
Placental and Renal Pathways Underlying Pre-Eclampsia


 , , , ,
, , , ,  and
and
Abstract
1. Introduction
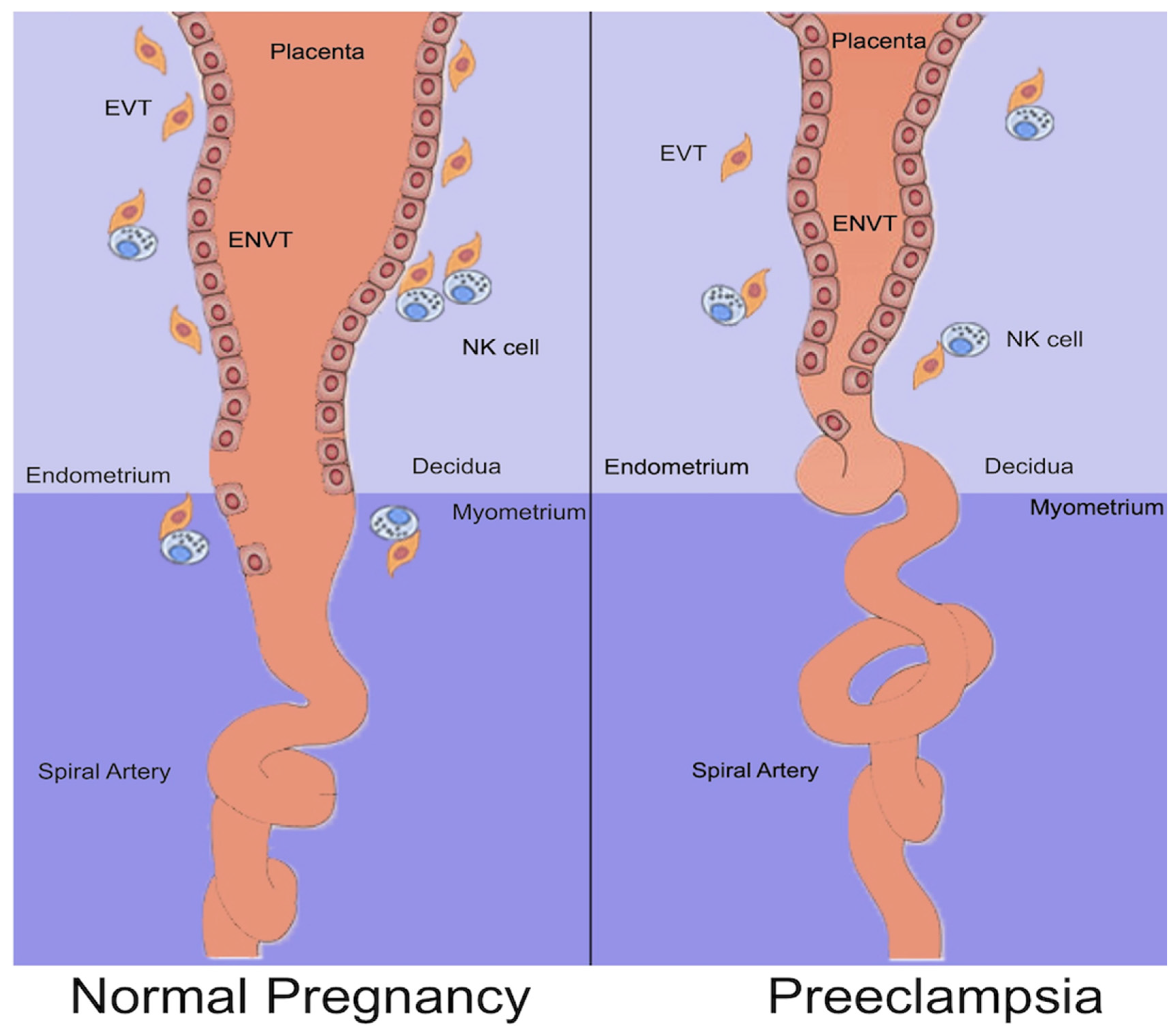
2. Normal Placentation
3. Definition of PE
- Proteinuria: 24 h urine protein ≥ 300 mg/day; spot urine protein/creatinine ratio ≥ 30 mg/mmoL or ≥ 0.3 mg/mg, or urine dipstick testing ≥ 2+;
- Other maternal organ dysfunctions:
- o
- Acute kidney injury (AKI) (creatinine ≥ 90 µmol/L; > 1.1 mg/dL);
- o
- Liver involvement (such as elevated liver transaminases > 40 IU/L) with or without the right upper quadrant or epigastric pain;
- o
- Neurological complications (including eclampsia, altered mental status, blindness, stroke, or, more commonly, hyperreflexia, when accompanied by clonus, severe headaches, and persistent visual scotomata);
- o
- Hematological complications (thrombocytopenia–platelet count < 150,000/µL, disseminated intravascular coagulation, and hemolysis);
- o
- Uteroplacental dysfunction (such as fetal growth restriction, abnormal umbilical artery Doppler waveform, or stillbirth).
4. Development of PE
4.1. Abnormal Placentation
4.2. Circulating Bioactive Agents
- VEGF is an endothelial-specific mitogen that promotes angiogenesis and induces vasopermeability and vasodilation in endothelial cells. PlGF is another member of the VEGF family that is predominantly produced in the placenta. VEGF interacts with two receptor tyrosine kinases, VEGFR-1 (VEGFR-1 or sFlt-1) and VEGFR-2 (kinase-insert domain region [KDR]/fetal liver kinase-1 [Flk-1]), which are selectively expressed on the vascular endothelial cell surface [35]. PlGF also binds to the VEGFR-1 receptor. sFlt-1 inhibits the pro-angiogenic effects of circulating VEGF and PlGF by binding to them and preventing them from interacting with their endogenous receptors in the body.
