
Sporadic Inclusion Body Myositis at the Crossroads between Muscle Degeneration, Inflammation, and Aging


Abstract
1. Introduction
2. Diagnosis of sIBM
2.1. Clinical Aspects
2.2. Laboratory Studies
2.3. Magnetic Resonance Imaging (MRI)
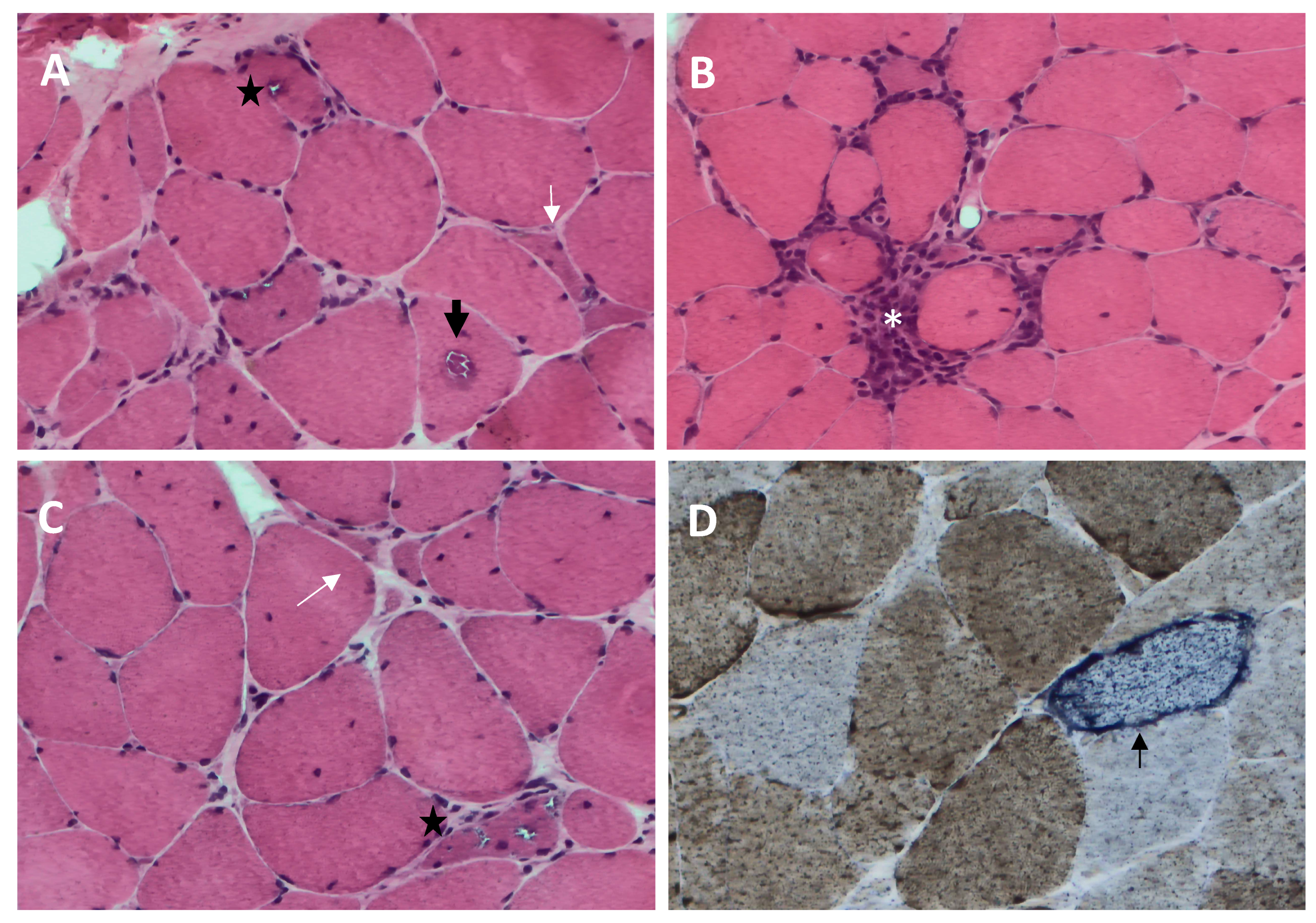
2.4. Pathological Characteristics
- (1)
- Rimmed vacuoles, namely irregular vacuoles of variable size and shape, bordered by basophilic granular deposits that occur in nonnecrotic muscle fibers. Rimmed vacuoles can be a rather rare finding and are visible in 0.4–6.4% of the fibers [9];
- (2)
- Eosinophilic cytoplasmic inclusions visible at hematoxylin and eosin (H&E) and modified Gomori trichrome staining [17];
- (3)
- Cytoplasmic accumulation of aggregated/misfolded proteins, referred to as amyloid deposits or protein inclusions, which occur in 60 to 80% of the sIBM vacuolated muscle fibers, usually in non-vacuolated areas of the fiber [1,17]. These β-pleated-sheet amyloid inclusions are detectable by fluorescence-enhanced Congo red staining. Several proteins have been found within these aggregates, including amyloid-β precursor protein (AβPP), amyloid-β (Aβ), phosphorylated-tau (p-tau), and ubiquitin, to name a few [8]. Notably, TDP-43 immunopositivity has been reported in over 60% of sIBM patients and is considered a hallmark of sIBM pathology [39,40];
- (4)
- Endomysial lymphocytic infiltrates consisting predominantly of macrophages and CD8+T cells that invade nonnecrotic muscle fibers expressing MHC class I on the sarcolemma [17];
- (5)
- Mitochondrial abnormalities consisting of the abnormal proliferation of mitochondrial leading to ragged red fibers (RRFs, muscle fibers containing excessive mitochondrial proliferation) and the impairment of mitochondrial function, as shown by increased cytochrome c oxidase (COX)-negative fibers [41,42];
- (6)
- Angulated muscle fibers of small caliber suggesting a neurogenic process [43].
2.5. Correlation of Pathological Features with Clinical Progression and Laboratory Findings
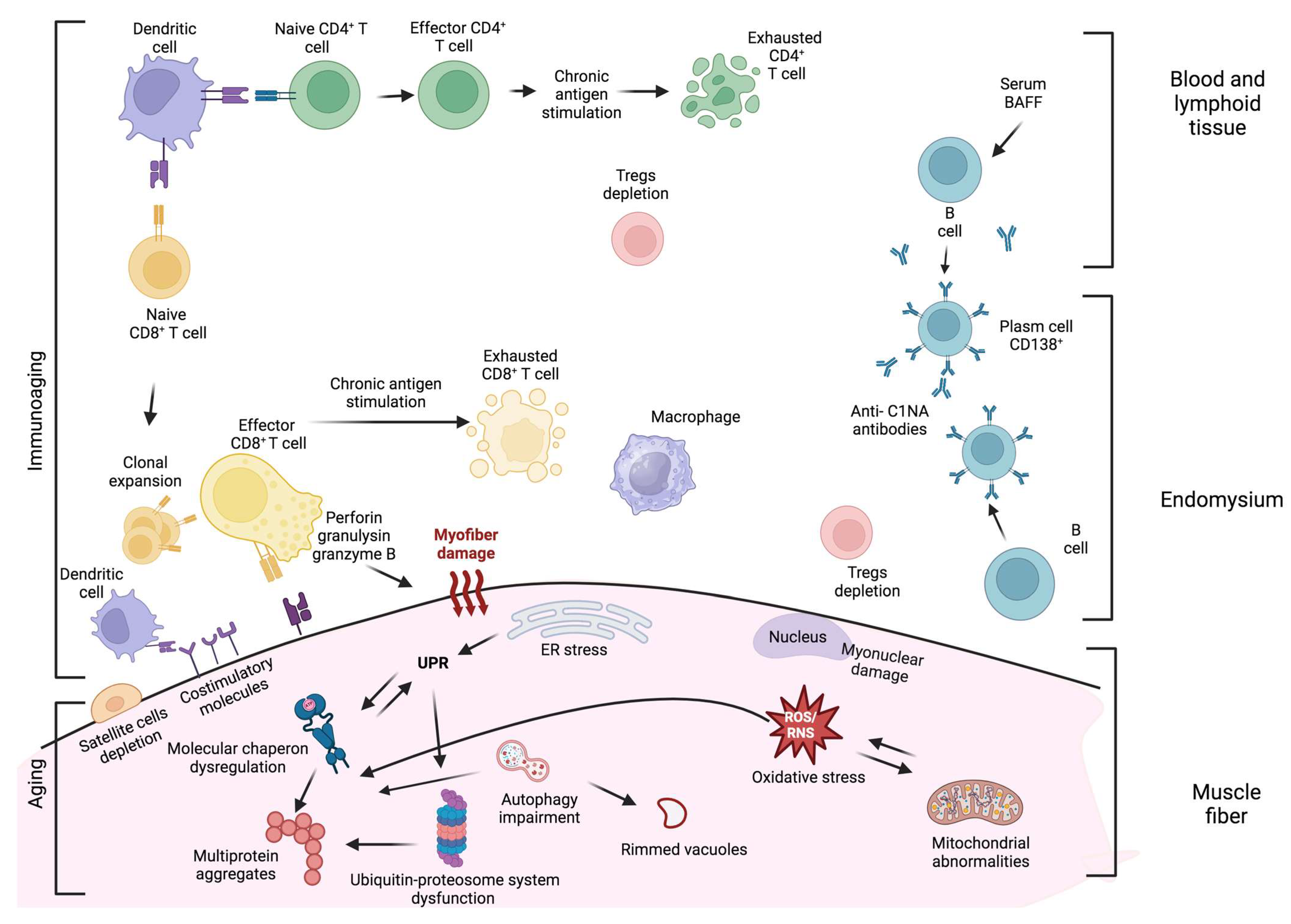
3. Degenerative Processes in sIBM Pathogenesis
3.1. Protein Aggregation
3.2. Impairment of Ubiquitin–Proteasome System (UPS) and Autophagy
3.3. Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response (UPR)
3.4. Mitochondrial Abnormalities
3.5. Oxidative Stress
3.6. Nuclear Degeneration
4. Aged Skeletal Muscle Milieu
5. Inflammation in sIBM Pathogenesis
5.1. T Cells
5.2. Plasma Cells and Antibody-Mediated Immune Response
5.3. Macrophages and Dendritic Cells
6. Aging of the Immune System and sIBM
7. Interplay between Degeneration and Inflammation
8. Therapeutic Approaches
9. Future Perspective in sIBM Research and Therapy
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Engel, W.K.; Askanas, V. Inclusion-body myositis: Clinical, diagnostic, and pathologic aspects. Neurology 2006, 66, S20–S29. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Karpati, G.; Heller, I.; Eisen, A. Inclusion body myositis: A distinct variety of idiopathic inflammatory myopathy. Neurology 1978, 28, 8–17. [Google Scholar] [CrossRef]
- Price, M.A.; Barghout, V.; Benveniste, O.; Christopher-Stine, L.; Corbett, A.; de Visser, M.; Hilton-Jones, D.; Kissel, J.T.; Lloyd, T.E.; Lundberg, I.E.; et al. Mortality and Causes of Death in Patients with Sporadic Inclusion Body Myositis: Survey Study Based on the Clinical Experience of Specialists in Australia, Europe and the USA. J. Neuromuscul. Dis. 2016, 3, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Needham, M.; Corbett, A.; Day, T.; Christiansen, F.; Fabian, V.; Mastaglia, F.L. Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J. Clin. Neurosci. 2008, 15, 1350–1353. [Google Scholar] [CrossRef] [PubMed]
- Callan, A.; Capkun, G.; Vasanthaprasad, V.; Freitas, R.; Needham, M. A Systematic Review and Meta-Analysis of Prevalence Studies of Sporadic Inclusion Body Myositis. J. Neuromuscul. Dis. 2017, 4, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Khan, A.; Machado, P.M.; Houlden, H. Inclusion body myositis: From genetics to clinical trials. J. Neurol. 2023, 270, 1787–1797. [Google Scholar] [CrossRef]
- Skolka, M.P.; Naddaf, E. Exploring challenges in the management and treatment of inclusion body myositis. Curr. Opin. Rheumatol. 2023, 35, 404–413. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K.; Nogalska, A. Sporadic inclusion-body myositis: A degenerative muscle disease associated with aging, impaired muscle protein homeostasis and abnormal mitophagy. Biochim. Biophys. Acta 2015, 1852, 633–643. [Google Scholar] [CrossRef]
- Greenberg, S.A. Inclusion body myositis: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2019, 15, 257–272. [Google Scholar] [CrossRef]
- Benveniste, O.; Guiguet, M.; Freebody, J.; Dubourg, O.; Squier, W.; Maisonobe, T.; Stojkovic, T.; Leite, M.I.; Allenbach, Y.; Herson, S.; et al. Long-term observational study of sporadic inclusion body myositis. Brain 2011, 134, 3176–3184. [Google Scholar] [CrossRef]
- Danon, M.J.; Reyes, M.G.; Perurena, O.H.; Masdeu, J.C.; Manaligod, J.R. Inclusion body myositis. A corticosteroid-resistant idiopathic inflammatory myopathy. Arch. Neurol. 1982, 39, 760–764. [Google Scholar] [CrossRef]
- Oh, T.H.; Brumfield, K.A.; Hoskin, T.L.; Kasperbauer, J.L.; Basford, J.R. Dysphagia in inclusion body myositis: Clinical features, management, and clinical outcome. Am. J. Phys. Med. Rehabil. 2008, 87, 883–889. [Google Scholar] [CrossRef]
- Goodman, B.P.; Liewluck, T.; Crum, B.A.; Spinner, R.J. Camptocormia due to inclusion body myositis. J. Clin. Neuromuscul. Dis. 2012, 14, 78–81. [Google Scholar] [CrossRef]
- Salam, S.; Morrow, J.M.; Howard, R.; Miller, J.A.L.; Quinlivan, R.M.; Machado, P.M. Two emerging phenotypes of atypical inclusion body myositis: Illustrative cases. Clin. Exp. Rheumatol. 2023, 41, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Naddaf, E. Inclusion body myositis: Update on the diagnostic and therapeutic landscape. Front. Neurol. 2022, 13, 1020113. [Google Scholar] [CrossRef] [PubMed]
- Beyenburg, S.; Zierz, S.; Jerusalem, F. Inclusion body myositis: Clinical and histopathological features of 36 patients. Clin. Investig. 1993, 71, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Dimachkie, M.M. Idiopathic inflammatory myopathies. J. Neuroimmunol. 2011, 231, 32–42. [Google Scholar] [CrossRef]
- Satoh, M.; Tanaka, S.; Ceribelli, A.; Calise, S.J.; Chan, E.K. A Comprehensive Overview on Myositis-Specific Antibodies: New and Old Biomarkers in Idiopathic Inflammatory Myopathy. Clin. Rev. Allergy Immunol. 2017, 52, 1–19. [Google Scholar] [CrossRef]
- Salajegheh, M.; Lam, T.; Greenberg, S.A. Autoantibodies against a 43 KDa muscle protein in inclusion body myositis. PLoS ONE 2011, 6, e20266. [Google Scholar] [CrossRef]
- Larman, H.B.; Salajegheh, M.; Nazareno, R.; Lam, T.; Sauld, J.; Steen, H.; Kong, S.W.; Pinkus, J.L.; Amato, A.A.; Elledge, S.J.; et al. Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann. Neurol. 2013, 73, 408–418. [Google Scholar] [CrossRef]
- Pluk, H.; van Hoeve, B.J.; van Dooren, S.H.; Stammen-Vogelzangs, J.; van der Heijden, A.; Schelhaas, H.J.; Verbeek, M.M.; Badrising, U.A.; Arnardottir, S.; Gheorghe, K.; et al. Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann. Neurol. 2013, 73, 397–407. [Google Scholar] [CrossRef]
- Rietveld, A.; van den Hoogen, L.L.; Bizzaro, N.; Blokland, S.L.M.; Dähnrich, C.; Gottenberg, J.E.; Houen, G.; Johannsen, N.; Mandl, T.; Meyer, A.; et al. Autoantibodies to Cytosolic 5′-Nucleotidase 1A in Primary Sjögren’s Syndrome and Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1200. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.K.; Stammen-Vogelzangs, J.; Verbeek, M.M.; Rietveld, A.; Lundberg, I.E.; Chinoy, H.; Lamb, J.A.; Cooper, R.G.; Roberts, M.; Badrising, U.A.; et al. Disease specificity of autoantibodies to cytosolic 5′-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann. Rheum. Dis. 2016, 75, 696–701. [Google Scholar] [CrossRef]
- Diederichsen, L.P.; Iversen, L.V.; Nielsen, C.T.; Jacobsen, S.; Hermansen, M.L.; Witting, N.; Cortes, R.; Korsholm, S.S.; Krogager, M.E.; Friis, T. Myositis-related autoantibody profile and clinical characteristics stratified by anti-cytosolic 5′-nucleotidase 1A status in connective tissue diseases. Muscle Nerve 2023, 68, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Lilleker, J.B.; Rietveld, A.; Pye, S.R.; Mariampillai, K.; Benveniste, O.; Peeters, M.T.; Miller, J.A.; Hanna, M.G.; Machado, P.M.; Parton, M.J.; et al. Cytosolic 5′-nucleotidase 1A autoantibody profile and clinical characteristics in inclusion body myositis. Ann. Rheum. Dis. 2017, 76, 862–868. [Google Scholar] [CrossRef]
