
Genomic and Epigenomic Changes in the Progeny of Cold-Stressed Arabidopsis thaliana Plants

 ,
,
Abstract
1. Introduction
2. Results
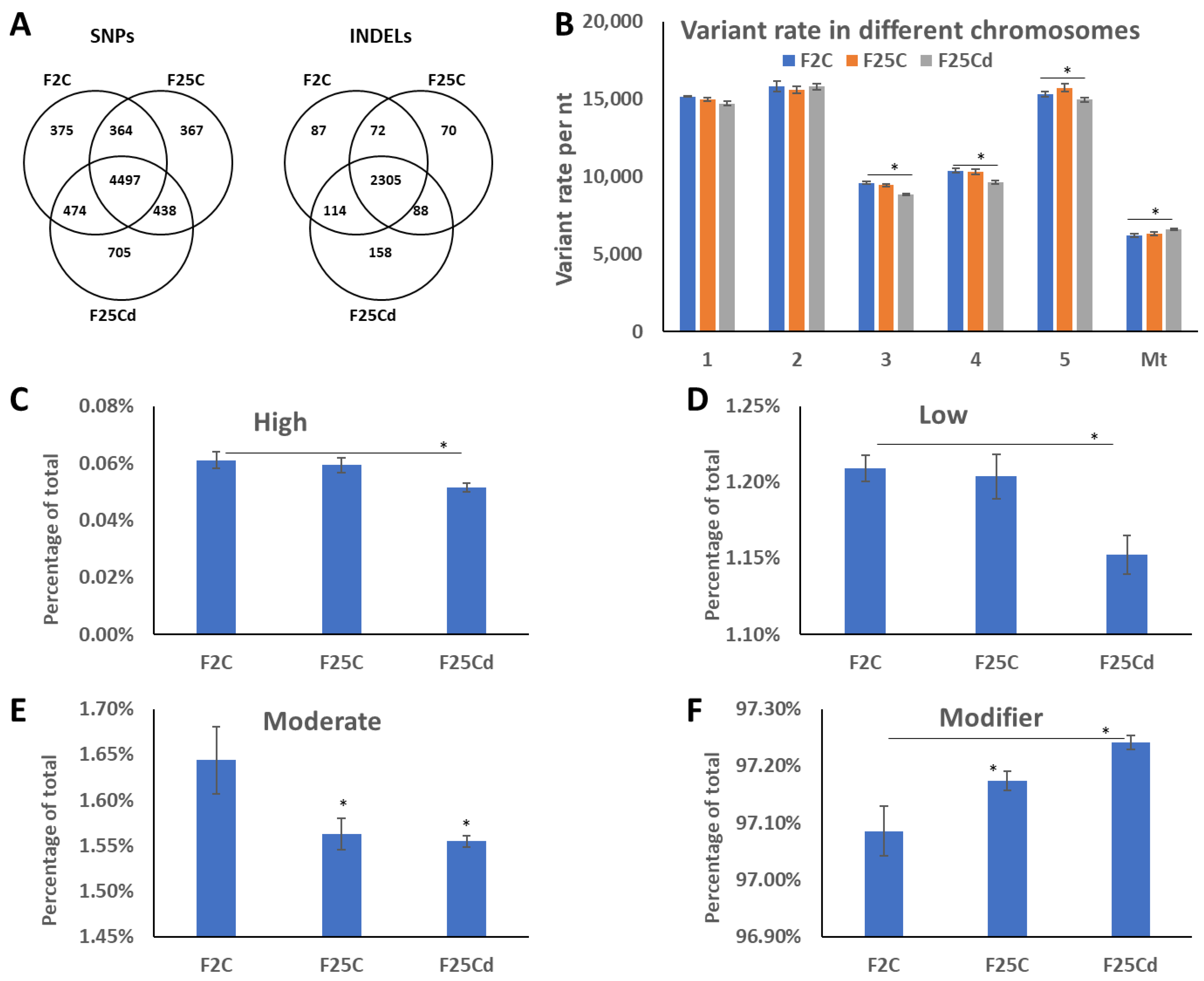
2.1. Comparison of Genomic Variants
2.1.1. Unique and Common SNPs and InDels
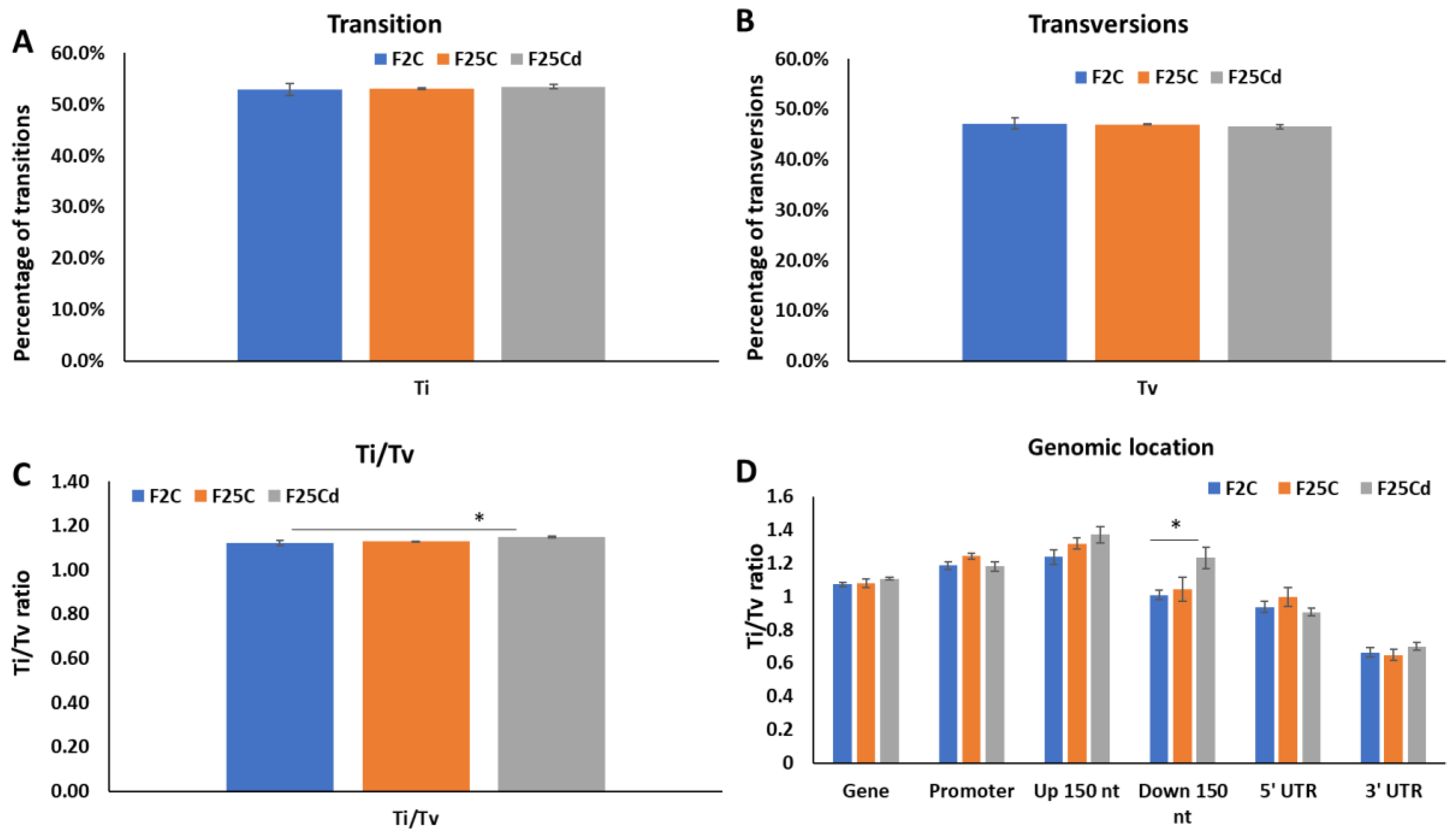
2.1.2. Analysis of Distribution of SNPs and Their Potential Impact
2.1.3. SNPs and InDels Associated with Transposons
2.1.4. The Functional Classification of SNPs and InDels
2.2. Epigenomic Profiling
2.2.1. The Percentage of Global DNA Methylation
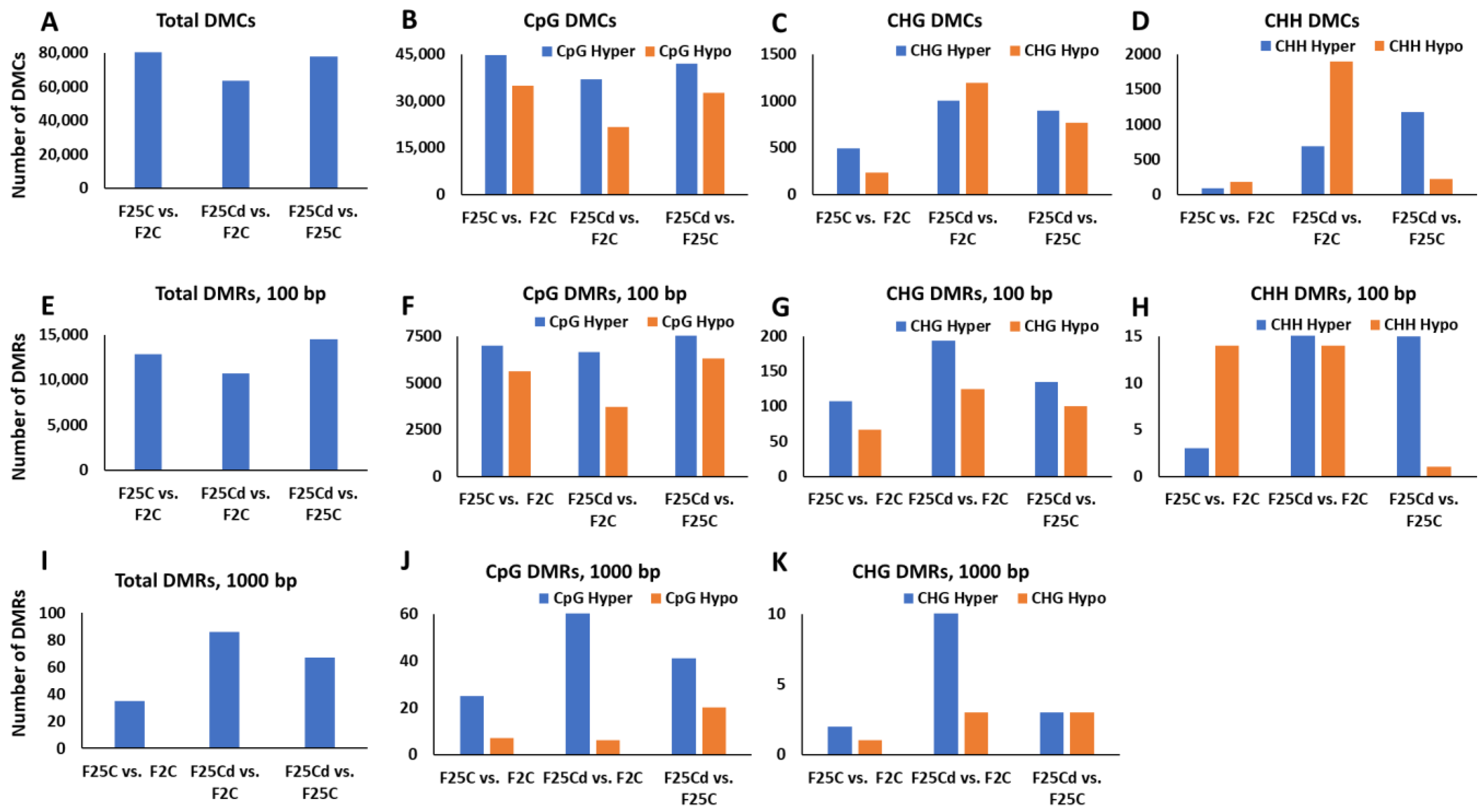
2.2.2. Analysis of DMCs and DMRs
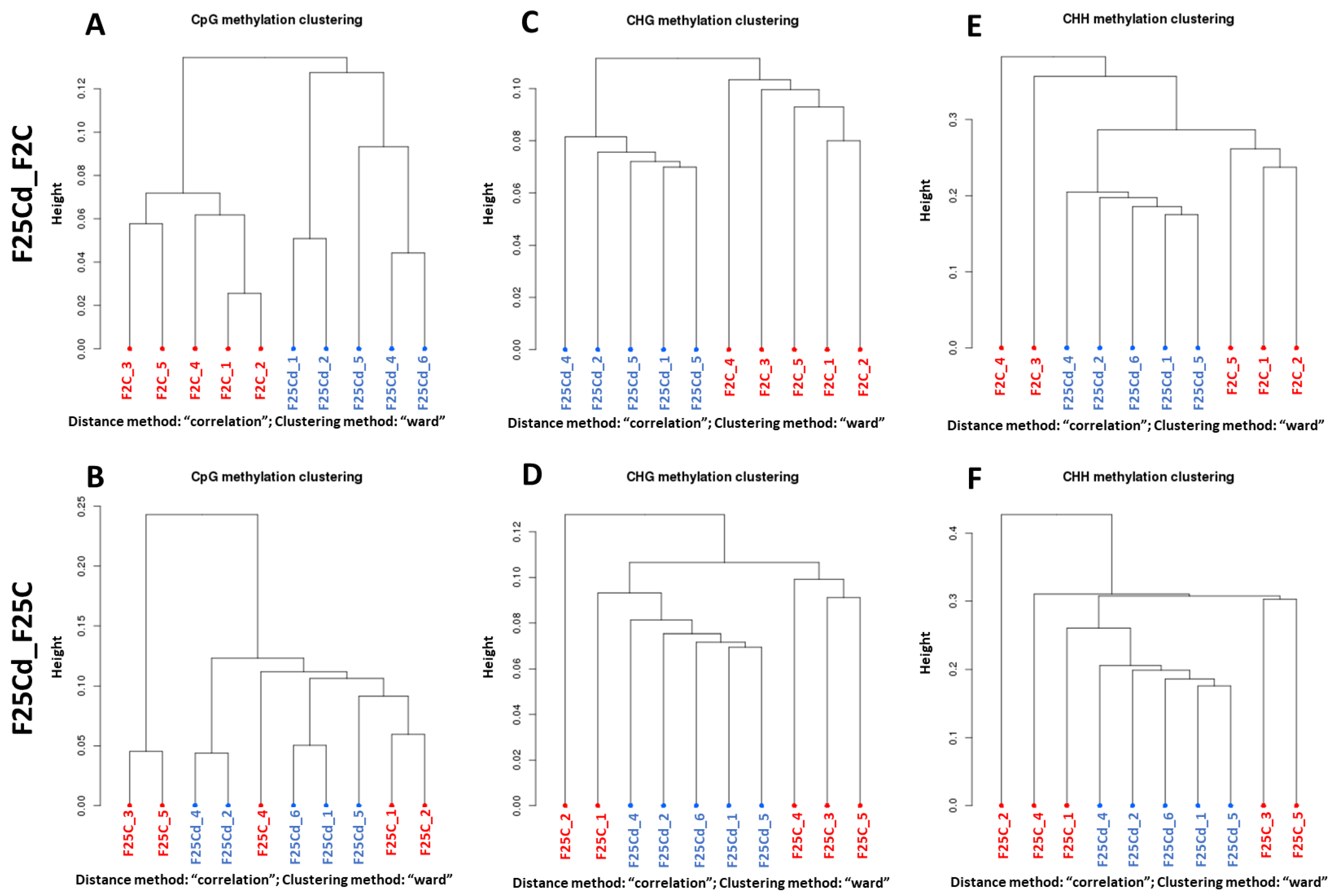
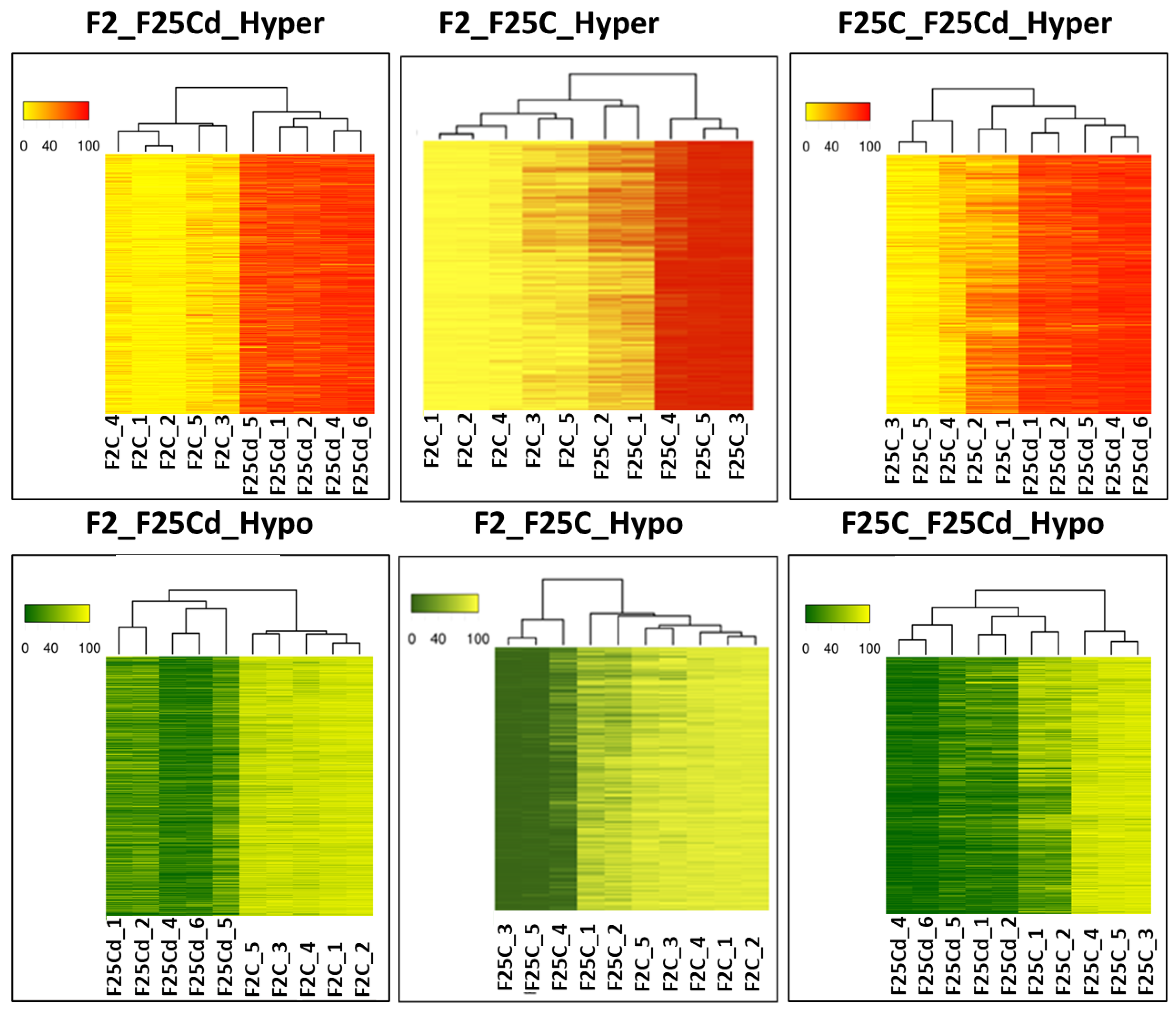
2.2.3. Hierarchical Clustering of Groups in DMCs and DMRs
2.2.4. The Analysis of the Distribution of DMCs across the Chromosomes
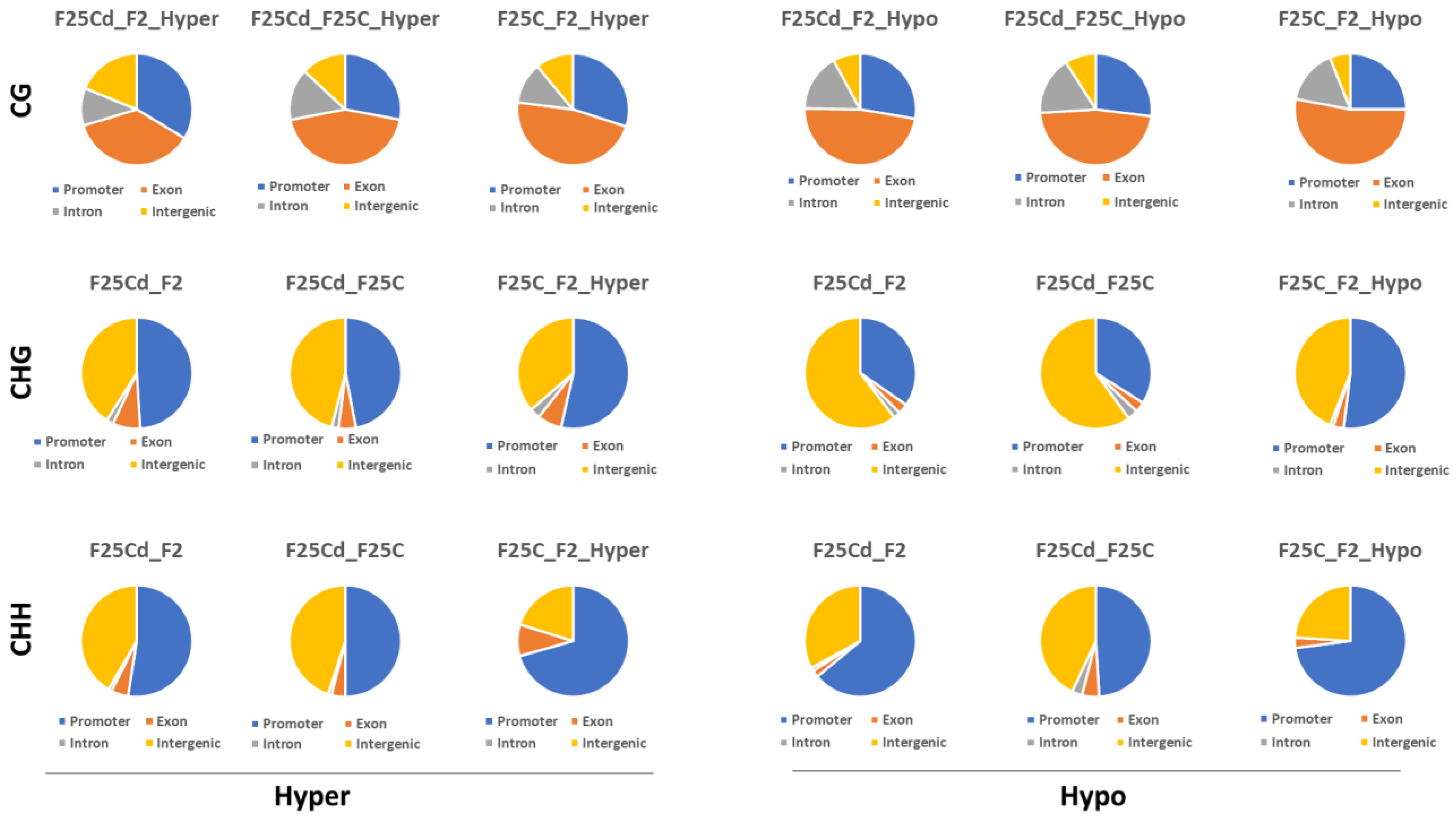
2.2.5. Distribution of DMCs and DMRs in the Genomic Regions
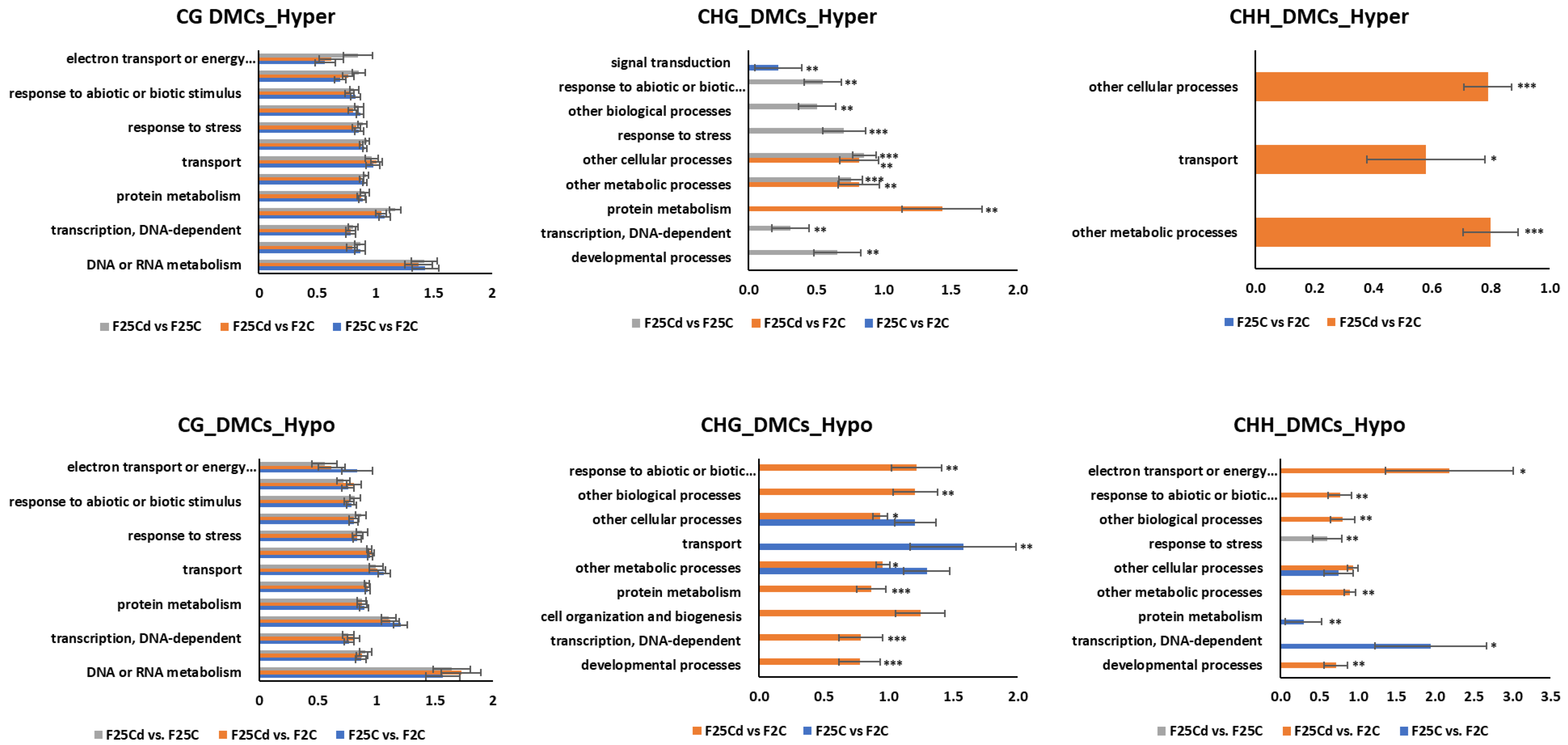
2.2.6. Biological Enrichment Analysis of DMCs and DMRs
3. Discussion
3.1. Genomic Analysis
3.1.1. Single Nucleotide Polymorphisms
3.1.2. Insertions and Deletions
3.1.3. The Functional Classification of SNPs and InDels
3.2. Analysis of Methylome
3.2.1. The Percentage of Global DNA Methylation
3.2.2. The Total Number of DMCs and DMRs
3.2.3. The Distribution of DMCs across Genic and Intergenic Regions
3.2.4. Biological Enrichment Analysis
4. Materials and Methods
4.1. Plant Material
4.2. Genomic and Epigenomic Profiling
4.3. DNA Isolation
4.4. Whole Genome Sequencing (WGS) and Whole Genome Bisulfite Sequencing (WGBS)
4.4.1. WGS Libraries Construction and Sequencing
4.4.2. WGBS Libraries Construction
4.4.3. WGS and WGBS Sequencing
4.5. Bioinformatics Analysis of WGS Data
4.6. Bioinformatics Analysis of WGBS Data
4.7. Analysis of Differentially Methylated Regions (DMRs)
4.8. Quality Control and Statistical Analysis of Sequencing Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rejeb, I.B.; Pastor, V.; Mauch-Mani, B. Plant Responses to Simultaneous Biotic and Abiotic Stress: Molecular Mechanisms. Plants 2014, 3, 458–475. [Google Scholar] [CrossRef]
- Lamke, J.; Baurle, I. Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 2017, 18, 124. [Google Scholar] [CrossRef]
