
The Single Toxin Origin of Alzheimer’s Disease and Other Neurodegenerative Disorders Enables Targeted Approach to Treatment and Prevention

Abstract
1. Introduction
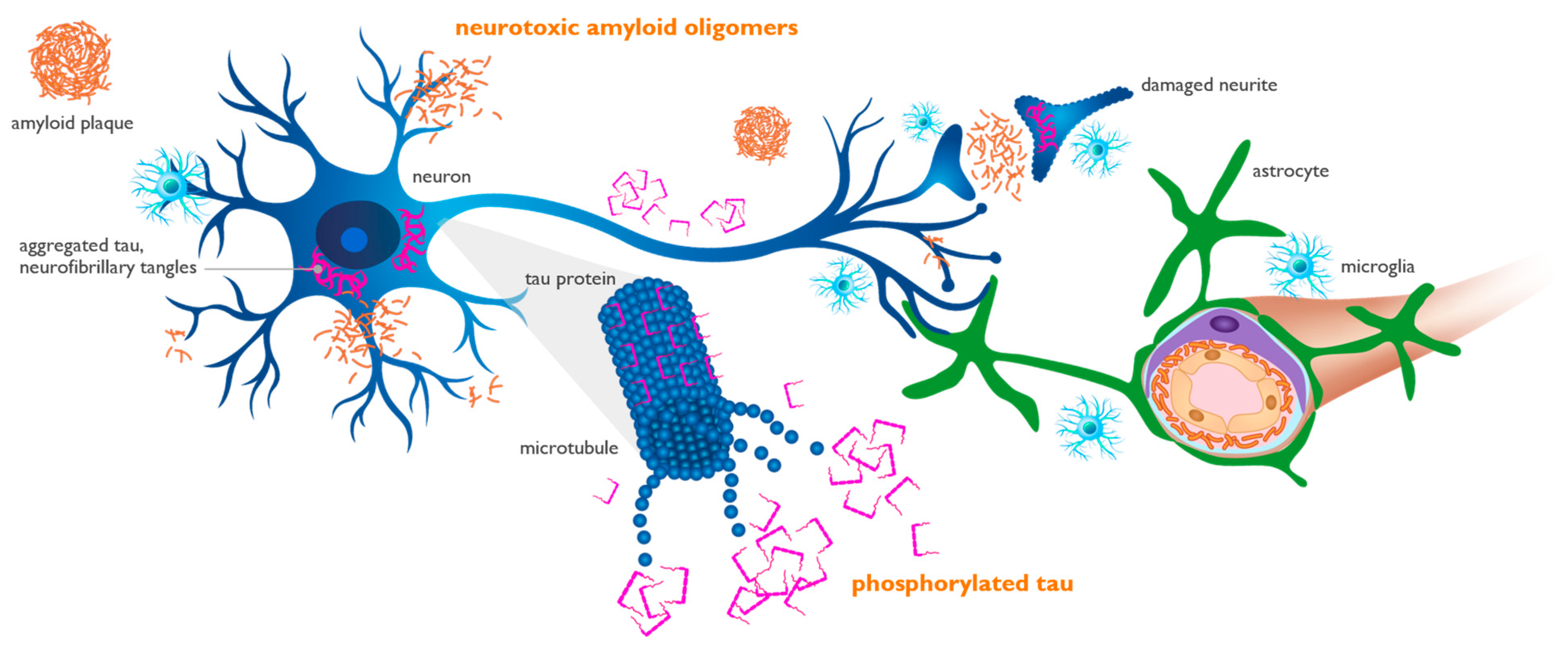
2. Physiologic and Pathogenic Role of Beta-Amyloid Protein
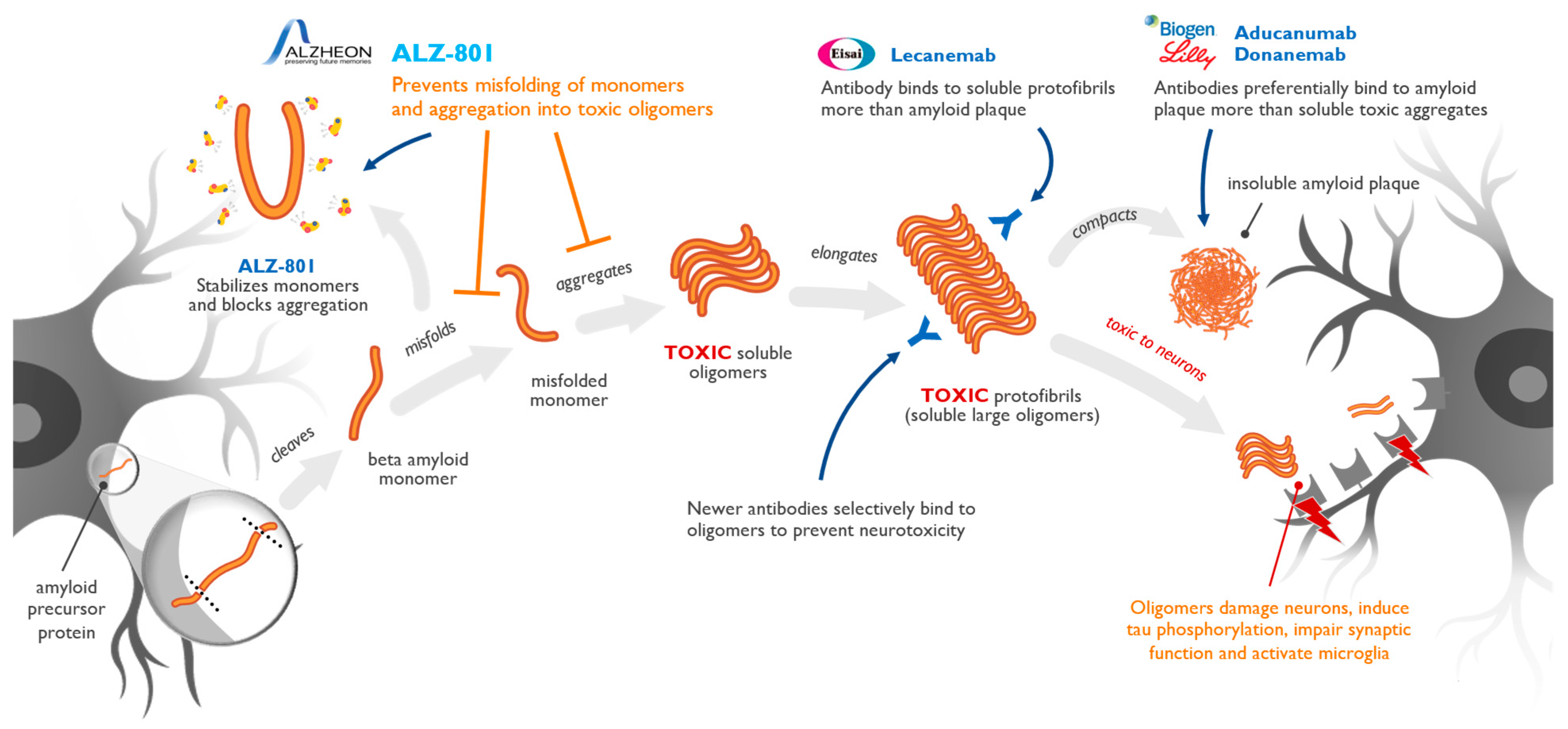
3. Amyloid Pathogenic Cascade
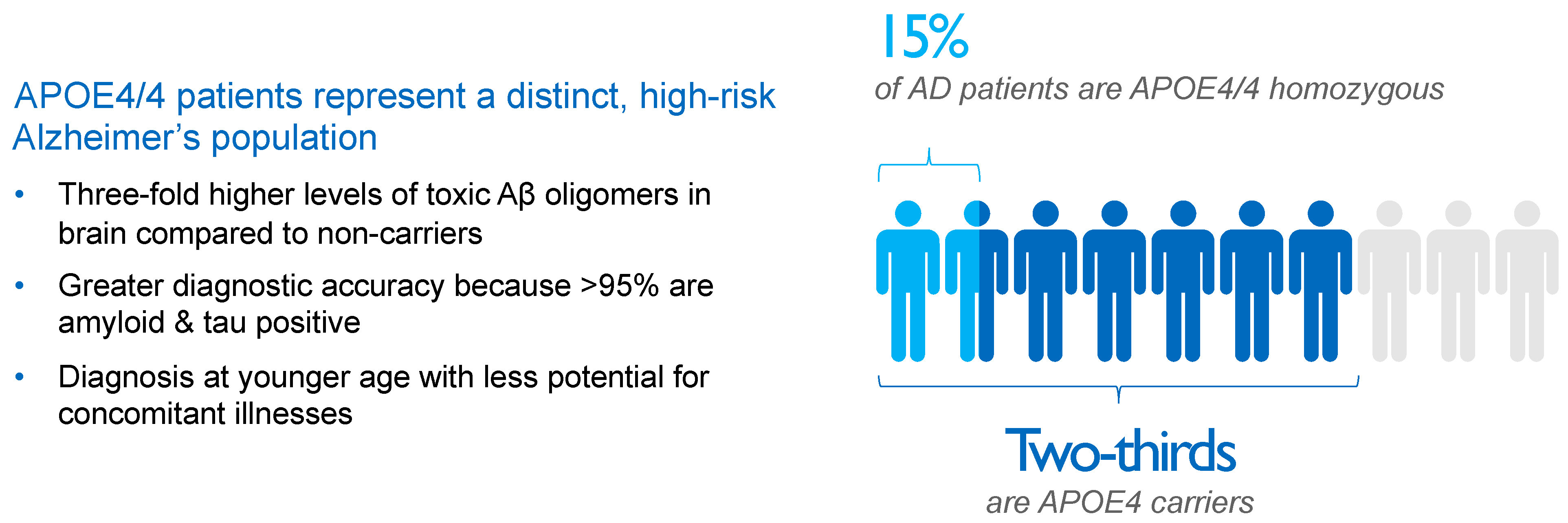
4. APOE4 Represents Main Genetic Risk Factor for Alzheimer’s Disease
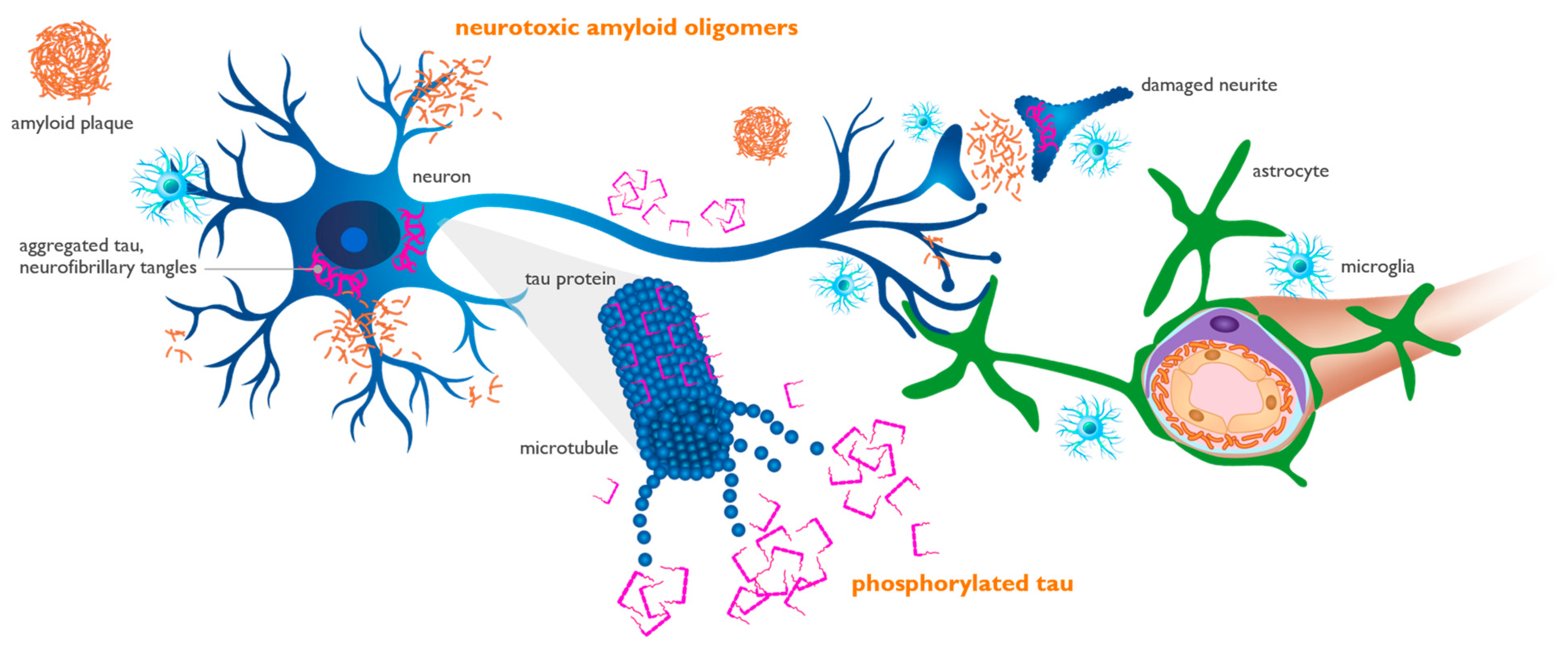
5. Interaction of Aβ and APOE4 with Other Molecules in AD Brain