- Goyal, N.A.; Cash, T.M.; Alam, U.; Enam, S.; Tierney, P.; Araujo, N.; Mozaffar, F.H.; Pestronk, A.; Mozaffar, T. Seropositivity for NT5c1A antibody in sporadic inclusion body myositis predicts more severe motor, bulbar and respiratory involvement. J. Neurol. Neurosurg. Psychiatry 2016, 87, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Lucchini, M.; Maggi, L.; Pegoraro, E.; Filosto, M.; Rodolico, C.; Antonini, G.; Garibaldi, M.; Valentino, M.L.; Siciliano, G.; Tasca, G.; et al. Anti-cN1A Antibodies Are Associated with More Severe Dysphagia in Sporadic Inclusion Body Myositis. Cells 2021, 10, 1146. [Google Scholar] [CrossRef] [PubMed]
- Felice, K.J.; Whitaker, C.H.; Wu, Q.; Larose, D.T.; Shen, G.; Metzger, A.L.; Barton, R.W. Sensitivity and clinical utility of the anti-cytosolic 5′-nucleotidase 1A (cN1A) antibody test in sporadic inclusion body myositis: Report of 40 patients from a single neuromuscular center. Neuromuscul. Disord. 2018, 28, 660–664. [Google Scholar] [CrossRef]
- Oyama, M.; Ohnuki, Y.; Inoue, M.; Uruha, A.; Yamashita, S.; Yutani, S.; Tanboon, J.; Nakahara, J.; Suzuki, S.; Shiina, T.; et al. HLA-DRB1 allele and autoantibody profiles in Japanese patients with inclusion body myositis. PLoS ONE 2020, 15, e0237890. [Google Scholar] [CrossRef]
- Tawara, N.; Yamashita, S.; Zhang, X.; Korogi, M.; Zhang, Z.; Doki, T.; Matsuo, Y.; Nakane, S.; Maeda, Y.; Sugie, K.; et al. Pathomechanisms of anti-cytosolic 5′-nucleotidase 1A autoantibodies in sporadic inclusion body myositis. Ann. Neurol. 2017, 81, 512–525. [Google Scholar] [CrossRef]
- Greenberg, S.A. Cytoplasmic 5′-nucleotidase autoantibodies in inclusion body myositis: Isotypes and diagnostic utility. Muscle Nerve 2014, 50, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Zubair, A.S.; Salam, S.; Dimachkie, M.M.; Machado, P.M.; Roy, B. Imaging biomarkers in the idiopathic inflammatory myopathies. Front. Neurol. 2023, 14, 1146015. [Google Scholar] [CrossRef] [PubMed]
- Dion, E.; Cherin, P.; Payan, C.; Fournet, J.C.; Papo, T.; Maisonobe, T.; Auberton, E.; Chosidow, O.; Godeau, P.; Piette, J.C.; et al. Magnetic resonance imaging criteria for distinguishing between inclusion body myositis and polymyositis. J. Rheumatol. 2002, 29, 1897–1906. [Google Scholar]
- Phillips, B.A.; Cala, L.A.; Thickbroom, G.W.; Melsom, A.; Zilko, P.J.; Mastaglia, F.L. Patterns of muscle involvement in inclusion body myositis: Clinical and magnetic resonance imaging study. Muscle Nerve 2001, 24, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, J.B.; Zanoteli, E.; Link, T.M.; de Camargo, L.V.; Facchetti, L.; Nardo, L.; Fernandes, A. Sporadic Inclusion Body Myositis: MRI Findings and Correlation With Clinical and Functional Parameters. Am. J. Roentgenol. 2017, 209, 1340–1347. [Google Scholar] [CrossRef]
- Ansari, B.; Salort-Campana, E.; Ogier, A.; Le Troter Ph, D.A.; De Sainte Marie, B.; Guye, M.; Delmont, E.; Grapperon, A.M.; Verschueren, A.; Bendahan, D.; et al. Quantitative muscle MRI study of patients with sporadic inclusion body myositis. Muscle Nerve 2020, 61, 496–503. [Google Scholar] [CrossRef]
- Tasca, G.; Monforte, M.; De Fino, C.; Kley, R.A.; Ricci, E.; Mirabella, M. Magnetic resonance imaging pattern recognition in sporadic inclusion-body myositis. Muscle Nerve 2015, 52, 956–962. [Google Scholar] [CrossRef]
- Cox, F.M.; Reijnierse, M.; van Rijswijk, C.S.; Wintzen, A.R.; Verschuuren, J.J.; Badrising, U.A. Magnetic resonance imaging of skeletal muscles in sporadic inclusion body myositis. Rheumatology 2011, 50, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Hiniker, A.; Daniels, B.H.; Lee, H.S.; Margeta, M. Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol. Commun. 2013, 1, 29. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K.; Nogalska, A. Pathogenic considerations in sporadic inclusion-body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J. Neuropathol. Exp. Neurol. 2012, 71, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, U.; Roos, S.; Hedberg Oldfors, C.; Moslemi, A.R.; Lindberg, C.; Oldfors, A. Mitochondrial pathology in inclusion body myositis. Neuromuscul. Disord. 2015, 25, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Oldfors, A.; Moslemi, A.R.; Jonasson, L.; Ohlsson, M.; Kollberg, G.; Lindberg, C. Mitochondrial abnormalities in inclusion-body myositis. Neurology 2006, 66, S49–S55. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; Mirabella, M.; Guglielmi, V.; Lucchini, M.; Tomelleri, G.; Ghirardello, A.; Doria, A. Muscle biopsy features of idiopathic inflammatory myopathies and differential diagnosis. Autoimmun. Highlights 2014, 5, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.V.; Laughlin, R.S.; Klein, C.J.; Mandrekar, J.; Naddaf, E. Inclusion body myositis: Correlation of clinical outcomes with histopathology, electromyography and laboratory findings. Rheumatology 2022, 61, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, T.E.; Christopher-Stine, L.; Pinal-Fernandez, I.; Tiniakou, E.; Petri, M.; Baer, A.; Danoff, S.K.; Pak, K.; Casciola-Rosen, L.A.; Mammen, A.L. Cytosolic 5′-Nucleotidase 1A As a Target of Circulating Autoantibodies in Autoimmune Diseases. Arthritis Care Res. 2016, 68, 66–71. [Google Scholar] [CrossRef]
- Ikenaga, C.; Findlay, A.R.; Goyal, N.A.; Robinson, S.; Cauchi, J.; Hussain, Y.; Wang, L.H.; Kershen, J.C.; Beson, B.A.; Wallendorf, M.; et al. Clinical utility of anti-cytosolic 5′-nucleotidase 1A antibody in idiopathic inflammatory myopathies. Ann. Clin. Transl. Neurol. 2021, 8, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Paul, P.; Liewluck, T.; Ernste, F.C.; Mandrekar, J.; Milone, M. Anti-cN1A antibodies do not correlate with specific clinical, electromyographic, or pathological findings in sporadic inclusion body myositis. Muscle Nerve 2021, 63, 490–496. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Gales, T.; Paul, L. Amyloid filaments in inclusion body myositis. Novel findings provide insight into nature of filaments. Arch. Neurol. 1991, 48, 1229–1234. [Google Scholar] [CrossRef]
- Askanas, V.; Serdaroglu, P.; Engel, W.K.; Alvarez, R.B. Immunolocalization of ubiquitin in muscle biopsies of patients with inclusion body myositis and oculopharyngeal muscular dystrophy. Neurosci. Lett. 1991, 130, 73–76. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K.; Alvarez, R.B. Light and electron microscopic localization of beta-amyloid protein in muscle biopsies of patients with inclusion-body myositis. Am. J. Pathol. 1992, 141, 31–36. [Google Scholar]
- Askanas, V.; Engel, W.K.; Bilak, M.; Alvarez, R.B.; Selkoe, D.J. Twisted tubulofilaments of inclusion body myositis muscle resemble paired helical filaments of Alzheimer brain and contain hyperphosphorylated tau. Am. J. Pathol. 1994, 144, 177–187. [Google Scholar]
- Askanas, V.; Alvarez, R.B.; Engel, W.K. beta-Amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann. Neurol. 1993, 34, 551–560. [Google Scholar] [CrossRef]
- Sarkozi, E.; Askanas, V.; Johnson, S.A.; Engel, W.K.; Alvarez, R.B. beta-Amyloid precursor protein mRNA is increased in inclusion-body myositis muscle. Neuroreport 1993, 4, 815–818. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. NeuroMolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef]
- Nogalska, A.; D’Agostino, C.; Engel, W.K.; Klein, W.L.; Askanas, V. Novel demonstration of amyloid-β oligomers in sporadic inclusion-body myositis muscle fibers. Acta Neuropathol. 2010, 120, 661–666. [Google Scholar] [CrossRef]
- Abdo, W.F.; van Mierlo, T.; Hengstman, G.J.; Schelhaas, H.J.; van Engelen, B.G.; Verbeek, M.M. Increased plasma amyloid-beta42 protein in sporadic inclusion body myositis. Acta Neuropathol. 2009, 118, 429–431. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vattemi, G.; Nogalska, A.; King Engel, W.; D’Agostino, C.; Checler, F.; Askanas, V. Amyloid-beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol. 2009, 117, 569–574. [Google Scholar] [CrossRef]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Buxbaum, J.D.; Pastorino, L.; Askanas, V. Presence of BACE1 and BACE2 in muscle fibres of patients with sporadic inclusion-body myositis. Lancet 2001, 358, 1962–1964. [Google Scholar] [CrossRef]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Pastorino, L.; Buxbaum, J.D.; Askanas, V. BACE1 and BACE2 in pathologic and normal human muscle. Exp. Neurol. 2003, 179, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Maurage, C.A.; Bussière, T.; Sergeant, N.; Ghesteem, A.; Figarella-Branger, D.; Ruchoux, M.M.; Pellissier, J.F.; Delacourte, A. Tau aggregates are abnormally phosphorylated in inclusion body myositis and have an immunoelectrophoretic profile distinct from other tauopathies. Neuropathol. Appl. Neurobiol. 2004, 30, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Wilczynski, G.M.; Engel, W.K.; Askanas, V. Association of active extracellular signal-regulated protein kinase with paired helical filaments of inclusion-body myositis muscle suggests its role in inclusion-body myositis tau phosphorylation. Am. J. Pathol. 2000, 156, 1835–1840. [Google Scholar] [CrossRef][Green Version]