- Kovalchuk, I. Role of Epigenetic Factors in Response to Stress and Establishment of Somatic Memory of Stress Exposure in Plants. Plants 2023, 12, 3667. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Vu, N.T.; Cheong, J.J. Transcriptional Stress Memory and Transgenerational Inheritance of Drought Tolerance in Plants. Int. J. Mol. Sci. 2022, 23, 2918. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; Verbon, E.H.; van Gurp, T.P.; Oplaat, C.; Ferreira de Carvalho, J.; Morse, A.M.; Stahl, M.; Macel, M.; McIntyre, L.M. Intergenerational environmental effects: Functional signals in offspring transcriptomes and metabolomes after parental jasmonic acid treatment in apomictic dandelion. New Phytol. 2018, 217, 871–882. [Google Scholar] [CrossRef]
- Lv, C.; Hu, Z.; Wei, J.; Wang, Y. Transgenerational effects of elevated CO(2) on rice photosynthesis and grain yield. Plant Mol. Biol. 2022, 110, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.S.; Titov, V.; Ayemere, I.; Byeon, B.; Ilnytskyy, Y.; Kovalchuk, I. Multigenerational Exposure to Heat Stress Induces Phenotypic Resilience, and Genetic and Epigenetic Variations in Arabidopsis thaliana Offspring. Front. Plant Sci. 2022, 13, 728167. [Google Scholar] [CrossRef] [PubMed]
- Pecinka, A.; Rosa, M.; Schikora, A.; Berlinger, M.; Hirt, H.; Luschnig, C.; Mittelsten Scheid, O. Transgenerational stress memory is not a general response in Arabidopsis. PLoS ONE 2009, 4, e5202. [Google Scholar] [CrossRef] [PubMed]
- Rasmann, S.; De Vos, M.; Casteel, C.L.; Tian, D.; Halitschke, R.; Sun, J.Y.; Agrawal, A.A.; Felton, G.W.; Jander, G. Herbivory in the previous generation primes plants for enhanced insect resistance. Plant Physiol. 2012, 158, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Wibowo, A.; Becker, C.; Marconi, G.; Durr, J.; Price, J.; Hagmann, J.; Papareddy, R.; Putra, H.; Kageyama, J.; Becker, J.; et al. Hyperosmotic stress memory in Arabidopsis is mediated by distinct epigenetically labile sites in the genome and is restricted in the male germline by DNA glycosylase activity. eLife 2016, 5, e13546. [Google Scholar] [CrossRef] [PubMed]
- Bilichak, A.; Ilnystkyy, Y.; Hollunder, J.; Kovalchuk, I. The progeny of Arabidopsis thaliana plants exposed to salt exhibit changes in DNA methylation, histone modifications and gene expression. PLoS ONE 2012, 7, e30515. [Google Scholar] [CrossRef]