- The role of sFLT1 in the development of PE has been suggested by several studies: sFLT1 mRNA expression was high in pre-eclamptic placentas [36]; injecting rodents with exogenous sFLT1 induced hypertension, proteinuria, and glomerular endotheliosis, a hallmark of PE observed by renal biopsy, among other pre-eclamptic features [37]; and reduction in, or antagonism of, sFLT1 in animal models of PE improved clinical symptoms [38]. Furthermore, it appears that sFlt1 mediates the migration of monocytes/macrophages and the expression of tissue factors induced by VEGF and PlGF [39].
- Endothelin 1 (ET-1) may be a mediator in the pathogenesis of PE syndrome through the release of anti-angiogenic factors by the placenta [40]. ET-1 is a member of the human endothelin system, which also includes ET-2 and ET-3, and is a peptide produced exclusively by the vascular endothelium. Of the endogenously produced molecules, it is the endothelium-derived peptide with the most potent vasoconstrictive effect [41]. However, the majority of studies show that there is no difference in serum ET-1 levels between women with PE and those with normal pregnancies [42]. High levels of ET-1 are mainly observed in cases of severe PE and HELLP syndrome [41].
- Endoglin (Eng), also referred to as CD105, is a 180 kDa homodimeric co-receptor for the TGF-b group that has been implicated in hematopoiesis, cardiovascular development, and angiogenesis as a type I integral membrane glycoprotein. Eng functions as a cell surface coreceptor for the TGF-b1 and TGF-b3 isoforms, is highly expressed in endothelial cells and syncytiotrophoblasts, and modulates the actions of TGF-b1 and TGF-b3 [43,44]. In pre-eclamptic patients, the soluble TGF-b co-receptor derived from the placenta, endoglin (sEng), increases and correlates with the severity of the syndrome and decreases after delivery. These events lead to sEng acting synergistically with sFlt1 to induce severe PE and fetal growth restriction in pregnant rats [45].
- Hypoxia-inducible factor (HIF)-1-alpha, a marker of cellular oxygen deprivation, is expressed at high levels in proliferative trophoblasts and the hypoxic placenta. Recent studies suggest that the increased expression of HIF-1α regulates forkhead box O transcription factor 3a (FOXO3a), which, in turn, increases trophoblastic apoptosis. This mechanism may be involved in the pathogenesis of PE [46]. Under hypoxic conditions, HIF-1α increases the expression and release of sFlt-1, sEng, ACE, and many mediators, including angiotensin II (Ag II), into the maternal circulation [47]. Many of these soluble factors cause systemic endothelial damage.
- A causal link has been established between syncytiotrophoblast stress and the development of PE. Increased Gαq signaling and mitochondrial reactive oxygen species have been identified as causing this stress [48]. The activation of Gαq in mouse placental syncytiotrophoblasts caused hypertension, renal damage, proteinuria, increased circulating proinflammatory factors, decreased placental vascularization, reduced spiral artery diameters, and increased responses to mitochondrial superoxide.
4.3. Genetic and Immunologic Factors
4.4. Inflammation
5. PE and the Kidney
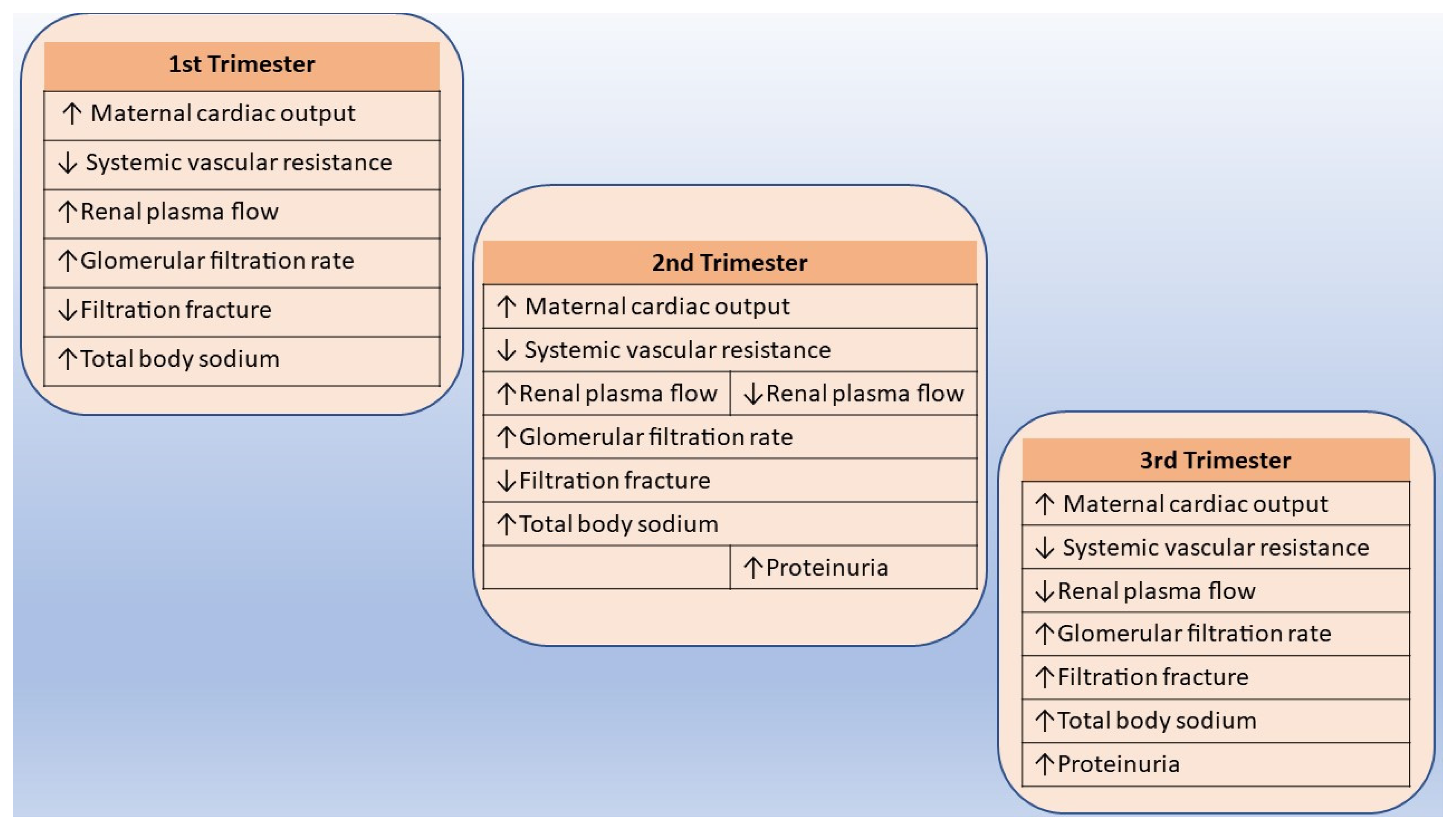
5.1. Normal Adjustments in Renal Function and Physiology during Pregnancy
5.2. The Role of Renin–Angiotensin–Aldosterone System
5.3. AKI and PE
5.4. CKD and PE
6. Management of PE—Therapy Perspectives
7. Diagnostic Tools and Novel Biomarkers
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallis, A.B.; Saftlas, A.F.; Hsia, J.; Atrash, H.K. Secular Trends in the Rates of Preeclampsia, Eclampsia, and Gestational Hypertension, United States, 1987–2004. Am. J. Hypertens. 2008, 21, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.; Gammill, H.S. Preeclampsia: Recent Insights. Hypertension 2005, 46, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Ghulmiyyah, L.; Sibai, B. Maternal Mortality from Preeclampsia/Eclampsia. Semin. Perinatol. 2012, 36, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Zhao, L.; Yu, S.; Zhou, J.; Li, J.; Zhang, N.; Xing, B.; Cui, X.; Yang, S. Differences in Epidemiology of Patients with Preeclampsia between China and the US (Review). Exp. Ther. Med. 2021, 22, 1012. [Google Scholar] [CrossRef] [PubMed]
