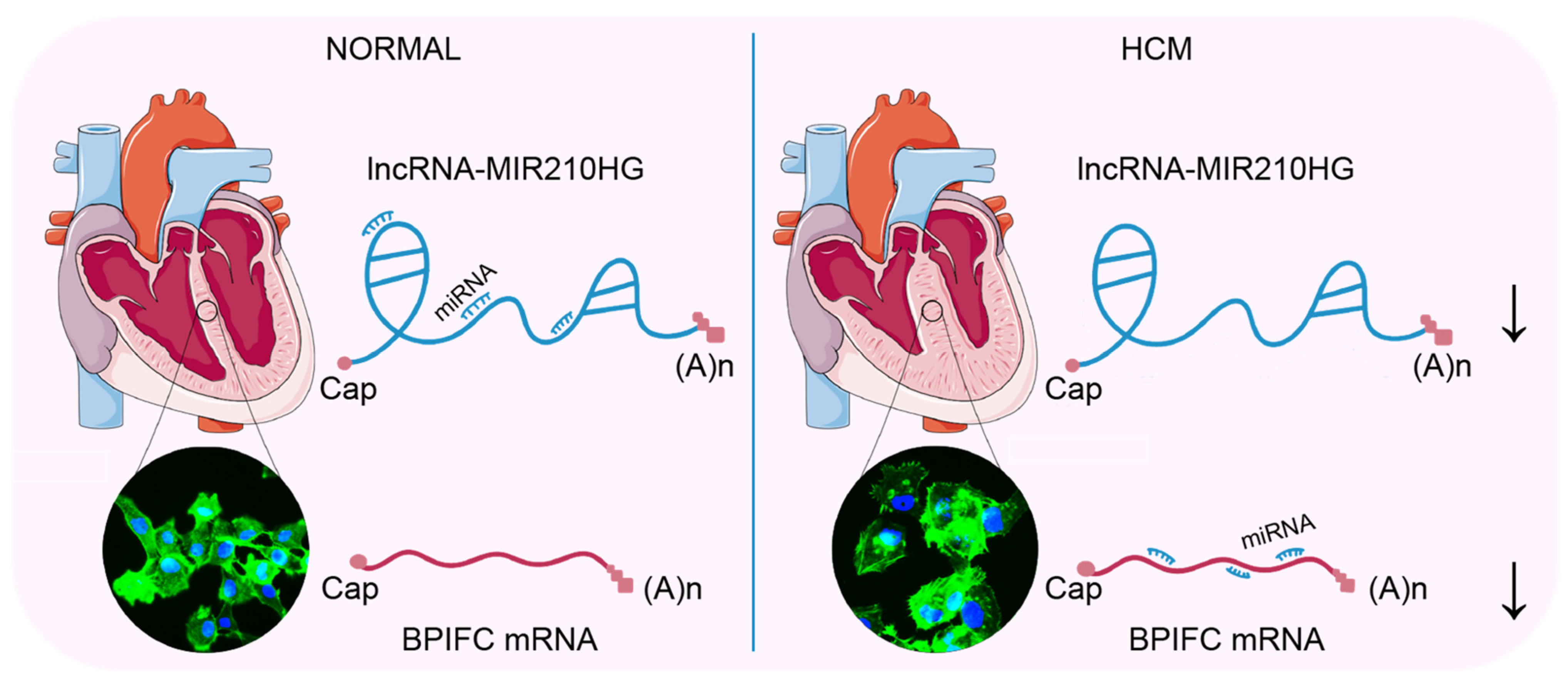
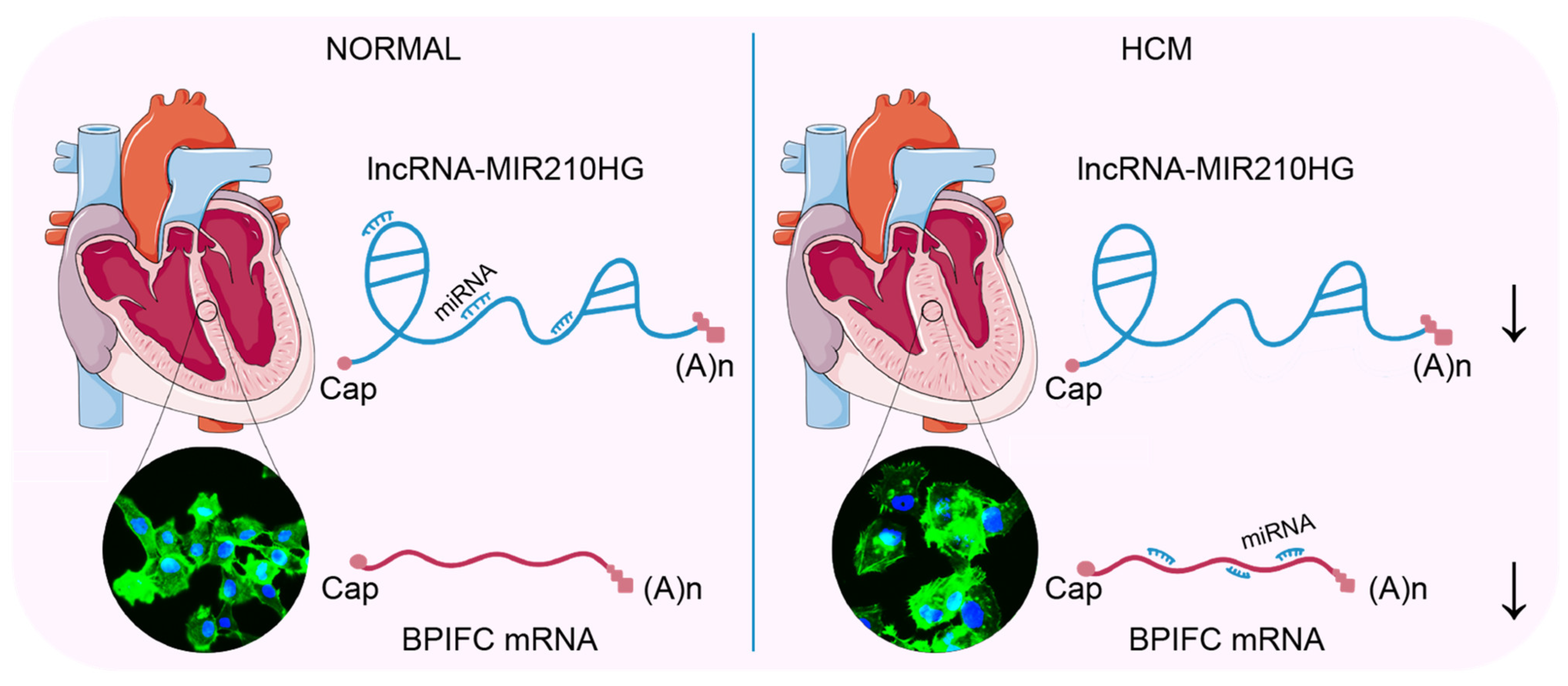
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Teekakirikul, P.; Zhu, W.; Huang, H.C.; Fung, E. Hypertrophic cardiomyopathy: An overview of genetics and management. Biomolecules 2019, 9, 878. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Firth, J. Cardiology: Hypertrophic cardiomyopathy. Clin. Med. 2019, 19, 61–63. [Google Scholar]
- Yu, H.; Gu, L.; Du, L.; Dong, Z.; Li, Z.; Yu, M.; Yin, Y.; Wang, Y.; Yu, L.; Ma, H. Identification and analysis of key hypoxia- and immune-related genes in hypertrophic cardiomyopathy. Biol. Res. 2023, 56, 45. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, W.; Dai, Y.; Chen, K. Identification and verification of IGFBP3 and YTHDC1 as biomarkers associated with immune infiltration and mitophagy in hypertrophic cardiomyopathy. Front. Genet. 2022, 13, 986995. [Google Scholar] [CrossRef]
- Viereck, J.; Kumarswamy, R.; Foinquinos, A.; Xiao, K.; Avramopoulos, P.; Kunz, M.; Dittrich, M.; Maetzig, T.; Zimmer, K.; Remke, J.; et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl. Med. 2016, 8, 326ra322. [Google Scholar] [CrossRef] [PubMed]
- Bazgir, F.; Nau, J.; Nakhaei-Rad, S.; Amin, E.; Wolf, M.J.; Saucerman, J.J.; Lorenz, K.; Ahmadian, M.R. The Microenvironment of the Pathogenesis of Cardiac Hypertrophy. Cells 2023, 12, 1780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Wang, G.; Zheng, N.; Cheng, W.; Ouyang, K.; Lin, H.; Liao, Y.; Liu, J. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension 2019, 73, 1058–1070. [Google Scholar] [CrossRef]
- Yang, R.; Masters, A.R.; Fortner, K.A.; Champagne, D.P.; Yanguas-Casás, N.; Silberger, D.J.; Weaver, C.T.; Haynes, L.; Rincon, M. IL-6 promotes the differentiation of a subset of naive CD8+ T cells into IL-21-producing B helper CD8+ T cells. J. Exp. Med. 2016, 213, 2281–2291. [Google Scholar] [CrossRef]
- Liang, H.; Pan, Z.; Zhao, X.; Liu, L.; Sun, J.; Su, X.; Xu, C.; Zhou, Y.; Zhao, D.; Xu, B.; et al. LncRNA PFL contributes to cardiac fibrosis by acting as a competing endogenous RNA of let-7d. Theranostics 2018, 8, 1180–1194. [Google Scholar] [CrossRef]
- Tao, L.; Shi, J.; Huang, X.; Hua, F.; Yang, L. Identification of a lncRNA-miRNA-mRNA network based on competitive endogenous RNA theory reveals functional lncRNAs in hypertrophic cardiomyopathy. Exp. Ther. Med. 2020, 20, 1176–1190. [Google Scholar] [CrossRef]