- Migicovsky, Z.; Kovalchuk, I. Transgenerational inheritance of epigenetic response to cold in Arabidopsis thaliana. Biocatal. Agric. Biotechnol. 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Migicovsky, Z.; Yao, Y.; Kovalchuk, I. Transgenerational phenotypic and epigenetic changes in response to heat stress in Arabidopsis thaliana. Plant Signal Behav. 2014, 9, e27971. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Kovalchuk, I. Abiotic stress leads to somatic and heritable changes in homologous recombination frequency, point mutation frequency and microsatellite stability in Arabidopsis plants. Mutat. Res. 2011, 707, 61–66. [Google Scholar] [CrossRef]
- Yao, Y.; Bilichak, A.; Titov, V.; Golubov, A.; Kovalchuk, I. Genome stability of Arabidopsis atm, ku80 and rad51b mutants: Somatic and transgenerational responses to stress. Plant Cell Physiol. 2013, 54, 982–989. [Google Scholar] [CrossRef]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollunder, J.; Meins, F., Jr.; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef]
- Molinier, J.; Ries, G.; Zipfel, C.; Hohn, B. Transgeneration memory of stress in plants. Nature 2006, 442, 1046–1049. [Google Scholar] [CrossRef]
- Donohue, K. Completing the cycle: Maternal effects as the missing link in plant life histories. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Panda, K.; Mohanasundaram, B.; Gutierrez, J.; McLain, L.; Castillo, S.E.; Sheng, H.; Casto, A.; Gratacos, G.; Chakrabarti, A.; Fahlgren, N.; et al. The plant response to high CO2 levels is heritable and orchestrated by DNA methylation. New Phytol. 2023, 238, 2427–2439. [Google Scholar] [CrossRef]
- Thellier, M.; Luttge, U. Plant memory: A tentative model. Plant Biol. 2013, 15, 1–12. [Google Scholar] [CrossRef]
- Muller-Xing, R.; Xing, Q.; Goodrich, J. Footprints of the sun: Memory of UV and light stress in plants. Front. Plant Sci. 2014, 5, 474. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, K.J.; Preite, V. Epigenetic variation in asexually reproducing organisms. Evolution 2014, 68, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.M.; Baulcombe, D.C. Small RNAs and heritable epigenetic variation in plants. Trends Cell Biol. 2014, 24, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Weng, M.L.; Becker, C.; Hildebrandt, J.; Neumann, M.; Rutter, M.T.; Shaw, R.G.; Weigel, D.; Fenster, C.B. Fine-Grained Analysis of Spontaneous Mutation Spectrum and Frequency in Arabidopsis thaliana. Genetics 2019, 211, 703–714. [Google Scholar] [CrossRef] [PubMed]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledo, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Weigel, D.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef]