- Melchiorre, K.; Giorgione, V.; Thilaganathan, B. The Placenta and Preeclampsia: Villain or Victim? Am. J. Obstet. Gynecol. 2022, 226, S954–S962. [Google Scholar] [CrossRef] [PubMed]
- Brosens, I.; Pijnenborg, R.; Vercruysse, L.; Romero, R. The “Great Obstetrical Syndromes” Are Associated with Disorders of Deep Placentation. Am. J. Obstet. Gynecol. 2011, 204, 193–201. [Google Scholar] [CrossRef]
- Gilbert, J.S.; Ryan, M.J.; LaMarca, B.B.; Sedeek, M.; Murphy, S.R.; Granger, J.P. Pathophysiology of Hypertension during Preeclampsia: Linking Placental Ischemia with Endothelial Dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H541–H550. [Google Scholar] [CrossRef]
- Srialluri, N.; Surapaneni, A.; Chang, A.; Mackeen, A.D.; Paglia, M.J.; Grams, M.E. Preeclampsia and Long-Term Kidney Outcomes: An Observational Cohort Study. Am. J. Kidney Dis. 2023, 82, 698–705. [Google Scholar] [CrossRef]
- Goetz, M.; Müller, M.; Gutsfeld, R.; Dijkstra, T.; Hassdenteufel, K.; Brucker, S.Y.; Bauer, A.; Joos, S.; Colombo, M.G.; Hawighorst-Knapstein, S.; et al. An Observational Claims Data Analysis on the Risk of Maternal Chronic Kidney Disease after Preterm Delivery and Preeclampsia. Sci. Rep. 2021, 11, 12596. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic Kidney Disease: Global Dimension and Perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Rolfo, A.; Attini, R.; Tavassoli, E.; Neve, F.V.; Nigra, M.; Cicilano, M.; Nuzzo, A.M.; Giuffrida, D.; Biolcati, M.; Nichelatti, M.; et al. Is It Possible to Differentiate Chronic Kidney Disease and Preeclampsia by Means of New and Old Biomarkers? A Prospective Study. Dis. Markers 2015, 2015, 127083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Ma, X.-X.; Hao, L.; Liu, L.-J.; Lv, J.-C.; Zhang, H. A Systematic Review and Meta-Analysis of Outcomes of Pregnancy in CKD and CKD Outcomes in Pregnancy. Clin. J. Am. Soc. Nephrol. 2015, 10, 1964–1978. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, G.B.; Attini, R.; Cabiddu, G.; Kooij, I.; Fassio, F.; Gerbino, M.; Maxia, S.; Biolcati, M.; Versino, E.; Todros, T. Maternal-Foetal Outcomes in Pregnant Women with Glomerulonephritides. Are All Glomerulonephritides Alike in Pregnancy? J. Autoimmun. 2017, 79, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Leaños-Miranda, A.; Campos-Galicia, I.; Ramírez-Valenzuela, K.L.; Berumen-Lechuga, M.G.; Isordia-Salas, I.; Molina-Pérez, C.J. Urinary IgM Excretion: A Reliable Marker for Adverse Pregnancy Outcomes in Women with Chronic Kidney Disease. J. Nephrol. 2019, 32, 241–251. [Google Scholar] [CrossRef]
- Kervella, D.; Torreggiani, M. Baseline Proteinuria Level and Adverse Outcomes in Pregnant Women with Chronic Kidney Disease: New Evidence and a Note of Caution. Clin. Kidney J. 2023, 16, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Conde-Agudelo, A.; Villar, J.; Lindheimer, M. Maternal Infection and Risk of Preeclampsia: Systematic Review and Metaanalysis. Am. J. Obstet. Gynecol. 2008, 198, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Webster, P.; Webster, L.M.; Cook, H.T.; Horsfield, C.; Seed, P.T.; Vaz, R.; Santos, C.; Lydon, I.; Homsy, M.; Lightstone, L.; et al. A Multicenter Cohort Study of Histologic Findings and Long-Term Outcomes of Kidney Disease in Women Who Have Been Pregnant. Clin. J. Am. Soc. Nephrol. 2017, 12, 408–416. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, X.; Zheng, J.; Liu, X.; Yan, T. Pregnancy Outcomes in Patients with Acute Kidney Injury during Pregnancy: A Systematic Review and Meta-Analysis. BMC Pregnancy Childbirth 2017, 17, 235. [Google Scholar] [CrossRef]
- Wiles, K.; Bramham, K.; Seed, P.T.; Nelson-Piercy, C.; Lightstone, L.; Chappell, L.C. Serum Creatinine in Pregnancy: A Systematic Review. Kidney Int. Rep. 2018, 4, 408–419. [Google Scholar] [CrossRef]