- Hu, X.; Shen, G.; Lu, X.; Ding, G.; Shen, L. Identification of key proteins and lncRNAs in hypertrophic cardiomyopathy by integrated network analysis. Arch. Med. Sci. 2019, 15, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Gong, Q.; Le, X.; He, J.; Zhuang, L. Bioinformatics and Experimental Analyses Reveal Immune-Related LncRNA-mRNA Pair AC011483.1-CCR7 as a Biomarker and Therapeutic Target for Ischemic Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 11994. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Aghajanian, H.; Epstein, J.A. Immune cells and immunotherapy for cardiac injury and repair. Circ. Res. 2021, 128, 1766–1779. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Steen, C.B.; Liu, C.L.; Alizadeh, A.A.; Newman, A.M. Profiling Cell Type Abundance and Expression in Bulk Tissues with CIBERSORTx. Methods Mol. Biol. 2020, 2117, 135–157. [Google Scholar] [PubMed]
- Wu, L.-D.; Xiao, F.; Sun, J.-Y.; Li, F.; Chen, Y.-J.; Chen, J.-Y.; Zhang, J.; Qian, L.-L.; Wang, R.-X. Integrated identification of key immune related genes and patterns of immune infiltration in calcified aortic valvular disease: A network based meta-analysis. Front. Genet. 2022, 13, 971808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Z.; Zhang, S.; Tang, T.-T.; Cheng, X. Bioinformatics and Immune Infiltration Analyses Reveal the Key Pathway and Immune Cells in the Pathogenesis of Hypertrophic Cardiomyopathy. Front. Cardiovasc. Med. 2021, 8, 696321. [Google Scholar] [CrossRef]
- Cuesta-Llavona, E.; Lorca, R.; Rolle, V.; Alonso, B.; Iglesias, S.; Rodríguez-Reguero, J.; Duarte-Herrera, I.D.; Pérez-Oliveira, S.; Junco-Vicente, A.; Lago, C.G.; et al. Association of the Genetic Variation in the Long Non-Coding RNA FENDRR with the Risk of Developing Hypertrophic Cardiomyopathy. Life 2022, 12, 818. [Google Scholar] [CrossRef]
- Cao, J.; Yuan, L. Identification of key genes for hypertrophic cardiomyopathy using integrated network analysis of differential lncRNA and gene expression. Front. Cardiovasc. Med. 2022, 9, 946229. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, Z.; Chen, Y.; Xu, D.; Chen, S. Susceptibility Modules and Genes in Hypertrophic Cardiomyopathy by WGCNA and ceRNA Network Analysis. Front. Cell. Dev. Biol. 2021, 9, 822465. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D. Calcineurin and beyond: Cardiac hypertrophic signaling. Circ. Res. 2000, 87, 731–738. [Google Scholar] [CrossRef]