- Migicovsky, Z.; Kovalchuk, I. Transgenerational changes in plant physiology and in transposon expression in response to UV-C stress in Arabidopsis thaliana. Plant Signal Behav. 2014, 9, e976490. [Google Scholar] [CrossRef]
- Yang, J.; Duan, G.; Li, C.; Liu, L.; Han, G.; Zhang, Y.; Wang, C. The Crosstalks between Jasmonic Acid and Other Plant Hormone Signaling Highlight the Involvement of Jasmonic Acid as a Core Component in Plant Response to Biotic and Abiotic Stresses. Front. Plant Sci. 2019, 10, 1349. [Google Scholar] [CrossRef]
- Saijo, Y.; Loo, E.P. Plant immunity in signal integration between biotic and abiotic stress responses. New Phytol. 2020, 225, 87–104. [Google Scholar] [CrossRef]
- Zhu, F.; Sun, Y.; Jadhav, S.S.; Cheng, Y.; Alseekh, S.; Fernie, A.R. The Plant Metabolic Changes and the Physiological and Signaling Functions in the Responses to Abiotic Stress. Methods Mol. Biol. 2023, 2642, 129–150. [Google Scholar] [CrossRef]
- Gill, S.S.; Anjum, N.A.; Gill, R.; Jha, M.; Tuteja, N. DNA damage and repair in plants under ultraviolet and ionizing radiations. Sci. World J. 2015, 2015, 250158. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Genome instability and epigenetic modification—Heritable responses to environmental stress? Curr. Opin. Plant Biol. 2011, 14, 260–266. [Google Scholar] [CrossRef]
- Migicovsky, Z.; Kovalchuk, I. Changes to DNA methylation and homologous recombination frequency in the progeny of stressed plants. Biochem. Cell Biol. 2013, 91, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Shastry, B.S. SNPs: Impact on gene function and phenotype. Methods Mol. Biol. 2009, 578, 3–22. [Google Scholar] [CrossRef]
- Filiault, D.L.; Wessinger, C.A.; Dinneny, J.R.; Lutes, J.; Borevitz, J.O.; Weigel, D.; Chory, J.; Maloof, J.N. Amino acid polymorphisms in Arabidopsis phytochrome B cause differential responses to light. Proc. Natl. Acad. Sci. USA 2008, 105, 3157–3162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Mao, X.; Zhang, J.; Chang, X.; Jing, R. Single-nucleotide polymorphisms and association analysis of drought-resistance gene TaSnRK2.8 in common wheat. Plant Physiol. Biochem. 2013, 70, 174–181. [Google Scholar] [CrossRef]