- Lopes van Balen, V.A.; van Gansewinkel, T.A.G.; de Haas, S.; Spaan, J.J.; Ghossein-Doha, C.; van Kuijk, S.M.J.; van Drongelen, J.; Cornelis, T.; Spaanderman, M.E.A. Maternal Kidney Function during Pregnancy: Systematic Review and Meta-analysis. Ultrasound Obstet. Gynecol. 2019, 54, 297–307. [Google Scholar] [CrossRef]
- Guibourdenche, J.; Fournier, T.; Malassiné, A.; Evain-Brion, D. Development and Hormonal Functions of the Human Placenta. Folia Histochem. Cytobiol. 2009, 47, S35–S40. [Google Scholar] [CrossRef][Green Version]
- DaSilva-Arnold, S.; James, J.L.; Al-Khan, A.; Zamudio, S.; Illsley, N.P. Differentiation of First Trimester Cytotrophoblast to Extravillous Trophoblast Involves an Epithelial-Mesenchymal Transition. Placenta 2015, 36, 1412–1418. [Google Scholar] [CrossRef]
- Fraser, R.; Whitley, G.S.J.; Thilaganathan, B.; Cartwright, J.E. Decidual Natural Killer Cells Regulate Vessel Stability: Implications for Impaired Spiral Artery Remodelling. J. Reprod. Immunol. 2015, 110, 54–60. [Google Scholar] [CrossRef]
- Staff, A.C.; Fjeldstad, H.E.; Fosheim, I.K.; Moe, K.; Turowski, G.; Johnsen, G.M.; Alnaes-Katjavivi, P.; Sugulle, M. Failure of Physiological Transformation and Spiral Artery Atherosis: Their Roles in Preeclampsia. Am. J. Obstet. Gynecol. 2022, 226 (Suppl. S2), S895–S906. [Google Scholar] [CrossRef]
- Craven, C.M.; Morgan, T.; Ward, K. Decidual Spiral Artery Remodelling Begins before Cellular Interaction with Cytotrophoblasts. Placenta 1998, 19, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Tranquilli, A.L.; Dekker, G.; Magee, L.; Roberts, J.; Sibai, B.M.; Steyn, W.; Zeeman, G.G.; Brown, M.A. The Classification, Diagnosis and Management of the Hypertensive Disorders of Pregnancy: A Revised Statement from the ISSHP. Pregnancy Hypertens. 2014, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, F.; Robertson, W.B.; Brosens, I. The Ultrastructure of Acute Atherosis in Hypertensive Pregnancy. Am. J. Obstet. Gynecol. 1975, 123, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Fetal Growth Restriction: Evaluation—UpToDate. Available online: https://www.uptodate.com/contents/fetal-growth-restriction-evaluation/print#! (accessed on 31 January 2024).
- Özgökçe, Ç.; Öcal, A.; Ermiş, I.S.; Deveci, E. Histopathological, Ultrastructural, and Immunohistochemical Examination of Changes in the Placenta as a Result of Severe Preeclampsia. Acta Cir. Bras. 2023, 38, e382023. [Google Scholar] [CrossRef] [PubMed]
- McElwain, C.J.; Tuboly, E.; McCarthy, F.P.; McCarthy, C.M. Mechanisms of Endothelial Dysfunction in Pre-Eclampsia and Gestational Diabetes Mellitus: Windows into Future Cardiometabolic Health? Front. Endocrinol. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Erez, O.; Romero, R.; Jung, E.; Chaemsaithong, P.; Bosco, M.; Suksai, M.; Gotsch, F. Preeclampsia/Eclampsia: The Conceptual Evolution of a Syndrome. Am. J. Obstet. Gynecol. 2022, 226 (Suppl. S2), S786–S803. [Google Scholar] [CrossRef] [PubMed]
- Magann, E.F.; Martin, J.N.; Isaacs, J.D.; Perry, K.G.; Martin, R.W.; Meydrech, E.F. Immediate Postpartum Curettage: Accelerated Recovery from Severe Preeclampsia. Obstet. Gynecol. 1993, 81, 502–506. [Google Scholar] [PubMed]
- Qu, H.; Khalil, R.A. Vascular Mechanisms and Molecular Targets in Hypertensive Pregnancy and Preeclampsia. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H661–H681. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Burke, S.D.; Karumanchi, S.A. Imbalances in Circulating Angiogenic Factors in the Pathophysiology of Preeclampsia and Related Disorders. Am. J. Obstet. Gynecol. 2022, 226 (Suppl. S2), S1019–S1034. [Google Scholar] [CrossRef] [PubMed]
- De Falco, S. The Discovery of Placenta Growth Factor and Its Biological Activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef]
- Tsatsaris, V.; Goffin, F.; Munaut, C.; Brichant, J.-F.; Pignon, M.-R.; Noel, A.; Schaaps, J.-P.; Cabrol, D.; Frankenne, F.; Foidart, J.-M. Overexpression of the Soluble Vascular Endothelial Growth Factor Receptor in Preeclamptic Patients: Pathophysiological Consequences. J. Clin. Endocrinol. Metab. 2003, 88, 5555–5563. [Google Scholar] [CrossRef]