- Wilczynski, G.M.; Engel, W.K.; Askanas, V. Cyclin-dependent kinase 5 colocalizes with phosphorylated tau in human inclusion-body myositis paired-helical filaments and may play a role in tau phosphorylation. Neurosci. Lett. 2000, 293, 33–36. [Google Scholar] [CrossRef]
- Nakano, S.; Shinde, A.; Kawashima, S.; Nakamura, S.; Akiguchi, I.; Kimura, J. Inclusion body myositis: Expression of extracellular signal-regulated kinase and its substrate. Neurology 2001, 56, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Kannanayakal, T.J.; Mendell, J.R.; Kuret, J. Casein kinase 1 alpha associates with the tau-bearing lesions of inclusion body myositis. Neurosci. Lett. 2008, 431, 141–145. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Terracciano, C.; Nogalska, A.; Engel, W.K.; Askanas, V. In AbetaPP-overexpressing cultured human muscle fibers proteasome inhibition enhances phosphorylation of AbetaPP751 and GSK3beta activation: Effects mitigated by lithium and apparently relevant to sporadic inclusion-body myositis. J. Neurochem. 2010, 112, 389–396. [Google Scholar] [CrossRef]
- Askanas, V.; Bilak, M.; Engel, W.K.; Alvarez, R.B.; Tomé, F.; Leclerc, A. Prion protein is abnormally accumulated in inclusion-body myositis. Neuroreport 1993, 5, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Plagnol, V.; Brady, S.; Simone, R.; Lashley, T.; Acevedo-Arozena, A.; de Silva, R.; Greensmith, L.; Holton, J.; Hanna, M.G.; et al. Widespread RNA metabolism impairment in sporadic inclusion body myositis TDP43-proteinopathy. Neurobiol. Aging 2014, 35, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Pinkus, J.L.; Amato, A.A.; Taylor, J.P.; Greenberg, S.A. Abnormal distribution of heterogeneous nuclear ribonucleoproteins in sporadic inclusion body myositis. Neuromuscul. Disord. 2014, 24, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Samant, R.S.; Frydman, J. Mechanisms and Functions of Spatial Protein Quality Control. Annu. Rev. Biochem. 2017, 86, 97–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Retzlaff, M.; Roos, T.; Frydman, J. Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 2011, 3, a004374. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Li, L.; Chin, L.S. Aggresome formation and neurodegenerative diseases: Therapeutic implications. Curr. Med. Chem. 2008, 15, 47–60. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K.; Nogalska, A. Inclusion body myositis: A degenerative muscle disease associated with intra-muscle fiber multi-protein aggregates, proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation. Brain Pathol. 2009, 19, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Banwell, B.L.; Engel, A.G. AlphaB-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology 2000, 54, 1033–1041. [Google Scholar] [CrossRef]
- Wojcik, S.; Engel, W.K.; McFerrin, J.; Paciello, O.; Askanas, V. AbetaPP-overexpression and proteasome inhibition increase alphaB-crystallin in cultured human muscle: Relevance to inclusion-body myositis. Neuromuscul. Disord. 2006, 16, 839–844. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef]
- Fratta, P.; Engel, W.K.; McFerrin, J.; Davies, K.J.; Lin, S.W.; Askanas, V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am. J. Pathol. 2005, 167, 517–526. [Google Scholar] [CrossRef]
- Ferrer, I.; Martín, B.; Castaño, J.G.; Lucas, J.J.; Moreno, D.; Olivé, M. Proteasomal expression, induction of immunoproteasome subunits, and local MHC class I presentation in myofibrillar myopathy and inclusion body myositis. J. Neuropathol. Exp. Neurol. 2004, 63, 484–498. [Google Scholar] [CrossRef][Green Version]
- McConkey, D.J.; White, M.; Yan, W. HDAC inhibitor modulation of proteotoxicity as a therapeutic approach in cancer. Adv. Cancer Res. 2012, 116, 131–163. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; Engel, W.K.; Askanas, V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010, 177, 1377–1387. [Google Scholar] [CrossRef]
- Kumamoto, T.; Ueyama, H.; Tsumura, H.; Toyoshima, I.; Tsuda, T. Expression of lysosome-related proteins and genes in the skeletal muscles of inclusion body myositis. Acta Neuropathol. 2004, 107, 59–65. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Schmidt, J.; Schmid, D.; Barthel, K.; Wrede, A.; Dalakas, M.C.; Münz, C. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann. Neurol. 2007, 61, 476–483. [Google Scholar] [CrossRef]
- Nogalska, A.; Terracciano, C.; D’Agostino, C.; King Engel, W.; Askanas, V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusions of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropathol. 2009, 118, 407–413. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- D’Agostino, C.; Nogalska, A.; Cacciottolo, M.; Engel, W.K.; Askanas, V. Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol. 2011, 122, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Girolamo, F.; Lia, A.; Amati, A.; Strippoli, M.; Coppola, C.; Virgintino, D.; Roncali, L.; Toscano, A.; Serlenga, L.; Trojano, M. Overexpression of autophagic proteins in the skeletal muscle of sporadic inclusion body myositis. Neuropathol. Appl. Neurobiol. 2013, 39, 736–749. [Google Scholar] [CrossRef]
- Cacciottolo, M.; Nogalska, A.; D’Agostino, C.; Engel, W.K.; Askanas, V. Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol. Appl. Neurobiol. 2013, 39, 750–761. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; Engel, W.K.; McFerrin, J.; Askanas, V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am. J. Pathol. 2004, 164, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nogalska, A.; D’Agostino, C.; Engel, W.K.; Cacciottolo, M.; Asada, S.; Mori, K.; Askanas, V. Activation of the Unfolded Protein Response in Sporadic Inclusion-Body Myositis but Not in Hereditary GNE Inclusion-Body Myopathy. J. Neuropathol. Exp. Neurol. 2015, 74, 538–546. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nogalska, A.; Engel, W.K.; McFerrin, J.; Kokame, K.; Komano, H.; Askanas, V. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J. Neurochem. 2006, 96, 1491–1499. [Google Scholar] [CrossRef]
- Nogalska, A.; Wojcik, S.; Engel, W.K.; McFerrin, J.; Askanas, V. Endoplasmic reticulum stress induces myostatin precursor protein and NF-kappaB in cultured human muscle fibers: Relevance to inclusion body myositis. Exp. Neurol. 2007, 204, 610–618. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wójcik, S.; Engel, W.K.; McFerrin, J.; Askanas, V. Myostatin is increased and complexes with amyloid-beta within sporadic inclusion-body myositis muscle fibers. Acta Neuropathol. 2005, 110, 173–177. [Google Scholar] [CrossRef]
- Sachdev, R.; Kappes-Horn, K.; Paulsen, L.; Duernberger, Y.; Pleschka, C.; Denner, P.; Kundu, B.; Reimann, J.; Vorberg, I. Endoplasmic Reticulum Stress Induces Myostatin High Molecular Weight Aggregates and Impairs Mature Myostatin Secretion. Mol. Neurobiol. 2018, 55, 8355–8373. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, B. Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras. Biomolecules 2019, 9, 15. [Google Scholar] [CrossRef]
- Oldfors, A.; Larsson, N.G.; Lindberg, C.; Holme, E. Mitochondrial DNA deletions in inclusion body myositis. Brain 1993, 116 Pt 2, 325–336. [Google Scholar] [CrossRef]
- Catalán-García, M.; Garrabou, G.; Morén, C.; Guitart-Mampel, M.; Hernando, A.; Díaz-Ramos, À.; González-Casacuberta, I.; Juárez, D.L.; Bañó, M.; Enrich-Bengoa, J.; et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin. Sci. 2016, 130, 1741–1751. [Google Scholar] [CrossRef]
- Oikawa, Y.; Izumi, R.; Koide, M.; Hagiwara, Y.; Kanzaki, M.; Suzuki, N.; Kikuchi, K.; Matsuhashi, T.; Akiyama, Y.; Ichijo, M.; et al. Mitochondrial dysfunction underlying sporadic inclusion body myositis is ameliorated by the mitochondrial homing drug MA-5. PLoS ONE 2020, 15, e0231064. [Google Scholar] [CrossRef]