- Guyon-Debast, A.; Lecureuil, A.; Bonhomme, S.; Guerche, P.; Gallois, J.L. A SNP associated with alternative splicing of RPT5b causes unequal redundancy between RPT5a and RPT5b among Arabidopsis thaliana natural variation. BMC Plant Biol. 2010, 10, 158. [Google Scholar] [CrossRef]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Hase, Y.; Satoh, K.; Kitamura, S.; Oono, Y. Physiological status of plant tissue affects the frequency and types of mutations induced by carbon-ion irradiation in Arabidopsis. Sci. Rep. 2018, 8, 1394. [Google Scholar] [CrossRef]
- Lu, Z.; Cui, J.; Wang, L.; Teng, N.; Zhang, S.; Lam, H.M.; Zhu, Y.; Xiao, S.; Ke, W.; Lin, J.; et al. Genome-wide DNA mutations in Arabidopsis plants after multigenerational exposure to high temperatures. Genome Biol. 2021, 22, 160. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Moharana, K.C.; Shankar, R.; Kumari, R.; Garg, R. Genomewide discovery of DNA polymorphisms in rice cultivars with contrasting drought and salinity stress response and their functional relevance. Plant Biotechnol. J. 2014, 12, 253–264. [Google Scholar] [CrossRef]
- Wheeler, B.S. Small RNAs, big impact: Small RNA pathways in transposon control and their effect on the host stress response. Chromosome Res. 2013, 21, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ye, C.; Wang, M.; Chu, G. Identification of cold-stress responsive proteins in Anabasis aphylla seedlings via the iTRAQ proteomics technique. J. Plant Interact. 2017, 12, 505–510. [Google Scholar] [CrossRef]
- Pikaard, C.S.; Mittelsten Scheid, O. Epigenetic regulation in plants. Cold Spring Harb. Perspect. Biol. 2014, 6, a019315. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.N.; Zhu, C.; Jiang, J.; Zhang, H.; Zhu, J.K.; Duan, C.G. Epigenetic regulation in plant abiotic stress responses. J. Integr. Plant Biol. 2020, 62, 563–580. [Google Scholar] [CrossRef]
- Remy, J.J. Stable inheritance of an acquired behavior in Caenorhabditis elegans. Curr. Biol. 2010, 20, R877–R878. [Google Scholar] [CrossRef]
- Groot, M.P.; Kooke, R.; Knoben, N.; Vergeer, P.; Keurentjes, J.J.; Ouborg, N.J.; Verhoeven, K.J. Effects of Multi-Generational Stress Exposure and Offspring Environment on the Expression and Persistence of Transgenerational Effects in Arabidopsis thaliana. PLoS ONE 2016, 11, e0151566. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, L.; Xia, H.; Wei, H.; Lou, Q.; Li, M.; Li, T.; Luo, L. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant’s adaptation to drought condition. Sci. Rep. 2017, 7, 39843. [Google Scholar] [CrossRef]