- Maynard, S.E.; Min, J.-Y.; Merchan, J.; Lim, K.-H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess Placental Soluble Fms-like Tyrosine Kinase 1 (sFlt1) May Contribute to Endothelial Dysfunction, Hypertension, and Proteinuria in Preeclampsia. J. Clin. Investig. 2003, 111, 649–658. [Google Scholar] [CrossRef]
- Bergmann, A.; Ahmad, S.; Cudmore, M.; Gruber, A.D.; Wittschen, P.; Lindenmaier, W.; Christofori, G.; Gross, V.; da Gonzalves, A.C.C.; Gröne, H.-J.; et al. Reduction of Circulating Soluble Flt-1 Alleviates Preeclampsia-like Symptoms in a Mouse Model. J. Cell Mol. Med. 2010, 14, 1857–1867. [Google Scholar] [CrossRef]
- Clauss, M.; Weich, H.; Breier, G.; Knies, U.; Röckl, W.; Waltenberger, J.; Risau, W. The Vascular Endothelial Growth Factor Receptor Flt-1 Mediates Biological Activities. Implications for a Functional Role of Placenta Growth Factor in Monocyte Activation and Chemotaxis. J. Biol. Chem. 1996, 271, 17629–17634. [Google Scholar] [CrossRef]
- Aggarwal, P.K.; Chandel, N.; Jain, V.; Jha, V. The Relationship between Circulating Endothelin-1, Soluble Fms-like Tyrosine Kinase-1 and Soluble Endoglin in Preeclampsia. J. Hum. Hypertens. 2012, 26, 236–241. [Google Scholar] [CrossRef]
- Karakus, S.; Bozoklu Akkar, O.; Yildiz, C.; Sancakdar, E.; Cetin, M.; Cetin, A. Serum Levels of ET-1, M30, and Angiopoietins-1 and -2 in HELLP Syndrome and Preeclampsia Compared to Controls. Arch. Gynecol. Obstet. 2016, 293, 351–359. [Google Scholar] [CrossRef]
- Celik, H.; Avci, B.; Işik, Y. Vascular Endothelial Growth Factor and Endothelin-1 Levels in Normal Pregnant Women and Pregnant Women with Pre-Eclampsia. J. Obstet. Gynaecol. 2013, 33, 355–358. [Google Scholar] [CrossRef]
- Gougos, A.; St Jacques, S.; Greaves, A.; O’Connell, P.J.; d’Apice, A.J.; Bühring, H.J.; Bernabeu, C.; van Mourik, J.A.; Letarte, M. Identification of Distinct Epitopes of Endoglin, an RGD-Containing Glycoprotein of Endothelial Cells, Leukemic Cells, and Syncytiotrophoblasts. Int. Immunol. 1992, 4, 83–92. [Google Scholar] [CrossRef]
- St-Jacques, S.; Forte, M.; Lye, S.J.; Letarte, M. Localization of Endoglin, a Transforming Growth Factor-Beta Binding Protein, and of CD44 and Integrins in Placenta during the First Trimester of Pregnancy. Biol. Reprod. 1994, 51, 405–413. [Google Scholar] [CrossRef]
- Stepan, H.; Krämer, T.; Faber, R. Maternal Plasma Concentrations of Soluble Endoglin in Pregnancies with Intrauterine Growth Restriction. J. Clin. Endocrinol. Metab. 2007, 92, 2831–2834. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, C.; Wang, P.; Gao, J.; Liu, X.; Li, Y.; Yan, S.; Shi, Y. HIF-1α Affects Trophoblastic Apoptosis Involved in the Onset of Preeclampsia by Regulating FOXO3a under Hypoxic Conditions. Mol. Med. Rep. 2020, 21, 2484–2492. [Google Scholar] [CrossRef]
- Tal, R. The Role of Hypoxia and Hypoxia-Inducible Factor-1alpha in Preeclampsia Pathogenesis. Biol. Reprod. 2012, 87, 134. [Google Scholar] [CrossRef]
- Opichka, M.A.; Livergood, M.C.; Balapattabi, K.; Ritter, M.L.; Brozoski, D.T.; Wackman, K.K.; Lu, K.-T.; Kozak, K.N.; Wells, C.; Fogo, A.B.; et al. Mitochondrial-Targeted Antioxidant Attenuates Preeclampsia-like Phenotypes Induced by Syncytiotrophoblast-Specific Gαq Signaling. Sci. Adv. 2023, 9, eadg8118. [Google Scholar] [CrossRef]
- Tyrmi, J.S.; Kaartokallio, T.; Lokki, A.I.; Jääskeläinen, T.; Kortelainen, E.; Ruotsalainen, S.; Karjalainen, J.; Ripatti, S.; Kivioja, A.; Laisk, T.; et al. Genetic Risk Factors Associated with Preeclampsia and Hypertensive Disorders of Pregnancy. JAMA Cardiol. 2023, 8, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Teng, X.; Zhao, J.; Feng, Y.; Wang, L. A Potential Autophagy-Related-Gene Based Signature in Patients with Preeclampsia. Front. Biosci. 2023, 28, 132. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.; Herrock, O.; Campbell, N.; Cornelius, D.; Fitzgerald, S.; Amaral, L.M.; LaMarca, B. The Role of Immune Cells and Mediators in Preeclampsia. Nat. Rev. Nephrol. 2023, 19, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W.; Vaka, V.R.; McMaster, K.; Ibrahim, T.; Cornelius, D.C.; Amaral, L.; Campbell, N.; Wallukat, G.; McDuffy, S.; Usry, N.; et al. Renal Natural Killer Cell Activation and Mitochondrial Oxidative Stress; New Mechanisms in AT1-AA Mediated Hypertensive Pregnancy. Pregnancy Hypertens. 2019, 15, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, S.; Deer, E.; Hogg, J.; Cornelius, D.C.; Turner, T.; Amaral, L.M.; Hoang, N.; Edwards, K.; Herrock, O.; Campbell, N.; et al. RUPP Th17s Cause Hypertension and Mitochondrial Dysfunction in the Kidney and Placenta during Pregnancy. Pregnancy Hypertens. 2023, 32, 50–56. [Google Scholar] [CrossRef]