- Su, Z.; Liu, S.; Zou, Y.; Shan, L.; Yu, M.; Xie, S.; Li, X.; Jin, Y. Trastuzumab-induced human cardiomyocyte damage through the Notch2/JAK2/STAT3 pathway. Clinics 2023, 78, 100268. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Wang, R.-S.; Shevtsov, S.; Drakos, S.G.; Arons, E.; Wever-Pinzon, O.; Huggins, G.S.; Samokhin, A.O.; Oldham, W.M.; Aguib, Y.; et al. Individualized interactomes for network-based precision medicine in hypertrophic cardiomyopathy with implications for other clinical pathophenotypes. Nat. Commun. 2021, 12, 873. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Gelb, B.D.; Zenker, M. Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 161–179. [Google Scholar] [CrossRef]
- Kuhn, E.; Addona, T.; Keshishian, H.; Burgess, M.; Mani, D.R.; Lee, R.T.; Sabatine, M.S.; E Gerszten, R.; A Carr, S. Developing multiplexed assays for troponin I and interleukin-33 in plasma by peptide immunoaffinity enrichment and targeted mass spectrometry. Clin. Chem. 2009, 55, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Min, W.; Dai, D.; Wang, J.; Zhang, D.; Zhang, Y.; Han, G.; Zhang, L.; Chen, C.; Li, X.; Li, Y.; et al. Long Noncoding RNA miR210HG as a Potential Biomarker for the Diagnosis of Glioma. PLoS ONE 2016, 11, e0160451. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Kong, F.-F.; Yang, D.; Yang, H.; Wang, C.; Cong, R.; Ma, X.-X. lncRNA MIR210HG promotes the progression of endometrial cancer by sponging miR-337-3p/137 via the HMGA2-TGF-β/Wnt pathway. Mol. Ther. Nucleic Acids 2021, 24, 905–922. [Google Scholar] [CrossRef]
- Shi, W.; Tang, Y.; Lu, J.; Zhuang, Y.; Wang, J. MIR210HG promotes breast cancer progression by IGF2BP1 mediated m6A modification. Cell Biosci. 2022, 12, 38. [Google Scholar] [CrossRef]
- Wu, X.; Liu, J.; Zhu, C.; Ma, M.; Chen, X.; Liu, Y.; Xia, N.; Dong, Q. Identification of Potential Biomarkers of Prognosis-Related Long Non-Coding RNA (lncRNA) in Pediatric Rhabdoid Tumor of the Kidney Based on ceRNA Networks. Med. Sci. Monit. 2020, 26, e927725. [Google Scholar] [CrossRef]
- Han, Z.; Wang, H.; Liu, Y.; Xing, X.-L. Establishment of a prognostic ferroptosis- and immune-related long noncoding RNAs profile in kidney renal clear cell carcinoma. Front. Genet. 2022, 13, 915372. [Google Scholar] [CrossRef] [PubMed]