- Hedberg-Oldfors, C.; Lindgren, U.; Basu, S.; Visuttijai, K.; Lindberg, C.; Falkenberg, M.; Larsson Lekholm, E.; Oldfors, A. Mitochondrial DNA variants in inclusion body myositis characterized by deep sequencing. Brain Pathol. 2021, 31, e12931. [Google Scholar] [CrossRef] [PubMed]
- Georgantas, R.W.; Streicher, K.; Greenberg, S.A.; Greenlees, L.M.; Zhu, W.; Brohawn, P.Z.; Higgs, B.W.; Czapiga, M.; Morehouse, C.A.; Amato, A.; et al. Inhibition of myogenic microRNAs 1, 133, and 206 by inflammatory cytokines links inflammation and muscle degeneration in adult inflammatory myopathies. Arthritis Rheumatol. 2014, 66, 1022–1033. [Google Scholar] [CrossRef]
- Buzkova, J.; Nikkanen, J.; Ahola, S.; Hakonen, A.H.; Sevastianova, K.; Hovinen, T.; Yki-Järvinen, H.; Pietiläinen, K.H.; Lönnqvist, T.; Velagapudi, V.; et al. Metabolomes of mitochondrial diseases and inclusion body myositis patients: Treatment targets and biomarkers. EMBO Mol. Med. 2018, 10, e9091. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Miller, J.; Grady, J.P.; Rocha, M.C.; Taylor, R.W.; Turnbull, D.M. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol. Appl. Neurobiol. 2015, 41, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Oldfors, A.; Moslemi, A.R.; Fyhr, I.M.; Holme, E.; Larsson, N.G.; Lindberg, C. Mitochondrial DNA deletions in muscle fibers in inclusion body myositis. J. Neuropathol. Exp. Neurol. 1995, 54, 581–587. [Google Scholar] [CrossRef]
- Bhatt, P.S.; Tzoulis, C.; Balafkan, N.; Miletic, H.; Tran, G.T.T.; Sanaker, P.S.; Bindoff, L.A. Mitochondrial DNA depletion in sporadic inclusion body myositis. Neuromuscul. Disord. 2019, 29, 242–246. [Google Scholar] [CrossRef]
- Peng, T.I.; Yu, P.R.; Chen, J.Y.; Wang, H.L.; Wu, H.Y.; Wei, Y.H.; Jou, M.J. Visualizing common deletion of mitochondrial DNA-augmented mitochondrial reactive oxygen species generation and apoptosis upon oxidative stress. Biochim. Biophys. Acta 2006, 1762, 241–255. [Google Scholar] [CrossRef]
- Askanas, V.; Engel, W.K. Newest approaches to diagnosis and pathogenesis of sporadic inclusion-body myositis and hereditary inclusion-body myopathies, including molecular-pathologic similarities to Alzheimer disease. In Inclusion-Body Myositis and Myopathies; Cambridge University Press: Cambridge, UK, 1998; Volume 3–78. [Google Scholar]
- Sabadashka, M.; Nagalievska, M.; Sybirna, N. Tyrosine nitration as a key event of signal transduction that regulates functional state of the cell. Cell Biol. Int. 2021, 45, 481–497. [Google Scholar] [CrossRef]
- Mattila, J.T.; Thomas, A.C. Nitric oxide synthase: Non-canonical expression patterns. Front. Immunol. 2014, 5, 478. [Google Scholar] [CrossRef]
- Yang, C.C.; Alvarez, R.B.; Engel, W.K.; Askanas, V. Increase of nitric oxide synthases and nitrotyrosine in inclusion-body myositis. Neuroreport 1996, 8, 153–158. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free. Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Tsuruta, Y.; Furuta, A.; Taniguchi, N.; Yamada, T.; Kira, J.; Iwaki, T. Increased expression of manganese superoxide dismutase is associated with that of nitrotyrosine in myopathies with rimmed vacuoles. Acta Neuropathol. 2002, 103, 59–65. [Google Scholar] [CrossRef]
- Askanas, V.; Sarkozi, E.; Alvares, R.B.; McFerrin, J.; Engel, W.K.; Siddique, T. Superoxide dismutase-1 gene and protein in vacuolated muscle fibers of sporadic inclusion-body myositis, hereditary inclusion-body myopathy, and cultured human muscle after B-amyloid precursor protein gene transfer. Neurology 1996, 46, A487. [Google Scholar]
- Broccolini, A.; Engel, W.K.; Alvarez, R.B.; Askanas, V. Redox factor-1 in muscle biopsies of patients with inclusion-body myositis. Neurosci. Lett. 2000, 287, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Broccolini, A.; Ricci, E.; Pescatori, M.; Papacci, M.; Gliubizzi, C.; D’Amico, A.; Servidei, S.; Tonali, P.; Mirabella, M. Insulin-like growth factor I in inclusion-body myositis and human muscle cultures. J. Neuropathol. Exp. Neurol. 2004, 63, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Chen, S. DJ-1 in neurodegenerative diseases: Pathogenesis and clinical application. Prog. Neurobiol. 2021, 204, 102114. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, C.; Nogalska, A.; Engel, W.K.; Wojcik, S.; Askanas, V. In inclusion-body myositis muscle fibers Parkinson-associated DJ-1 is increased and oxidized. Free. Radic. Biol. Med. 2008, 45, 773–779. [Google Scholar] [CrossRef][Green Version]
- Askanas, V.; Engel, W.K. Molecular pathology and pathogenesis of inclusion-body myositis. Microsc. Res. Tech. 2005, 67, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G. Inclusion body myositis. In Myology, 3rd ed.; Engel, A.G., Franzini-Armstrong, C., Eds.; McGraw-Hill: New York, NY, USA, 2004; pp. 1367–1388. [Google Scholar]
- Nalbantoglu, J.; Karpati, G.; Carpenter, S. Conspicuous accumulation of a single-stranded DNA binding protein in skeletal muscle fibers in inclusion body myositis. Am. J. Pathol. 1994, 144, 874–882. [Google Scholar] [PubMed]
- Greenberg, S.A.; Pinkus, J.L.; Amato, A.A. Nuclear membrane proteins are present within rimmed vacuoles in inclusion-body myositis. Muscle Nerve 2006, 34, 406–416. [Google Scholar] [CrossRef]
- Nakano, S.; Shinde, A.; Fujita, K.; Ito, H.; Kusaka, H. Histone H1 is released from myonuclei and present in rimmed vacuoles with DNA in inclusion body myositis. Neuromuscul. Disord. 2008, 18, 27–33. [Google Scholar] [CrossRef]
- Greenberg, S.A. Inflammatory myopathies: Disease mechanisms. Curr. Opin. Neurol. 2009, 22, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Lain, A.; Millecamps, S.; Dubourg, O.; Salachas, F.; Bruneteau, G.; Lacomblez, L.; LeGuern, E.; Seilhean, D.; Duyckaerts, C.; Meininger, V.; et al. Abnormal TDP-43 and FUS proteins in muscles of sporadic IBM: Similarities in a TARDBP-linked ALS patient. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1414–1416. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Toth, M.J.; Matthews, D.E.; Tracy, R.P.; Previs, M.J. Age-related differences in skeletal muscle protein synthesis: Relation to markers of immune activation. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E883–E891. [Google Scholar] [CrossRef]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free. Radic. Biol. Med. 2019, 132, 58–66. [Google Scholar] [CrossRef]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Malatesta, M.; Perdoni, F.; Muller, S.; Zancanaro, C.; Pellicciari, C. Nuclei of aged myofibres undergo structural and functional changes suggesting impairment in RNA processing. Eur. J. Histochem. 2009, 53, e12. [Google Scholar] [CrossRef]
- Iyer, S.R.; Hsia, R.C.; Folker, E.S.; Lovering, R.M. Age-dependent changes in nuclear-cytoplasmic signaling in skeletal muscle. Exp. Gerontol. 2021, 150, 111338. [Google Scholar] [CrossRef] [PubMed]
- Day, K.; Shefer, G.; Shearer, A.; Yablonka-Reuveni, Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev. Biol. 2010, 340, 330–343. [Google Scholar] [CrossRef]
- Jackson, M.J.; McArdle, A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J. Physiol. 2011, 589, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Tann, A.W.; Mitra, S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: Susceptibility of skeletal muscles to oxidative injury. Mech. Ageing Dev. 2010, 131, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Perandini, L.A.; Chimin, P.; Lutkemeyer, D.D.S.; Câmara, N.O.S. Chronic inflammation in skeletal muscle impairs satellite cells function during regeneration: Can physical exercise restore the satellite cell niche? FEBS J. 2018, 285, 1973–1984. [Google Scholar] [CrossRef]
- Walston, J.D. Sarcopenia in older adults. Curr Opin Rheumatol 2012, 24, 623–627. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Pathak, R.U.; Soujanya, M.; Mishra, R.K. Deterioration of nuclear morphology and architecture: A hallmark of senescence and aging. Ageing Res. Rev. 2021, 67, 101264. [Google Scholar] [CrossRef]
- Conboy, I.M.; Rando, T.A. Aging, stem cells and tissue regeneration: Lessons from muscle. Cell Cycle 2005, 4, 407–410. [Google Scholar] [CrossRef]
- Wanschitz, J.V.; Dubourg, O.; Lacene, E.; Fischer, M.B.; Höftberger, R.; Budka, H.; Romero, N.B.; Eymard, B.; Herson, S.; Butler-Browne, G.S.; et al. Expression of myogenic regulatory factors and myo-endothelial remodeling in sporadic inclusion body myositis. Neuromuscul. Disord. 2013, 23, 75–83. [Google Scholar] [CrossRef]