- Scharfe-Nugent, A.; Corr, S.C.; Carpenter, S.B.; Keogh, L.; Doyle, B.; Martin, C.; Fitzgerald, K.A.; Daly, S.; O’Leary, J.J.; O’Neill, L.A.J. TLR9 Provokes Inflammation in Response to Fetal DNA: Mechanism for Fetal Loss in Preterm Birth and Preeclampsia. J. Immunol. 2012, 188, 5706–5712. [Google Scholar] [CrossRef] [PubMed]
- Germain, S.J.; Sacks, G.P.; Sooranna, S.R.; Sargent, I.L.; Redman, C.W. Systemic Inflammatory Priming in Normal Pregnancy and Preeclampsia: The Role of Circulating Syncytiotrophoblast Microparticles. J. Immunol. 2007, 178, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Lafayette, R.A. Renal Physiology of Pregnancy. Adv. Chronic Kidney Dis. 2013, 20, 209–214. [Google Scholar] [CrossRef]
- Conrad, K.P.; Davison, J.M. The Renal Circulation in Normal Pregnancy and Preeclampsia: Is There a Place for Relaxin? Am. J. Physiol. Ren. Physiol. 2014, 306, F1121–F1135. [Google Scholar] [CrossRef]
- Faúndes, A.; Brícola-Filho, M.; Pinto e Silva, J.L. Dilatation of the Urinary Tract during Pregnancy: Proposal of a Curve of Maximal Caliceal Diameter by Gestational Age. Am. J. Obstet. Gynecol. 1998, 178, 1082–1086. [Google Scholar] [CrossRef]
- Au, K.K.; Woo, J.S.; Tang, L.C.; Liang, S.T. Aetiological Factors in the Genesis of Pregnancy Hydronephrosis. Aust. N. Z. J. Obstet. Gynaecol. 1985, 25, 248–251. [Google Scholar] [CrossRef]
- Brown, M.A.; Gallery, E.D. Volume Homeostasis in Normal Pregnancy and Pre-Eclampsia: Physiology and Clinical Implications. Baillieres Clin. Obstet. Gynaecol. 1994, 8, 287–310. [Google Scholar] [CrossRef]
- Chapman, A.B.; Abraham, W.T.; Zamudio, S.; Coffin, C.; Merouani, A.; Young, D.; Johnson, A.; Osorio, F.; Goldberg, C.; Moore, L.G.; et al. Temporal Relationships between Hormonal and Hemodynamic Changes in Early Human Pregnancy. Kidney Int. 1998, 54, 2056–2063. [Google Scholar] [CrossRef]
- Weisinger, R.S.; Burns, P.; Eddie, L.W.; Wintour, E.M. Relaxin Alters the Plasma Osmolality-Arginine Vasopressin Relationship in the Rat. J. Endocrinol. 1993, 137, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.P. Maternal Vasodilation in Pregnancy: The Emerging Role of Relaxin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R267–R275. [Google Scholar] [CrossRef]
- Post Uiterweer, E.D.; Koster, M.P.H.; Jeyabalan, A.; Kuc, S.; Siljee, J.E.; Stewart, D.R.; Conrad, K.P.; Franx, A. Circulating Pregnancy Hormone Relaxin as a First Trimester Biomarker for Preeclampsia. Pregnancy Hypertens. 2020, 22, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Lumbers, E.R.; Pringle, K.G. Roles of the Circulating Renin-Angiotensin-Aldosterone System in Human Pregnancy. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2014, 306, R91–R101. [Google Scholar] [CrossRef]
- Gathiram, P.; Moodley, J. The Role of the Renin-Angiotensin-Aldosterone System in Preeclampsia: A Review. Curr. Hypertens. Rep. 2020, 22, 89. [Google Scholar] [CrossRef]
- Shoemaker, R.; Poglitsch, M.; Huang, H.; Vignes, K.; Srinivasan, A.; Cockerham, C.; Schadler, A.; Bauer, J.A.; O’Brien, J.M. Activation of the Renin–Angiotensin–Aldosterone System Is Attenuated in Hypertensive Compared with Normotensive Pregnancy. Int. J. Mol. Sci. 2023, 24, 12728. [Google Scholar] [CrossRef]
- Jung, E.; Romero, R.; Yeo, L.; Gomez-Lopez, N.; Chaemsaithong, P.; Jaovisidha, A.; Erez, O. The Etiology of Preeclampsia. Am. J. Obstet. Gynecol. 2022, 226 (Suppl. S2), S844–S866. [Google Scholar] [CrossRef] [PubMed]
- Gaber, L.W.; Spargo, B.H.; Lindheimer, M.D. Renal Pathology in Pre-Eclampsia. Baillieres Clin. Obstet. Gynaecol. 1987, 1, 971–995. [Google Scholar] [CrossRef]