- Mulero, J.J.; Boyle, B.J.; Bradley, S.; Bright, J.M.; Nelken, S.T.; Ho, T.T.; Mize, N.K.; Childs, J.D.; Ballinger, D.G.; Ford, J.E.; et al. Three new human members of the lipid transfer/lipopolysaccharide binding protein family (lt/lbp). Immunogenetics 2002, 54, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Earp, M.A.; Australian Cancer Study; Kelemen, L.E.; Magliocco, A.M.; Swenerton, K.D.; Chenevix-Trench, G.; Lu, Y.; Hein, A.; Ekici, A.B.; Beckmann, M.W.; et al. Genome-wide association study of subtype-specific epithelial ovarian cancer risk alleles using pooled DNA. Hum. Genet. 2014, 133, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Shiota, N.; Rysä, J.; Kovanen, P.T.; Ruskoaho, H.; O Kokkonen, J.; A Lindstedt, K. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 2003, 21, 1935–1944. [Google Scholar] [CrossRef]
- Dvorak, A.M. Mast-cell degranulation in human hearts. N. Engl. J. Med. 1986, 315, 969–970. [Google Scholar]
- Wang, H.; da Silva, J.; Alencar, A.; Zapata-Sudo, G.; Lin, M.R.; Sun, X.; Ahmad, S.; Ferrario, C.M.; Groban, L. Mast Cell Inhibition Attenuates Cardiac Remodeling and Diastolic Dysfunction in Middle-aged, Ovariectomized Fischer 344 × Brown Norway Rats. J. Cardiovasc. Pharmacol. 2016, 68, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Santhosh, S.; Bahl, A.; Saikia, U.N.; Lad, D.; Mittal, B.R.; Malhotra, P.; Varma, S. FDG PET/CT in the staging and follow-up of primary cardiac ‘T’ cell lymphoma presenting as hypertrophic cardiomyopathy. J. Nucl. Cardiol. 2016, 23, 581–584. [Google Scholar] [CrossRef]
- Chaffin, M.; Papangeli, I.; Simonson, B.; Akkad, A.D.; Hill, M.C.; Arduini, A.; Fleming, S.J.; Melanson, M.; Hayat, S.; Kost-Alimova, M.; et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature 2022, 608, 174–180. [Google Scholar] [CrossRef]
- Ni, Y.; Deng, J.; Liu, X.; Li, Q.; Zhang, J.; Bai, H.; Zhang, J. Echinacoside reverses myocardial remodeling and improves heart function via regulating SIRT1/FOXO3a/MnSOD axis in HF rats induced by isoproterenol. J. Cell. Mol. Med. 2021, 25, 203–216. [Google Scholar] [CrossRef]
- Cai, J.; Guan, H.; Li, D.; Shi, B.; Jiang, Y.; Qiao, S.; Liu, Q.; Fang, C.; Zhang, Z. New insights into Microalgal astaxanthin’s effect on Lambda-cyhalothrin-induced lymphocytes immunotoxicity in Cyprinus carpio: Involving miRNA-194-5p-FoxO1-mediated-mitophagy and pyroptosis. Fish Shellfish Immunol. 2023, 141, 109046. [Google Scholar] [CrossRef]