- Uthup, T.K.; Ravindran, M.; Bini, K.; Thakurdas, S. Divergent DNA methylation patterns associated with abiotic stress in Hevea brasiliensis. Mol. Plant 2011, 4, 996–1013. [Google Scholar] [CrossRef]
- Suter, L.; Widmer, A. Phenotypic effects of salt and heat stress over three generations in Arabidopsis thaliana. PLoS ONE 2013, 8, e80819. [Google Scholar] [CrossRef]
- Jiang, C.; Mithani, A.; Belfield, E.J.; Mott, R.; Hurst, L.D.; Harberd, N.P. Environmentally responsive genome-wide accumulation of de novo Arabidopsis thaliana mutations and epimutations. Genome Res. 2014, 24, 1821–1829. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Zhang, Y.; Xu, C.; Lin, X.; Zang, Q.; Zhuang, T.; Jiang, L.; von Wettstein, D.; Liu, B. Transgenerational inheritance of modified DNA methylation patterns and enhanced tolerance induced by heavy metal stress in rice (Oryza sativa L.). PLoS ONE 2012, 7, e41143. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef]
- Cramer, G.R.; Urano, K.; Delrot, S.; Pezzotti, M.; Shinozaki, K. Effects of abiotic stress on plants: A systems biology perspective. BMC Plant Biol. 2011, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Harrison, J.; Paul, C.L.; Frommer, M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994, 22, 2990–2997. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.D.; Schmitz, R.J.; Ecker, J.R. ‘Leveling’ the playing field for analyses of single-base resolution DNA methylomes. Trends Genet. 2012, 28, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Franke, V.; Vlahovicek, K.; Mason, C.E.; Schubeler, D. Genomation: A toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015, 31, 1127–1129. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Provart, N.J.; Zhu, T. A browser-based functional classification SuperViewer for Arabidopsis genomics. Curr. Comp. Mol. Biol. 2003, 2003, 271–272. [Google Scholar]
- Iwasaki, M.; Paszkowski, J. Identification of genes preventing transgenerational transmission of stress-induced epigenetic states. Proc. Natl. Acad. Sci. USA 2014, 111, 8547–8552. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A Role for Epigenetic Regulation in the Adaptation and Stress Responses of Non-Model Plants. Front. Plant Sci. 2019, 10, 246. [Google Scholar] [CrossRef]
- Song, J.; Angel, A.; Howard, M.; Dean, C. Vernalization—A cold-induced epigenetic switch. J. Cell Sci. 2012, 125, 3723–3731. [Google Scholar] [CrossRef]