- Hollemann, D.; Budka, H.; Löscher, W.N.; Yanagida, G.; Fischer, M.B.; Wanschitz, J.V. Endothelial and myogenic differentiation of hematopoietic progenitor cells in inflammatory myopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 711–719. [Google Scholar] [CrossRef]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA repair in aging and disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Lax, N.Z.; Turnbull, D.M.; Reeve, A.K. Mitochondrial mutations: Newly discovered players in neuronal degeneration. Neuroscientist 2011, 17, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Tanaka, M.; Yamamoto, H.; Ohbayashi, T.; Nimura, Y.; Ozawa, T. Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem. Int. 1991, 25, 47–56. [Google Scholar] [PubMed]
- Chung, S.S.; Weindruch, R.; Schwarze, S.R.; McKenzie, D.I.; Aiken, J.M. Multiple age-associated mitochondrial DNA deletions in skeletal muscle of mice. Aging 1994, 6, 193–200. [Google Scholar] [CrossRef]
- Kadenbach, B.; Münscher, C.; Frank, V.; Müller-Höcker, J.; Napiwotzki, J. Human aging is associated with stochastic somatic mutations of mitochondrial DNA. Mutat. Res. 1995, 338, 161–172. [Google Scholar] [CrossRef]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal muscle mitochondria and aging: A review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef]
- Wei, Y.H.; Lu, C.Y.; Lee, H.C.; Pang, C.Y.; Ma, Y.S. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann. N. Y. Acad. Sci. 1998, 854, 155–170. [Google Scholar] [CrossRef]
- Nelke, C.; Schroeter, C.B.; Theissen, L.; Preusse, C.; Pawlitzki, M.; Räuber, S.; Dobelmann, V.; Cengiz, D.; Kleefeld, F.; Roos, A.; et al. Senescent fibro-adipogenic progenitors are potential drivers of pathology in inclusion body myositis. Acta Neuropathol. 2023, 146, 725–745. [Google Scholar] [CrossRef]
- Engel, A.G.; Arahata, K. Monoclonal antibody analysis of mononuclear cells in myopathies. II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann. Neurol. 1984, 16, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, M.; Rakocevic, G.; Raju, R.; Shatunov, A.; Goldfarb, L.G.; Dalakas, M.C. T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology 2007, 69, 1672–1679. [Google Scholar] [CrossRef]
- Hofbauer, M.; Wiesener, S.; Babbe, H.; Roers, A.; Wekerle, H.; Dornmair, K.; Hohlfeld, R.; Goebels, N. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 4090–4095. [Google Scholar] [CrossRef]
- Müntzing, K.; Lindberg, C.; Moslemi, A.R.; Oldfors, A. Inclusion body myositis: Clonal expansions of muscle-infiltrating T cells persist over time. Scand. J. Immunol. 2003, 58, 195–200. [Google Scholar] [CrossRef]
- Fyhr, I.M.; Moslemi, A.R.; Tarkowski, A.; Lindberg, C.; Oldfors, A. Limited T-cell receptor V gene usage in inclusion body myositis. Scand. J. Immunol. 1996, 43, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Fyhr, I.M.; Moslemi, A.R.; Lindberg, C.; Oldfors, A. T cell receptor beta-chain repertoire in inclusion body myositis. J. Neuroimmunol. 1998, 91, 129–134. [Google Scholar] [CrossRef]
- Lindberg, C.; Oldfors, A.; Tarkowski, A. Restricted use of T cell receptor V genes in endomysial infiltrates of patients with inflammatory myopathies. Eur. J. Immunol. 1994, 24, 2659–2663. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, T.P.; Dalakas, M.C.; Plotz, P.H.; Miller, F.W. The alpha beta T-cell receptor repertoire in inclusion body myositis: Diverse patterns of gene expression by muscle-infiltrating lymphocytes. J. Autoimmun. 1994, 7, 321–333. [Google Scholar] [CrossRef]
- Fyhr, I.M.; Moslemi, A.R.; Mosavi, A.A.; Lindberg, C.; Tarkowski, A.; Oldfors, A. Oligoclonal expansion of muscle infiltrating T cells in inclusion body myositis. J. Neuroimmunol. 1997, 79, 185–189. [Google Scholar] [CrossRef]
- Bender, A.; Behrens, L.; Engel, A.G.; Hohlfeld, R. T-cell heterogeneity in muscle lesions of inclusion body myositis. J. Neuroimmunol. 1998, 84, 86–91. [Google Scholar] [CrossRef]
- Amemiya, K.; Granger, R.P.; Dalakas, M.C. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain 2000, 123 Pt 10, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Dalakas, M.C. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am. J. Pathol. 1999, 155, 453–460. [Google Scholar] [CrossRef]
- Behrens, L.; Kerschensteiner, M.; Misgeld, T.; Goebels, N.; Wekerle, H.; Hohlfeld, R. Human muscle cells express a functional costimulatory molecule distinct from B7.1 (CD80) and B7.2 (CD86) in vitro and in inflammatory lesions. J. Immunol. 1998, 161, 5943–5951. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Rakocevic, G.; Raju, R.; Dalakas, M.C. Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: Significance for CD8+ T cell cytotoxicity. Brain 2004, 127, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Waschbisch, A.; Wintterle, S.; Lochmüller, H.; Walter, M.C.; Wischhusen, J.; Kieseier, B.C.; Wiendl, H. Human muscle cells express the costimulatory molecule B7-H3, which modulates muscle-immune interactions. Arthritis Rheumatol. 2008, 58, 3600–3608. [Google Scholar] [CrossRef] [PubMed]
- Pandya, J.M.; Fasth, A.E.; Zong, M.; Arnardottir, S.; Dani, L.; Lindroos, E.; Malmström, V.; Lundberg, I.E. Expanded T cell receptor Vβ-restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheumatol. 2010, 62, 3457–3466. [Google Scholar] [CrossRef]
- Lindberg, C.; Oldfors, A.; Tarkowski, A. Local T-cell proliferation and differentiation in inflammatory myopathies. Scand. J. Immunol. 1995, 41, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Allenbach, Y.; Chaara, W.; Rosenzwajg, M.; Six, A.; Prevel, N.; Mingozzi, F.; Wanschitz, J.; Musset, L.; Charuel, J.L.; Eymard, B.; et al. Th1 response and systemic treg deficiency in inclusion body myositis. PLoS ONE 2014, 9, e88788. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Pinkus, J.L.; Kong, S.W.; Baecher-Allan, C.; Amato, A.A.; Dorfman, D.M. Highly differentiated cytotoxic T cells in inclusion body myositis. Brain 2019, 142, 2590–2604. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, K.; Ohshima, S.; Osoegawa, M.; Tanaka, M.; Ogawa, K.; Nagata, K.; Kira, J.I. Expression of granulysin in polymyositis and inclusion-body myositis. J. Neurol. Neurosurg. Psychiatry 2006, 77, 1187–1190. [Google Scholar] [CrossRef]
- Orimo, S.; Koga, R.; Goto, K.; Nakamura, K.; Arai, M.; Tamaki, M.; Sugita, H.; Nonaka, I.; Arahata, K. Immunohistochemical analysis of perforin and granzyme A in inflammatory myopathies. Neuromuscul. Disord. 1994, 4, 219–226. [Google Scholar] [CrossRef]
- Goebels, N.; Michaelis, D.; Engelhardt, M.; Huber, S.; Bender, A.; Pongratz, D.; Johnson, M.A.; Wekerle, H.; Tschopp, J.; Jenne, D.; et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J. Clin. Invest. 1996, 97, 2905–2910. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Suzuki, S.; Komori, T. Immunohistochemical Phenotype of T Cells Invading Muscle in Inclusion Body Myositis. J. Neuropathol. Exp. Neurol. 2022, 81, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Dzangué-Tchoupou, G.; Mariampillai, K.; Bolko, L.; Amelin, D.; Mauhin, W.; Corneau, A.; Blanc, C.; Allenbach, Y.; Benveniste, O. CD8+(T-bet+) cells as a predominant biomarker for inclusion body myositis. Autoimmun. Rev. 2019, 18, 325–333. [Google Scholar] [CrossRef]
- Gao, Z.; Feng, Y.; Xu, J.; Liang, J. T-cell exhaustion in immune-mediated inflammatory diseases: New implications for immunotherapy. Front. Immunol. 2022, 13, 977394. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Knauss, S.; Preusse, C.; Allenbach, Y.; Leonard-Louis, S.; Touat, M.; Fischer, N.; Radbruch, H.; Mothes, R.; Matyash, V.; Böhmerle, W.; et al. PD1 pathway in immune-mediated myopathies: Pathogenesis of dysfunctional T cells revisited. Neurol. Neuroimmunol. Neuroinflammation 2019, 6, e558. [Google Scholar] [CrossRef]
- Eggenhuizen, P.J.; Ng, B.H.; Ooi, J.D. Treg Enhancing Therapies to Treat Autoimmune Diseases. Int. J. Mol. Sci. 2020, 21, 7015. [Google Scholar] [CrossRef]
- Valentini, N.; Requejo Cier, C.J.; Lamarche, C. Regulatory T-cell dysfunction and its implication for cell therapy. Clin. Exp. Immunol. 2023, 213, 40–49. [Google Scholar] [CrossRef]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef]