- Roberts, J.M.; Redman, C.W.G. Pre-Eclampsia: More than Pregnancy-Induced Hypertension. Lancet 1993, 341, 1447–1451. [Google Scholar] [CrossRef]
- Muhammad, N.; Liaqat, N. Causes and Outcome of Pregnancy Related Acute Kidney Injury. Pak. J. Med. Sci. 2024, 40 Pt I, 64–67. [Google Scholar] [CrossRef]
- Mei, J.Y.; Afshar, Y. Hypertensive Complications of Pregnancy: Hepatic Consequences of Preeclampsia through HELLP Syndrome. Clin Liver Dis (Hoboken) 2023, 22, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Shu, H.; Yu, Y.; Li, H.; Chen, L.; Liu, J.; Li, X.-M. Acute Kidney Injury in Patients with HELLP Syndrome. Int. Urol. Nephrol. 2019, 51, 1199–1206. [Google Scholar] [CrossRef]
- Nelson, D.B.; Byrne, J.J.; Cunningham, F.G. Acute Fatty Liver of Pregnancy. Clin. Obstet. Gynecol. 2020, 63, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Ibdah, J.A. Mitochondrial Dysfunction and Acute Fatty Liver of Pregnancy. Int. J. Mol. Sci. 2022, 23, 3595. [Google Scholar] [CrossRef] [PubMed]
- Botero, J.P.; McIntosh, J.J. Labor and Delivery: DIC, HELLP, Preeclampsia. Hematol. Am. Soc. Hematol. Educ. Program. 2023, 2023, 737–744. [Google Scholar] [CrossRef]
- Jiang, Y.; McIntosh, J.J.; Reese, J.A.; Deford, C.C.; Kremer Hovinga, J.A.; Lämmle, B.; Terrell, D.R.; Vesely, S.K.; Knudtson, E.J.; George, J.N. Pregnancy Outcomes Following Recovery from Acquired Thrombotic Thrombocytopenic Purpura. Blood 2014, 123, 1674–1680. [Google Scholar] [CrossRef]
- Fakhouri, F.; Scully, M.; Ardissino, G.; Al-Dakkak, I.; Miller, B.; Rondeau, E. Pregnancy-Triggered Atypical Hemolytic Uremic Syndrome (aHUS): A Global aHUS Registry Analysis. J. Nephrol. 2021, 34, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Hladunewich, M.A. Chronic Kidney Disease and Pregnancy. Semin. Nephrol. 2017, 37, 337–346. [Google Scholar] [CrossRef]
- Hou, S. Pregnancy in Chronic Renal Insufficiency and End-Stage Renal Disease. Am. J. Kidney Dis. 1999, 33, 235–252. [Google Scholar] [CrossRef]
- Vellanki, K. Pregnancy in Chronic Kidney Disease. Adv. Chronic Kidney Dis. 2013, 20, 223–228. [Google Scholar] [CrossRef]
- Ribeiro, C.I.; Silva, N. Pregnancy and Dialysis. J. Bras. Nefrol. 2020, 42, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; McCormick, C.A.; Krelle, A.; Champion de Crespigny, P.; Unterscheider, J. Pregnancy Outcomes Post-Kidney Transplantation across 23 Years. Aust. N. Z. J. Obstet. Gynaecol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Kovač, D.; Kovač, L.; Mertelj, T.; Steblovnik, L. Pregnancy After Kidney Transplantation. Transplant. Proc. 2021, 53, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.W.; Strauss, J.F. The Road to Low-Dose Aspirin Therapy for the Prevention of Preeclampsia Began with the Placenta. Int. J. Mol. Sci. 2021, 22, 6985. [Google Scholar] [CrossRef] [PubMed]
- Wiles, K.; Chappell, L.C.; Lightstone, L.; Bramham, K. Updates in Diagnosis and Management of Preeclampsia in Women with CKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 1371–1380. [Google Scholar] [CrossRef]
- Hypertension in Pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [CrossRef]
- Poon, L.C.; Magee, L.A.; Verlohren, S.; Shennan, A.; von Dadelszen, P.; Sheiner, E.; Hadar, E.; Visser, G.; Da Silva Costa, F.; Kapur, A.; et al. A Literature Review and Best Practice Advice for Second and Third Trimester Risk Stratification, Monitoring, and Management of Pre-eclampsia. Int. J. Gynaecol. Obstet. 2021, 154 (Suppl. S1), 3–31. [Google Scholar] [CrossRef]
- Eddy, A.C.; Chiang, C.Y.; Rajakumar, A.; Spradley, F.T.; Dauer, P.; Granger, J.P.; Rana, S. Bioflavonoid Luteolin Prevents sFlt-1 Release via HIF-1α Inhibition in Cultured Human Placenta. FASEB J. 2023, 37, e23078. [Google Scholar] [CrossRef]
- Kalousová, M.; Muravská, A.; Zima, T. Pregnancy-Associated Plasma Protein A (PAPP-A) and Preeclampsia. Adv. Clin. Chem. 2014, 63, 169–209. [Google Scholar] [CrossRef]
- Early Pregnancy Prediction of Preeclampsia—UpToDate. Available online: https://www.uptodate.com/contents/early-pregnancy-prediction-of-preeclampsia/print#! (accessed on 31 January 2024).