- Baulcombe, D.C.; Dean, C. Epigenetic regulation in plant responses to the environment. Cold Spring Harb. Perspect. Biol. 2014, 6, a019471. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrichment | F2C | F25C | F25Cd |
|---|---|---|---|
| Biological process | |||
| transcription, DNA-dependent | 1.3 ± 0.26 | - | - |
| other metabolic processes | 0.87 ± 0.08 | 0.82 ± 0.13 | 0.88 ± 0.08 |
| other cellular processes | 0.82 ± 0.08 | 0.79 ± 0.13 | 0.81 ± 0.08 |
| cell organization and biogenesis | 0.47 ± 0.16 | - | 0.51 ± 0.16 |
| response to stress | 0.69 ± 0.16 | - | - |
| other biological processes | - | 0.33 ± 0.19 | - |
| protein metabolism | - | - | 0.76 ± 0.17 |
| Molecular function | |||
| transcription factor activity | 1.53 ± 0.33 | - | - |
| protein binding | 0.74 ± 0.16 | - | 0.74 ± 0.16 |
| nucleotide binding | 0.7 ± 0.19 | 0.13 ± 0.11 * | 0.61 ± 0.19 |
| other binding | - | 0.73 ± 0.19 | 1.1 ± 0.12 * |
| transferase activity | - | 0.32 ± 0.17 | 0.72 ± 0.19 * |
| hydrolase activity | - | 0.23 ± 0.16 | - |
| other enzyme activity | - | 0.21 ± 0.14 | - |
| Cellular component | |||
| other membranes | 0.8 ± 0.13 | 0.55 ± 0.17 | 0.85 ± 0.12 |
| other cytoplasmic components | 0.77 ± 0.11 | 0.53 ± 0.16 | 0.79 ± 0.12 |
| chloroplast | 0.72 ± 0.16 | - | 0.8 ± 0.16 |
| other intracellular components | 0.71 ± 0.12 | 0.64 ± 0.19 | 0.72 ± 0.13 |
| cytosol | 0.49 ± 0.20 | - | - |
| plasma membrane | - | 0.5 ± 0.23 | - |
| nucleus | - | 1.26 ± 0.17 | - |
| other cellular components | - | - | 0.46 ± 0.23 |
| extracellular | - | - | 1.38 ± 0.24 |
| mitochondria | - | - | 0.72 ± 0.18 |
| Enrichment | F2C | F25C | F25Cd |
|---|---|---|---|
| Biological process | |||
| signal transduction | 1.23 ± 0.22 | 0.62 ± 0.18 | 1.20 ± 0.15 |
| transport | 1.20 ± 0.20 | - | 1.08 ± 0.14 |
| transcription, DNA-dependent | 1.12 ± 0.15 | 1.38 ± 0.2 | 1.38 ± 0.13 |
| protein metabolism | 1.10 ± 0.14 | 0.89 ± 0.13 | 0.91 ± 0.08 |
| other metabolic processes | 0.99 ± 0.06 | 1.00 ± 0.07 | 1.01 ± 0.04 |
| other cellular processes | 0.99 ± 0.05 | 0.93 ± 0.07 | 1.00 ± 0.04 |
| developmental processes | 0.87 ± 0.14 | - | 1.19 ± 0.12 * |
| response to abiotic or biotic stimulus | - | 0.72 ± 0.13 | 1.09 ± 0.11 * |
| response to stress | - | - | 1.25 ± 0.11 |
| cell organization and biogenesis | - | - | 0.85 ± 0.12 |
| other biological processes | - | - | 1.15 ± 0.13 |
| Molecular function | |||
| other molecular functions | 1.54 ± 0.33 | - | 1.28 ± 0.23 |
| nucleic acid binding | 1.39 ± 0.24 | 1.36 ± 0.26 | - |