- Schiaffino, S.; Pereira, M.G.; Ciciliot, S.; Rovere-Querini, P. Regulatory T cells and skeletal muscle regeneration. FEBS J. 2017, 284, 517–524. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Bradshaw, E.M.; Pinkus, J.L.; Pinkus, G.S.; Burleson, T.; Due, B.; Bregoli, L.; O’Connor, K.C.; Amato, A.A. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 2005, 65, 1782–1787. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Orihuela, A.; McArdel, S.L.; Salajegheh, M.; Amato, A.A.; Hafler, D.A.; Greenberg, S.A.; O’Connor, K.C. A local antigen-driven humoral response is present in the inflammatory myopathies. J. Immunol. 2007, 178, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, M.; Pinkus, J.L.; Amato, A.A.; Morehouse, C.; Jallal, B.; Yao, Y.; Greenberg, S.A. Permissive environment for B-cell maturation in myositis muscle in the absence of B-cell follicles. Muscle Nerve 2010, 42, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Krystufková, O.; Vallerskog, T.; Helmers, S.B.; Mann, H.; Putová, I.; Belácek, J.; Malmström, V.; Trollmo, C.; Vencovsky, J.; Lundberg, I.E. Increased serum levels of B cell activating factor (BAFF) in subsets of patients with idiopathic inflammatory myopathies. Ann. Rheum. Dis. 2009, 68, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Roy, B.; Wu, Q.; Mohanty, S.; Nowak, R.J.; Shaw, A.C.; Kleinstein, S.H.; O’Connor, K.C. The Plasma Cell Infiltrate Populating the Muscle Tissue of Patients with Inclusion Body Myositis Features Distinct B Cell Receptor Repertoire Properties. Immunohorizons 2023, 7, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, O.; Stenzel, W.; Hilton-Jones, D.; Sandri, M.; Boyer, O.; van Engelen, B.G. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: The inflammatory egg comes before the degenerative chicken. Acta Neuropathol. 2015, 129, 611–624. [Google Scholar] [CrossRef]
- Roos, A.; Preusse, C.; Hathazi, D.; Goebel, H.H.; Stenzel, W. Proteomic Profiling Unravels a Key Role of Specific Macrophage Subtypes in Sporadic Inclusion Body Myositis. Front. Immunol. 2019, 10, 1040. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Pinkus, G.S.; Amato, A.A.; Pinkus, J.L. Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve 2007, 35, 17–23. [Google Scholar] [CrossRef]
- de Padilla, C.M.; Reed, A.M. Dendritic cells and the immunopathogenesis of idiopathic inflammatory myopathies. Curr. Opin. Rheumatol. 2008, 20, 669–674. [Google Scholar] [CrossRef]
- Das, L.; Blumbergs, P.C.; Manavis, J.; Limaye, V.S. Major histocompatibility complex class I and II expression in idiopathic inflammatory myopathy. Appl. Immunohistochem. Mol. Morphol. 2013, 21, 539–542. [Google Scholar] [CrossRef]
- Englund, P.; Lindroos, E.; Nennesmo, I.; Klareskog, L.; Lundberg, I.E. Skeletal muscle fibers express major histocompatibility complex class II antigens independently of inflammatory infiltrates in inflammatory myopathies. Am. J. Pathol. 2001, 159, 1263–1273. [Google Scholar] [CrossRef]
- Bartoccioni, E.; Gallucci, S.; Scuderi, F.; Ricci, E.; Servidei, S.; Broccolini, A.; Tonali, P. MHC class I, MHC class II and intercellular adhesion molecule-1 (ICAM-1) expression in inflammatory myopathies. Clin. Exp. Immunol. 1994, 95, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Spits, H. Development of alphabeta T cells in the human thymus. Nat. Rev. Immunol. 2002, 2, 760–772. [Google Scholar] [CrossRef]
- Fagnoni, F.F.; Vescovini, R.; Passeri, G.; Bologna, G.; Pedrazzoni, M.; Lavagetto, G.; Casti, A.; Franceschi, C.; Passeri, M.; Sansoni, P. Shortage of circulating naive CD8(+) T cells provides new insights on immunodeficiency in aging. Blood 2000, 95, 2860–2868. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Weyand, C.M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 2019, 19, 573–583. [Google Scholar] [CrossRef]
- Decman, V.; Laidlaw, B.J.; Dimenna, L.J.; Abdulla, S.; Mozdzanowska, K.; Erikson, J.; Ertl, H.C.; Wherry, E.J. Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J. Immunol. 2010, 184, 5151–5159. [Google Scholar] [CrossRef]
- Saule, P.; Trauet, J.; Dutriez, V.; Lekeux, V.; Dessaint, J.P.; Labalette, M. Accumulation of memory T cells from childhood to old age: Central and effector memory cells in CD4(+) versus effector memory and terminally differentiated memory cells in CD8(+) compartment. Mech. Ageing Dev. 2006, 127, 274–281. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Lee, W.W.; Weyand, C.M. Aging and T-cell diversity. Exp. Gerontol. 2007, 42, 400–406. [Google Scholar] [CrossRef]
- Briceño, O.; Lissina, A.; Wanke, K.; Afonso, G.; von Braun, A.; Ragon, K.; Miquel, T.; Gostick, E.; Papagno, L.; Stiasny, K.; et al. Reduced naïve CD8(+) T-cell priming efficacy in elderly adults. Aging Cell 2016, 15, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Gómez de Las Heras, M.M.; Escrig-Larena, J.I.; Mittelbrunn, M. Extremely Differentiated T Cell Subsets Contribute to Tissue Deterioration During Aging. Annu. Rev. Immunol. 2023, 41, 181–205. [Google Scholar] [CrossRef]
- Wu, J.; Ren, B.; Wang, D.; Lin, H. Regulatory T cells in skeletal muscle repair and regeneration: Recent insights. Cell Death Dis. 2022, 13, 680. [Google Scholar] [CrossRef]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef]
- Salam, N.; Rane, S.; Das, R.; Faulkner, M.; Gund, R.; Kandpal, U.; Lewis, V.; Mattoo, H.; Prabhu, S.; Ranganathan, V.; et al. T cell ageing: Effects of age on development, survival & function. Indian J. Med. Res. 2013, 138, 595–608. [Google Scholar] [PubMed]
- Cui, C.Y.; Driscoll, R.K.; Piao, Y.; Chia, C.W.; Gorospe, M.; Ferrucci, L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 2019, 18, e13032. [Google Scholar] [CrossRef] [PubMed]
- McCord, B.; Day, R.M. Cytotoxic immune cells do not affect TDP-43 and p62 sarcoplasmic aggregation but influence TDP-43 localisation. Sci. Rep. 2023, 13, 15935. [Google Scholar] [CrossRef]
- Schmidt, J.; Barthel, K.; Wrede, A.; Salajegheh, M.; Bähr, M.; Dalakas, M.C. Interrelation of inflammation and APP in sIBM: IL-1 beta induces accumulation of beta-amyloid in skeletal muscle. Brain 2008, 131, 1228–1240. [Google Scholar] [CrossRef]
- Adams, V.; Nehrhoff, B.; Späte, U.; Linke, A.; Schulze, P.C.; Baur, A.; Gielen, S.; Hambrecht, R.; Schuler, G. Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation: An in vitro and in vivo study. Cardiovasc. Res. 2002, 54, 95–104. [Google Scholar] [CrossRef]
- Williams, G.; Brown, T.; Becker, L.; Prager, M.; Giroir, B.P. Cytokine-induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes. Am. J. Physiol. 1994, 267, R1020–R1025. [Google Scholar] [CrossRef]
- Kitazawa, M.; Trinh, D.N.; LaFerla, F.M. Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase-3beta. Ann. Neurol. 2008, 64, 15–24. [Google Scholar] [CrossRef]
- Britson, K.A.; Ling, J.P.; Braunstein, K.E.; Montagne, J.M.; Kastenschmidt, J.M.; Wilson, A.; Ikenaga, C.; Tsao, W.; Pinal-Fernandez, I.; Russell, K.A.; et al. Loss of TDP-43 function and rimmed vacuoles persist after T cell depletion in a xenograft model of sporadic inclusion body myositis. Sci. Transl. Med. 2022, 14, eabi9196. [Google Scholar] [CrossRef]
- Kummer, K.; Bertram, I.; Zechel, S.; Hoffmann, D.B.; Schmidt, J. Inflammasome in Skeletal Muscle: NLRP3 Is an Inflammatory Cell Stress Component in Inclusion Body Myositis. Int. J. Mol. Sci. 2023, 24, 10675. [Google Scholar] [CrossRef]
- Yao, J.; Sterling, K.; Wang, Z.; Zhang, Y.; Song, W. The role of inflammasomes in human diseases and their potential as therapeutic targets. Signal Transduct. Target. Ther. 2024, 9, 10. [Google Scholar] [CrossRef]
- Masters, S.L.; O’Neill, L.A. Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol. Med. 2011, 17, 276–282. [Google Scholar] [CrossRef]
- Shi, F.; Kouadir, M.; Yang, Y. NALP3 inflammasome activation in protein misfolding diseases. Life Sci. 2015, 135, 9–14. [Google Scholar] [CrossRef]
- Group, M.S. Randomized pilot trial of high-dose betaINF-1a in patients with inclusion body myositis. Neurology 2004, 63, 718–720. [Google Scholar] [CrossRef]
- Badrising, U.A.; Maat-Schieman, M.L.; Ferrari, M.D.; Zwinderman, A.H.; Wessels, J.A.; Breedveld, F.C.; van Doorn, P.A.; van Engelen, B.G.; Hoogendijk, J.E.; Höweler, C.J.; et al. Comparison of weakness progression in inclusion body myositis during treatment with methotrexate or placebo. Ann. Neurol. 2002, 51, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Leff, R.L.; Miller, F.W.; Hicks, J.; Fraser, D.D.; Plotz, P.H. The Treatment of Inclusion Body Myositis: A Retrospective Review and a Randomized, Prospective Trial of Immunosuppressive. Medicine 1993, 72, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Joffe, M.M.; Love, L.A.; Leff, R.L.; Fraser, D.D.; Targoff, I.N.; Hicks, J.E.; Plotz, P.H.; Miller, F.W. Drug therapy of the idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am. J. Med. 1993, 94, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Sonies, B.; Dambrosia, J.; Sekul, E.; Cupler, E.; Sivakumar, K. Treatment of inclusion-body myositis with IVIg: A double-blind, placebo-controlled study. Neurology 1997, 48, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Koffman, B.; Fujii, M.; Spector, S.; Sivakumar, K.; Cupler, E. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology 2001, 56, 323–327. [Google Scholar] [CrossRef]