- Giardini, V.; Grilli, L.; Terzaghi, A.; Todyrenchuk, L.; Zavettieri, C.; Mazzoni, G.; Cozzolino, S.; Casati, M.; Vergani, P.; Locatelli, A. sFlt-1 Levels as a Predicting Tool in Placental Dysfunction Complications in Multiple Pregnancies. Biomedicines 2023, 11, 2917. [Google Scholar] [CrossRef] [PubMed]
- Magee, L.A.; Brown, M.A.; Hall, D.R.; Gupte, S.; Hennessy, A.; Karumanchi, S.A.; Kenny, L.C.; McCarthy, F.; Myers, J.; Poon, L.C.; et al. The 2021 International Society for the Study of Hypertension in Pregnancy Classification, Diagnosis & Management Recommendations for International Practice. Pregnancy Hypertens. 2022, 27, 148–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ke, Y.; Chen, W.; Wu, S.; Zhuang, X.; Lin, Q.; Shi, Q.; Wu, Z. Association of the LEP Gene with Immune Infiltration as a Diagnostic Biomarker in Preeclampsia. Front. Mol. Biosci. 2023, 10, 1209144. [Google Scholar] [CrossRef] [PubMed]
- Ramdin, S.; Naicker, T.; Baijnath, S.; Govender, N. Kidney Injury Molecule-1 and Podocalyxin Dysregulation in an Arginine Vasopressin Induced Rodent Model of Preeclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2023, 284, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Santillan, M.K.; Santillan, D.A.; Scroggins, S.M.; Min, J.Y.; Sandgren, J.A.; Pearson, N.A.; Leslie, K.K.; Hunter, S.K.; Zamba, G.K.D.; Gibson-Corley, K.N.; et al. Vasopressin in Preeclampsia: A Novel Very-Early Human Pregnancy Biomarker and Clinically-Relevant Mouse Model. Hypertension 2014, 64, 852–859. [Google Scholar] [CrossRef]
- Akalın, S.A.; Öcal, E.; Deveci, E. Role of SOX9 and Hif-1α Expression in Placentas of Patients with HELLP. Acta Cir. Bras. 2023, 38, e388023. [Google Scholar] [CrossRef]
- Latha, A.P.; Haripriya, V.; Ramya Raj, P. Mid-Trimester Spot Urinary Albumin/Creatinine Ratio as a Screening Tool in Prediction of Pre-Eclampsia. J. Obstet. Gynaecol. India 2023, 73 (Suppl. S2), 234–239. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Authors | Article Type/Year of Publication | Aim/Purpose | Conclusions/Results |
|---|---|---|---|
| Conde Agudelo et al. [16] | Systematic review and meta-analysis/2008 | Examine the relationship between maternal infection and the risk of PE. | Both urinary tract infection and periodontal disease during pregnancy are associated with an increased risk of PE. |
| Zhang et al. [12] | Systematic review/2015 | Estimation of (1) the risk of pregnancy complications among patients with CKD versus those without CKD and (2) the risk of CKD progression among pregnant patients versus nonpregnant controls with CKD. | The risks of adverse maternal and fetal outcomes in pregnancy are higher for women with CKD versus pregnant women without CKD. However, pregnancy was not a risk factor for the progression of renal disease in women with CKD before pregnancy. |
| Webster et al. [17] | Multicenter cohort study/2017 | Comparison of causes and long-term renal outcomes of biopsy-proven renal disease identified during pregnancy or within 1 year postpartum, with nonpregnant women. | FSGS is more common in women who have been pregnant than in controls, and the disease identified in pregnancy or within 1 year postpartum is more likely to show a subsequent decline in renal function. |
| Liu et al. [18] | Systematic review and meta-analysis/2017 | Evaluate the impact of pregnancy-related AKI on pregnancy outcomes. | Pregnancy-related AKI remains a grave complication and has been associated with increased maternal and fetal mortality. |
| Wiles et al. [19] | Systematic review/2018 | Define the difference in serum creatinine in a healthy pregnancy compared with concentrations in nonpregnant women to facilitate the identification of abnormal kidney function in pregnancy. | Based on a nonpregnant reference interval of 45–90 μmol/L (0.51–1.02 mg/dL), a serum creatinine of >77 μmol/L (0.87 mg/dL) should be considered outside the normal range for pregnancy. |
| Lopes van Balen et al. [20] | Systematic review and meta-analysis/2019 | Estimate the extent of adaptation over the course of both healthy physiological and complicated singleton pregnancies, and determine healthy pregnancy reference values. | In healthy pregnancy, GFR is increased as early as the first trimester compared with non-pregnant values, and the kidneys continue to function at a higher rate throughout gestation. In contrast, kidney function is decreased in hypertensive pregnancy. |
| Goetz et al. [9] | Observational cohort study/2021 | Examine the risk of CKD after preterm delivery and PE in a large obstetric cohort in Germany, considering pre-existing comorbidities, potential confounders, and the severity of CKD. | Preterm delivery and PE were identified as independent risk factors for all CKD stages. A joint exposure or preterm birth and PE was associated with an excessive maternal risk burden for CKD in the first decade after pregnancy. |
| Srialluri et al. [8] | Observational cohort study/2023 | Evaluate the long-term association between PE and the risk of developing chronic hypertension and kidney disease. | Individuals with a pregnancy complicated by PE had a higher risk of hypertension, reduced eGFR, and albuminuria compared with individuals without PE. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andronikidi, P.E.; Orovou, E.; Mavrigiannaki, E.; Athanasiadou, V.; Tzitiridou-Chatzopoulou, M.; Iatrakis, G.; Grapsa, E. Placental and Renal Pathways Underlying Pre-Eclampsia. Int. J. Mol. Sci. 2024, 25, 2741. https://doi.org/10.3390/ijms25052741
Andronikidi PE, Orovou E, Mavrigiannaki E, Athanasiadou V, Tzitiridou-Chatzopoulou M, Iatrakis G, Grapsa E. Placental and Renal Pathways Underlying Pre-Eclampsia. International Journal of Molecular Sciences. 2024; 25(5):2741. https://doi.org/10.3390/ijms25052741
Chicago/Turabian StyleAndronikidi, Paraskevi Eva, Eirini Orovou, Eleftheria Mavrigiannaki, Virginia Athanasiadou, Maria Tzitiridou-Chatzopoulou, George Iatrakis, and Eirini Grapsa. 2024. "Placental and Renal Pathways Underlying Pre-Eclampsia" International Journal of Molecular Sciences 25, no. 5: 2741. https://doi.org/10.3390/ijms25052741
APA StyleAndronikidi, P. E., Orovou, E., Mavrigiannaki, E., Athanasiadou, V., Tzitiridou-Chatzopoulou, M., Iatrakis, G., & Grapsa, E. (2024). Placental and Renal Pathways Underlying Pre-Eclampsia. International Journal of Molecular Sciences, 25(5), 2741. https://doi.org/10.3390/ijms25052741