- Cai, J.; Liu, P.; Zhang, X.; Shi, B.; Jiang, Y.; Qiao, S.; Liu, Q.; Fang, C.; Zhang, Z. Micro-algal astaxanthin improves lambda-cyhalothrin-induced necroptosis and inflammatory responses via the ROS-mediated NF-κB signaling in lymphocytes of carp (Cyprinus carpio L.). Fish Shellfish Immunol. 2023, 139, 108929. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immune-Related mRNA | Immune-Related lncRNA | Correlation Coefficient |
|---|---|---|
| OAS1 | KTN1-AS1 | 0.789029469 |
| JAK2 | KTN1-AS1 | 0.752564718 |
| BPIFB1 | KTN1-AS1 | 0.858522985 |
| RHOA | TMEM72-AS1 | 0.758943191 |
| NGF | TMEM72-AS1 | 0.773519315 |
| IL33 | TMEM72-AS1 | 0.783806615 |
| COLEC12 | TMEM72-AS1 | 0.761881119 |
| PPP4C | LINC00092 | 0.75444876 |
| TG | C4B-AS1 | 0.835504137 |
| GZMB | C4B-AS1 | 0.791438674 |
| NGF | C4B-AS1 | 0.763980651 |
| IL33 | C4B-AS1 | 0.786158581 |
| COLEC12 | C4B-AS1 | 0.792932936 |
| UBR1 | C4B-AS1 | 0.798578035 |
| XCR1 | C4B-AS1 | 0.767467957 |
| BRAF | HHATL-AS1 | 0.855393248 |
| JAK2 | ZNF503-AS2 | 0.899336755 |
| NPR3 | ZNF503-AS2 | 0.831443126 |
| BRAF | ZNF503-AS2 | 0.871300503 |
| NGF | PLCXD2-AS1 | 0.77646062 |
| COLEC12 | PLCXD2-AS1 | 0.799703687 |
| CD8B | PLCXD2-AS1 | 0.761688487 |
| BPIFC | MIR210HG | 0.799484632 |
| FYN | AC091078.1 | 0.780037819 |
| TG | AC091078.1 | 0.833008855 |
| XCR1 | AC091078.1 | 0.766529515 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhao, J.; Jin, Q.; Zhuang, L. Transcriptomic Analyses and Experimental Validation Identified Immune-Related lncRNA–mRNA Pair MIR210HG–BPIFC Regulating the Progression of Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2024, 25, 2816. https://doi.org/10.3390/ijms25052816
Zhang Y, Zhao J, Jin Q, Zhuang L. Transcriptomic Analyses and Experimental Validation Identified Immune-Related lncRNA–mRNA Pair MIR210HG–BPIFC Regulating the Progression of Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences. 2024; 25(5):2816. https://doi.org/10.3390/ijms25052816
Chicago/Turabian StyleZhang, Yuan, Jiuxiao Zhao, Qiao Jin, and Lenan Zhuang. 2024. "Transcriptomic Analyses and Experimental Validation Identified Immune-Related lncRNA–mRNA Pair MIR210HG–BPIFC Regulating the Progression of Hypertrophic Cardiomyopathy" International Journal of Molecular Sciences 25, no. 5: 2816. https://doi.org/10.3390/ijms25052816
APA StyleZhang, Y., Zhao, J., Jin, Q., & Zhuang, L. (2024). Transcriptomic Analyses and Experimental Validation Identified Immune-Related lncRNA–mRNA Pair MIR210HG–BPIFC Regulating the Progression of Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences, 25(5), 2816. https://doi.org/10.3390/ijms25052816