- Walter, M.C.; Lochmüller, H.; Toepfer, M.; Schlotter, B.; Reilich, P.; Schröder, M.; Müller-Felber, W.; Pongratz, D. High-dose immunoglobulin therapy in sporadic inclusion body myositis: A double-blind, placebo-controlled study. J. Neurol. 2000, 247, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Dastmalchi, M.; Grundtman, C.; Alexanderson, H.; Mavragani, C.P.; Einarsdottir, H.; Helmers, S.B.; Elvin, K.; Crow, M.K.; Nennesmo, I.; Lundberg, I.E. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann. Rheum. Dis. 2008, 67, 1670–1677. [Google Scholar] [CrossRef]
- Zhen, C.; Hou, Y.; Zhao, B.; Ma, X.; Dai, T.; Yan, C. Efficacy and safety of rituximab treatment in patients with idiopathic inflammatory myopathies: A systematic review and meta-analysis. Front. Immunol. 2022, 13, 1051609. [Google Scholar] [CrossRef]
- Kosmidis, M.L.; Pikazis, D.; Vlachoyiannopoulos, P.; Tzioufas, A.G.; Dalakas, M.C. Trial of canakinumab, an IL-1β receptor antagonist, in patients with inclusion body myositis. Neurol. Neuroimmunol. Neuroinflammation 2019, 6, e581. [Google Scholar] [CrossRef]
- Sultan, S.M.; Ng, K.P.; Edwards, J.C.; Isenberg, D.A.; Cambridge, G. Clinical outcome following B cell depletion therapy in eight patients with refractory idiopathic inflammatory myopathy. Clin. Exp. Rheumatol. 2008, 26, 887–893. [Google Scholar] [PubMed]
- Saperstein, D.; Levine, T. Interim Analysis of a Pilot Trial of Natalizumab in Inclusion Body Myositis. Neurology 2016, 86, P3.161. [Google Scholar] [CrossRef]
- Schmidt, K.; Kleinschnitz, K.; Rakocevic, G.; Dalakas, M.C.; Schmidt, J. Molecular treatment effects of alemtuzumab in skeletal muscles of patients with IBM. BMC Neurol. 2016, 16, 48. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Rakocevic, G.; Schmidt, J.; Salajegheh, M.; McElroy, B.; Harris-Love, M.O.; Shrader, J.A.; Levy, E.W.; Dambrosia, J.; Kampen, R.L.; et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain 2009, 132, 1536–1544. [Google Scholar] [CrossRef]
- Barohn, R.J.; Herbelin, L.; Kissel, J.T.; King, W.; McVey, A.L.; Saperstein, D.S.; Mendell, J.R. Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology 2006, 66, S123–S124. [Google Scholar] [CrossRef]
- Kosmidis, M.L.; Alexopoulos, H.; Tzioufas, A.G.; Dalakas, M.C. The effect of anakinra, an IL1 receptor antagonist, in patients with sporadic inclusion body myositis (sIBM): A small pilot study. J. Neurol. Sci. 2013, 334, 123–125. [Google Scholar] [CrossRef]
- Lindberg, C.; Trysberg, E.; Tarkowski, A.; Oldfors, A. Anti-T-lymphocyte globulin treatment in inclusion body myositis: A randomized pilot study. Neurology 2003, 61, 260–262. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Kremer, J.M.; Bankhurst, A.D.; Bulpitt, K.J.; Fleischmann, R.M.; Fox, R.I.; Jackson, C.G.; Lange, M.; Burge, D.J. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N. Engl. J. Med. 1999, 340, 253–259. [Google Scholar] [CrossRef]
- Pars, K.; Garde, N.; Skripuletz, T.; Pul, R.; Dengler, R.; Stangel, M. Subcutaneous immunoglobulin treatment of inclusion-body myositis stabilizes dysphagia. Muscle Nerve 2013, 48, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Cherin, P.; Pelletier, S.; Teixeira, A.; Laforet, P.; Simon, A.; Herson, S.; Eymard, B. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology 2002, 58, 326. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, O.; Hogrel, J.Y.; Belin, L.; Annoussamy, M.; Bachasson, D.; Rigolet, A.; Laforet, P.; Dzangué-Tchoupou, G.; Salem, J.E.; Nguyen, L.S.; et al. Sirolimus for treatment of patients with inclusion body myositis: A randomised, double-blind, placebo-controlled, proof-of-concept, phase 2b trial. Lancet Rheumatol. 2021, 3, e40–e48. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Machado, P.M.; Miller, A.; Spicer, C.; Herbelin, L.; He, J.; Noel, J.; Wang, Y.; McVey, A.L.; Pasnoor, M.; et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci. Transl. Med. 2016, 8, 331ra341. [Google Scholar] [CrossRef] [PubMed]
- Rutkove, S.B.; Parker, R.A.; Nardin, R.A.; Connolly, C.E.; Felice, K.J.; Raynor, E.M. A pilot randomized trial of oxandrolone in inclusion body myositis. Neurology 2002, 58, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.A.; Hanna, M.G.; Machado, P.M.; Badrising, U.A.; Chinoy, H.; Benveniste, O.; Karanam, A.K.; Wu, M.; Tankó, L.B.; Schubert-Tennigkeit, A.A.; et al. Efficacy and Safety of Bimagrumab in Sporadic Inclusion Body Myositis: Long-term Extension of RESILIENT. Neurology 2021, 96, e1595–e1607. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G.; Badrising, U.A.; Benveniste, O.; Lloyd, T.E.; Needham, M.; Chinoy, H.; Aoki, M.; Machado, P.M.; Liang, C.; Reardon, K.A.; et al. Safety and efficacy of intravenous bimagrumab in inclusion body myositis (RESILIENT): A randomised, double-blind, placebo-controlled phase 2b trial. Lancet Neurol. 2019, 18, 834–844. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Al-Zaidy, S.; Rodino-Klapac, L.R.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Miller, N.; Yalvac, M.; Dvorchik, I.; et al. Follistatin Gene Therapy for Sporadic Inclusion Body Myositis Improves Functional Outcomes. Mol. Ther. 2017, 25, 870–879. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef]
- Amthor, H.; Nicholas, G.; McKinnell, I.; Kemp, C.F.; Sharma, M.; Kambadur, R.; Patel, K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev. Biol. 2004, 270, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.K.; Hicok, K.C.; Shanahan, R.; Zhu, M.; Miller, S.; Arm, D.M. The Celution® System: Automated Processing of Adipose-Derived Regenerative Cells in a Functionally Closed System. Adv. Wound Care 2014, 3, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, A.N.; Jensen, K.Y.; Nielsen, J.L.; Frandsen, U.; Hvid, L.G.; Bjørnshauge, M.; Diederichsen, L.P.; Aagaard, P. Effects of blood-flow restricted resistance training on mechanical muscle function and thigh lean mass in sIBM patients. Scand. J. Med. Sci. Sports 2022, 32, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Lemmer, J.T.; Koffman, B.M.; Fleisher, T.A.; Feuerstein, I.M.; Hurley, B.F.; Dalakas, M.C. Safety and efficacy of strength training in patients with sporadic inclusion body myositis. Muscle Nerve 1997, 20, 1242–1248. [Google Scholar] [CrossRef]
- Johnson, L.G.; Collier, K.E.; Edwards, D.J.; Philippe, D.L.; Eastwood, P.R.; Walters, S.E.; Thickbroom, G.W.; Mastaglia, F.L. Improvement in aerobic capacity after an exercise program in sporadic inclusion body myositis. J. Clin. Neuromuscul. Dis. 2009, 10, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.; Pietrusz, A.; Dewar, E.; Dudziec, M.; Jones, K.; Hennis, P.; Sterr, A.; Baio, G.; Machado, P.M.; Laurá, M.; et al. Community exercise is feasible for neuromuscular diseases and can improve aerobic capacity. Neurology 2019, 92, e1773–e1785. [Google Scholar] [CrossRef] [PubMed]
- Coudert, J.D.; Slater, N.; Sooda, A.; Beer, K.; Lim, E.M.; Boyder, C.; Zhang, R.; Mastaglia, F.L.; Learmonth, Y.C.; Fairchild, T.J.; et al. Immunoregulatory effects of testosterone supplementation combined with exercise training in men with Inclusion Body Myositis: A double-blind, placebo-controlled, cross-over trial. Clin. Transl. Immunol. 2022, 11, e1416. [Google Scholar] [CrossRef] [PubMed]
- Connor, S.G.; Fairchild, T.J.; Learmonth, Y.C.; Beer, K.; Cooper, I.; Boardman, G.; Teo, S.Y.M.; Shatahmasseb, B.; Zhang, R.; Hiscock, K.; et al. Testosterone treatment combined with exercise to improve muscle strength, physical function and quality of life in men affected by inclusion body myositis: A randomised, double-blind, placebo-controlled, crossover trial. PLoS ONE 2023, 18, e0283394. [Google Scholar] [CrossRef]
- Afzali, A.M.; Ruck, T.; Wiendl, H.; Meuth, S.G. Animal models in idiopathic inflammatory myopathies: How to overcome a translational roadblock? Autoimmun. Rev. 2017, 16, 478–494. [Google Scholar] [CrossRef]
- Ching, J.K.; Elizabeth, S.V.; Ju, J.S.; Lusk, C.; Pittman, S.K.; Weihl, C.C. mTOR dysfunction contributes to vacuolar pathology and weakness in valosin-containing protein associated inclusion body myopathy. Hum. Mol. Genet. 2013, 22, 1167–1179. [Google Scholar] [CrossRef]