| transporter activity | 1.32 ± 0.28 | - | - |
| transcription factor activity | 1.22 ± 0.21 | 1.62 ± 0.26 | 1.53 ± 0.17 |
| DNA or RNA binding | 1.18 ± 0.13 | 1.19 ± 0.15 | 1.17 ± 0.09 |
| protein binding | 1.17 ± 0.12 | - | 1.11 ± 0.09 |
| other binding | 0.99 ± 0.07 | 0.87 ± 0.09 | 1.00 ± 0.07 |
| transferase activity | 0.89 ± 0.12 | 0.70 ± 0.13 | 0.90 ± 0.08 |
| other enzyme activity | 0.86 ± 0.08 | 0.89 ± 0.11 | 0.87 ± 0.07 |
| hydrolase activity | - | 0.81 ± 0.15 | 0.87 ± 0.09 |
| nucleotide binding | - | 0.58 ± 0.14 | 0.91 ± 0.10 * |
| kinase activity | - | - | 1.15 ± 0.17 |
| Cellular component | |||
| other cellular components | 1.22 ± 0.26 | 1.63 ± 0.31 | 1.34 ± 0.2 |
| mitochondria | 1.13 ± 0.13 | - | 1.08 ± 0.1 |
| nucleus | 1.07 | 1.03 | 1.03 |
| other intracellular components | 1.01 ± 0.1 | 0.87 ± 0.1 | 0.90 ± 0.07 |
| other cytoplasmic components | 0.94 ± 0.07 | 0.96 ± 0.08 | 0.97 ± 0.06 |
| extracellular | 0.84 ± 0.14 | 0.83 ± 0.14 | 1.05 ± 0.11 |
| other membranes | 0.83 ± 0.09 | 0.91 ± 0.1 | 0.89 ± 0.06 |
| chloroplast | 0.78 ± 0.11 | 0.76 ± 0.12 | 0.83 ± 0.09 |
| Golgi apparatus | 0.66 ± 0.19 | - | 0.67 ± 0.15 |
| plasma membrane | - | 0.77 ± 0.13 | 0.87 ± 0.08 |
| cytosol | - | 0.56 ± 0.17 | 0.85 ± 0.11 * |
| plastid | - | - | 0.79 ± 0.14 |
| cell wall | - | - | 1.27 ± 0.23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, A.; Yadav, N.S.; Byeon, B.; Ilnytskyy, Y.; Kovalchuk, I. Genomic and Epigenomic Changes in the Progeny of Cold-Stressed Arabidopsis thaliana Plants. Int. J. Mol. Sci. 2024, 25, 2795. https://doi.org/10.3390/ijms25052795
Rahman A, Yadav NS, Byeon B, Ilnytskyy Y, Kovalchuk I. Genomic and Epigenomic Changes in the Progeny of Cold-Stressed Arabidopsis thaliana Plants. International Journal of Molecular Sciences. 2024; 25(5):2795. https://doi.org/10.3390/ijms25052795
Chicago/Turabian StyleRahman, Ashif, Narendra Singh Yadav, Boseon Byeon, Yaroslav Ilnytskyy, and Igor Kovalchuk. 2024. "Genomic and Epigenomic Changes in the Progeny of Cold-Stressed Arabidopsis thaliana Plants" International Journal of Molecular Sciences 25, no. 5: 2795. https://doi.org/10.3390/ijms25052795
APA StyleRahman, A., Yadav, N. S., Byeon, B., Ilnytskyy, Y., & Kovalchuk, I. (2024). Genomic and Epigenomic Changes in the Progeny of Cold-Stressed Arabidopsis thaliana Plants. International Journal of Molecular Sciences, 25(5), 2795. https://doi.org/10.3390/ijms25052795