- Nalbandian, A.; Llewellyn, K.J.; Nguyen, C.; Yazdi, P.G.; Kimonis, V.E. Rapamycin and chloroquine: The in vitro and in vivo effects of autophagy-modifying drugs show promising results in valosin containing protein multisystem proteinopathy. PLoS ONE 2015, 10, e0122888. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Tawara, N.; Zhang, Z.; Nakane, S.; Sugie, K.; Suzuki, N.; Nishino, I.; Aoki, M. Pathogenic role of anti-cN1A autoantibodies in sporadic inclusion body myositis. J. Neurol. Neurosurg. Psychiatry 2023, 94, 1018–1024. [Google Scholar] [CrossRef]
- Cantó-Santos, J.; Valls-Roca, L.; Tobías, E.; Oliva, C.; García-García, F.J.; Guitart-Mampel, M.; Andújar-Sánchez, F.; Esteve-Codina, A.; Martín-Mur, B.; Padrosa, J.; et al. Integrated Multi-Omics Analysis for Inferring Molecular Players in Inclusion Body Myositis. Antioxidants 2023, 12, 1639. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Noda, S.; Kazuta, T.; Hirano, S.; Kimura, S.; Nakanishi, H.; Matsuo, K.; Tsujikawa, K.; Iida, M.; Koike, H.; et al. Metabolome and transcriptome analysis on muscle of sporadic inclusion body myositis. Ann. Clin. Transl. Neurol. 2022, 9, 1602–1615. [Google Scholar] [CrossRef] [PubMed]
- Güttsches, A.K.; Brady, S.; Krause, K.; Maerkens, A.; Uszkoreit, J.; Eisenacher, M.; Schreiner, A.; Galozzi, S.; Mertens-Rill, J.; Tegenthoff, M.; et al. Proteomics of rimmed vacuoles define new risk allele in inclusion body myositis. Ann. Neurol. 2017, 81, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Pu, C.; Huang, X.; Liu, J.; Mao, Y.; Lu, X. Proteomic study of sporadic inclusion body myositis. Proteome Sci. 2014, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Derfoul, A.; Pak, K.; Plotz, P.; Miller, F.W.; Milisenda, J.C.; Grau-Junyent, J.M.; Selva-O’Callaghan, A.; Paik, J.; et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology 2019, 93, e1193–e1204. [Google Scholar] [CrossRef] [PubMed]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Derfoul, A.; Pak, K.; Miller, F.W.; Milisenda, J.C.; Grau-Junyent, J.M.; Selva-O’Callaghan, A.; Carrion-Ribas, C.; Paik, J.J.; et al. Machine learning algorithms reveal unique gene expression profiles in muscle biopsies from patients with different types of myositis. Ann. Rheum. Dis. 2020, 79, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Cantó-Santos, J.; Valls-Roca, L.; Tobías, E.; García-García, F.J.; Guitart-Mampel, M.; Esteve-Codina, A.; Martín-Mur, B.; Casado, M.; Artuch, R.; Solsona-Vilarrasa, E.; et al. Unravelling inclusion body myositis using a patient-derived fibroblast model. J. Cachexia Sarcopenia Muscle 2023, 14, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.; Horuluoglu, B.; Galindo-Feria, A.S.; Diaz-Boada, J.S.; Sijbranda, M.; Notarnicola, A.; Dani, L.; van Vollenhoven, A.; Ramsköld, D.; Nennesmo, I.; et al. Single-cell profiling of muscle-infiltrating T cells in idiopathic inflammatory myopathies. EMBO Mol. Med. 2023, 15, e17240. [Google Scholar] [CrossRef]
- Muñoz-Braceras, S.; Pinal-Fernandez, I.; Casal-Dominguez, M.; Pak, K.; Milisenda, J.C.; Lu, S.; Gadina, M.; Naz, F.; Gutierrez-Cruz, G.; Dell’Orso, S.; et al. Identification of Unique microRNA Profiles in Different Types of Idiopathic Inflammatory Myopathy. Cells 2023, 12, 2198. [Google Scholar] [CrossRef]
- Lucchini, M.; De Arcangelis, V.; Santoro, M.; Morosetti, R.; Broccolini, A.; Mirabella, M. Serum-Circulating microRNAs in Sporadic Inclusion Body Myositis. Int. J. Mol. Sci. 2023, 24, 11139. [Google Scholar] [CrossRef]
- McLeish, E.; Slater, N.; Mastaglia, F.L.; Needham, M.; Coudert, J.D. From data to diagnosis: How machine learning is revolutionizing biomarker discovery in idiopathic inflammatory myopathies. Brief. Bioinform. 2023, 25, bbad514. [Google Scholar] [CrossRef]
- Gonzalez-Chapa, J.A.; Macêdo, M.B.; Lood, C. The Emerging Role of Mitochondrial Dysfunction in the Pathogenesis of Idiopathic Inflammatory Myopathies. Rambam Maimonides Med. J. 2023, 14, e0006. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Y.; Qi, Z.; Cao, L.; Ding, S. Mitochondrial transfer/transplantation: An emerging therapeutic approach for multiple diseases. Cell Biosci. 2022, 12, 66. [Google Scholar] [CrossRef]
- Gonzalez Chapa, J.A.; Barguil Macêdo, M.; Naddaf, E.; Saketkoo, L.A.; Lood, C. Mitochondrial transfer and implications for muscle function in idiopathic inflammatory myopathies. Clin. Exp. Rheumatol. 2024. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Process | Molecular Players and Their Alterations in sIBM |
|---|---|
| Degenerative Features | |
| Protein aggregation in muscle fibers |
|
| Impairment of ubiquitin–proteasome system (UPS) and autophagy |
|
| Endoplasmic reticulum (ER) stress and Unfolded Protein Response (UPR) |
|
| Mitochondrial abnormalities |
|
| Oxidative stress |
|
| Nuclear degeneration |
|
| Aged skeletal muscle milieu |
|
| |
| T cells |
|
| Plasma cells and antibody-mediated immune response |
|
| Macrophages and dendritic cells |
|
| Aging of the immune system and sIBM |
|
| Agent | Description |
|---|---|
| Immunosuppressive | |
| Prednisolone | Glucocorticoid binds to glucocorticoid receptor, inhibits pro-inflammatory signals, decreases leukocyte migration to the site of inflammation, promotes anti-inflammatory effects |
| Azathioprine | Inhibits purine synthesis with consequent decrease in DNA, RNA, and protein synthesis. Inhibits CD28 signals in T and B cells |
| Methotrexate | Inhibition enzymes are responsible for nucleotide syntheses such as dihydrofolate reductase, thymidylate synthase, aminoimidazole caboxamide ribonucleotide transformylase (AICART), and amido phosphoribosyltransferase. Prevents cell division |
| IVIg | Pooled of polyclonal IgG from healthy donors. Bind pathogenic autoantibodies or cross-react with various antigenic peptides |
| Anti-T lymphocyte globulin (ATGAM) | Purified rabbit anti-human thymocyte antibodies. Blocks T cell-mediated immune reactions |
| β-Interferon 1a | Cytokine. Immunomodulation function. Promote anti-inflammatory immune response |
| Etanercept | TNF receptor fusion protein with Fc portion of human IgG. Binds to and inhibits TNFα-mediated immune responses |
| Infliximab | TNFα-inhibiting monoclonal-antibody. Block TNFα-mediated immune responses |
| Alemtuzumab | Anti-CD52 monoclonal antibody. Depletes circulating T and B lymphocytes |
| Rituximab | Anti-CD20 monoclonal antibody. B cell-depletion |
| Anakinra | IL1 receptor antagonist blocks IL1β binding to its receptors |
| Sirolimus | Mammalian target of rapamycin (mTOR) inhibition. Promotes autophagy. Blocks effector T cells |
| ABC008 | Anti-KLRG1 antibody. Depletion of highly cytotoxic T cells, without affecting regulatory and central memory T cells |
| Simvastatin | Statin, HMG-CoA reductase inhibitor. Cholesterol-lowering drug with multiple other actions, including inhibition/modulation of inflammatory responses, enhancement of endothelial function, modulation of progenitor cells, antioxidant and neuroprotective effects |
| Non-Immunosuppressive | |
| Lithium | Glycogen synthase kinase-3 (GSK) inhibition. Modulates proteostasis and immune responses |
| Oxandrolone | Synthetic anabolic androgen steroid. Skeletal muscle mass enhancer |
| Follistatin gene therapy | Natural inhibitor of the myostatin receptor. Skeletal muscle mass enhancer |
| Bimagrumab | Monoclonal antibody. Binds competitively to activin type II receptors (ActRII, the myostatin receptor). Skeletal muscle mass enhancer |
| Arimoclomol | Heat shock protein inducer. Promotes protein folding |
| Adipose-Derived Regenerative Cells | Autologous adipose tissue-derived stem cells |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guglielmi, V.; Cheli, M.; Tonin, P.; Vattemi, G. Sporadic Inclusion Body Myositis at the Crossroads between Muscle Degeneration, Inflammation, and Aging. Int. J. Mol. Sci. 2024, 25, 2742. https://doi.org/10.3390/ijms25052742
Guglielmi V, Cheli M, Tonin P, Vattemi G. Sporadic Inclusion Body Myositis at the Crossroads between Muscle Degeneration, Inflammation, and Aging. International Journal of Molecular Sciences. 2024; 25(5):2742. https://doi.org/10.3390/ijms25052742
Chicago/Turabian StyleGuglielmi, Valeria, Marta Cheli, Paola Tonin, and Gaetano Vattemi. 2024. "Sporadic Inclusion Body Myositis at the Crossroads between Muscle Degeneration, Inflammation, and Aging" International Journal of Molecular Sciences 25, no. 5: 2742. https://doi.org/10.3390/ijms25052742
APA StyleGuglielmi, V., Cheli, M., Tonin, P., & Vattemi, G. (2024). Sporadic Inclusion Body Myositis at the Crossroads between Muscle Degeneration, Inflammation, and Aging. International Journal of Molecular Sciences, 25(5), 2742. https://doi.org/10.3390/ijms25052742