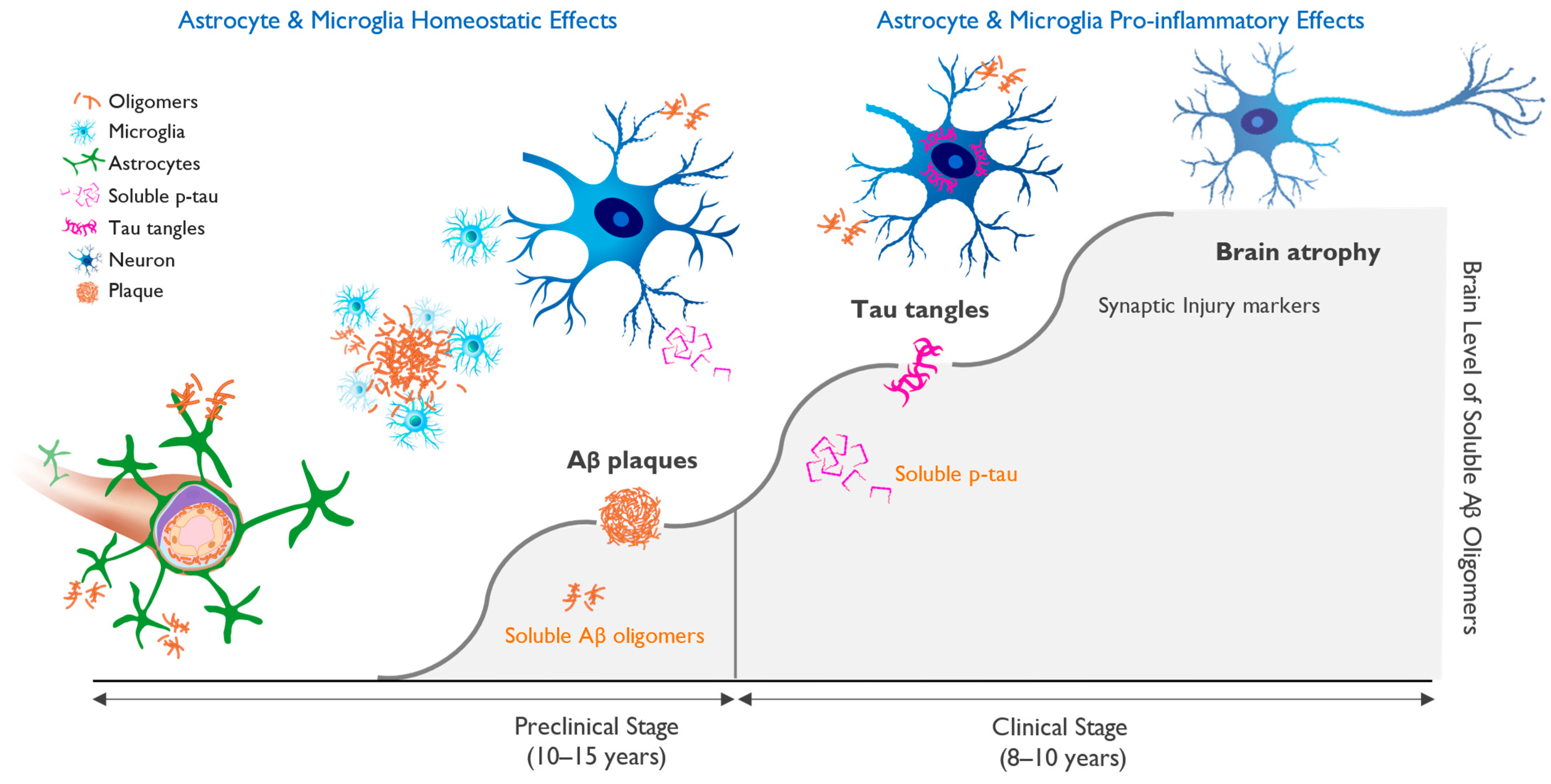
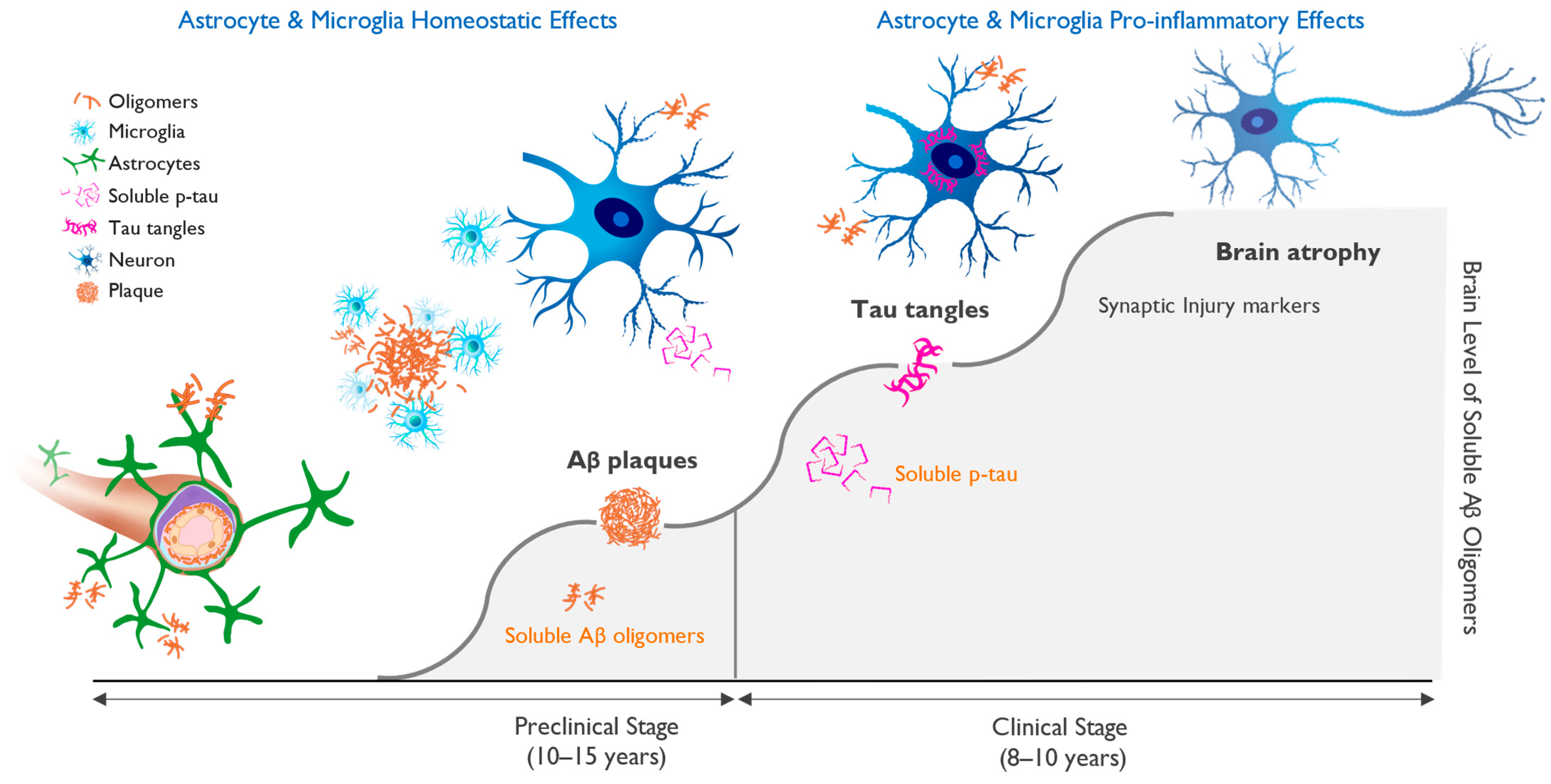
6. Biomarkers and Biological Definition of Alzheimer’s Disease
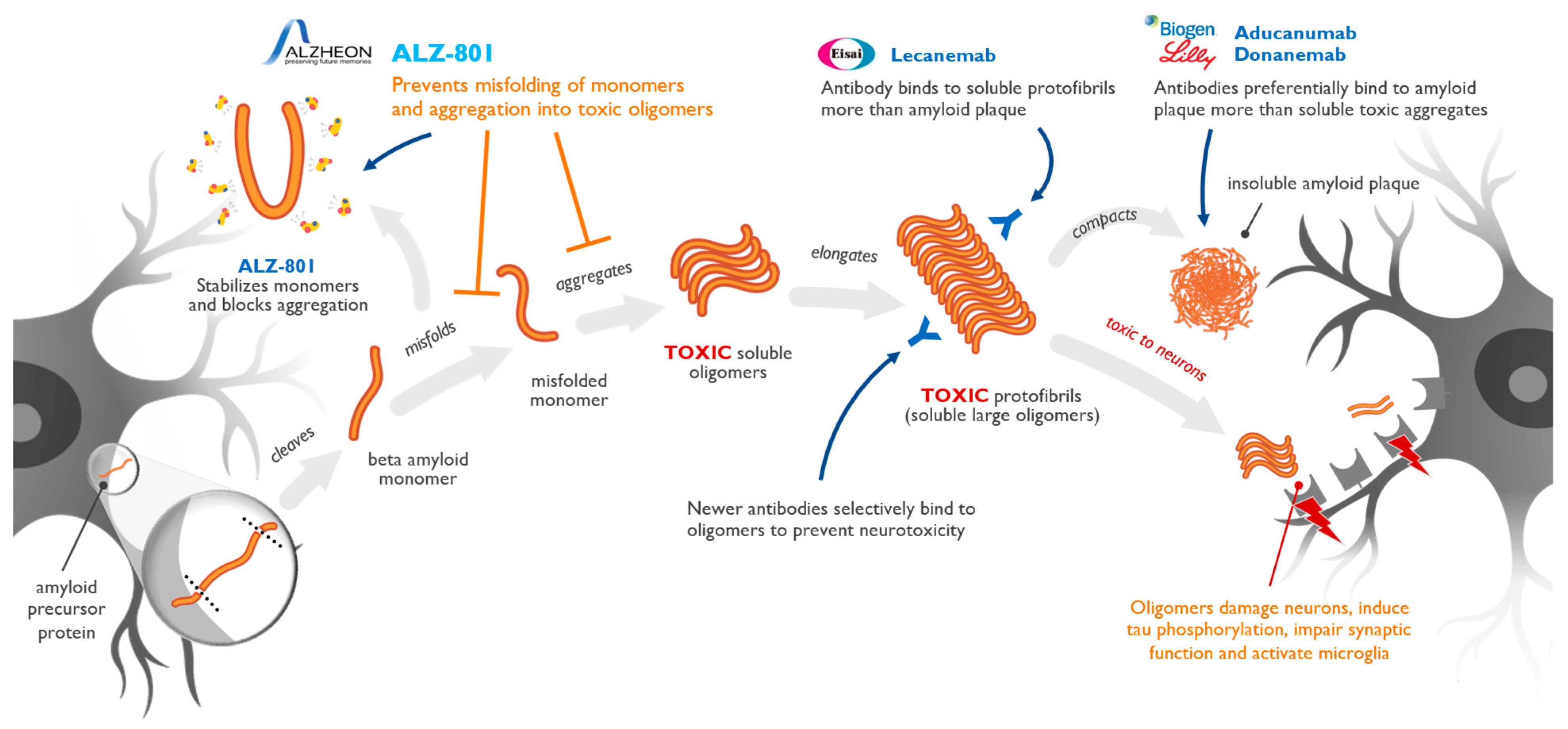
7. Targeting of Amyloid Oligomers Necessary for Efficacy in Alzheimer’s Disease
8. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—The first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res. Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J. Prev. Alzheimers Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in early Alzheimer’s disease. N. Eng. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in early symptomatic Alzheimer disease The TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Mora, P.; Luna, R.; Colín-Barenque, L. Amyloid beta: Multiple mechanisms of toxicity and only some protective effects? Oxid. Med. Cell. Longev. 2014, 2014, 795375. [Google Scholar] [CrossRef]
- Hefter, D.; Draguhn, A. APP as a protective factor in acute neuronal insults. Front. Mol. Neurosci. 2017, 10, 22. [Google Scholar] [CrossRef]
- Linse, S.; Scheidt, T.; Bernfur, K.; Vendruscolo, M.; Dobson, C.M.; Cohen, S.I.A.; Sileikis, E.; Lundqvist, M.; Qian, F.; O’Malley, T.; et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat. Struct. Mol. Biol. 2020, 27, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Duran-Aniotz, C.; Moreno-Gonzalez, I.; Gamez, N.; Perez-Urrutia, N.; Vegas-Gomez, L.; Soto, C.; Morales, R. Amyloid pathology arrangements in Alzheimer’s disease brains modulate in vivo seeding capability. Acta Neuropathol. Commun. 2021, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and a-synuclein in neurodegeneration. Brain 2017, 140, 266–278. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic soluble amyloid oligomers drive Alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Weaver, D.F. Microglia and microglial-based receptors in the pathogenesis and treatment of Alzheimer’s disease. Int. Immunopharmacol. 2022, 110, 109070. [Google Scholar] [CrossRef] [PubMed]
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J. Neurosci. 2020, 40, 1956–1974. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Wang, M.X.; Ismail, O.; Braun, M.; Schindler, A.G.; Reemmer, J.; Wan, Z.; Haveliwala, M.A.; O’Boyle, R.P.; Han, W.Y.; et al. Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid β plaque formation in mice. Alzheimers Res. Ther. 2022, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.; Visser, P.J.; Amyloid Biomarker Study Group; Aalten, P.; Aarsland, D.; et al. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef]
- Ossenkoppele, R.; Jansen, W.J.; Rabinovici, G.D.; Knol, D.L.; van der Flier, W.M.; van Berckel, B.N.; Scheltens, P.; Visser, P.J.; Amyloid PET Study Group; Verfaillie, S.C.; et al. Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 2015, 313, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Tijms, B.M.; Vromen, E.M.; Mjaavatten, O.; Holstege, H.; Reus, L.M.; van der Lee, S.; Wesenhagen, K.E.J.; Lorenzini, L.; Vermunt, L.; Venkatraghavan, V.; et al. Cerebrospinal fluid proteomics in patients with Alzheimer’s disease reveals five molecular subtypes with distinct genetic risk profiles. Nat. Aging 2024, 4, 33–47. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Eng. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; Lott, I.; Klunk, W.; Head, E.; Christian, B.; et al. The AT(N) framework for Alzheimer’s disease in adults with Down syndrome. Alzheimers Dement. 2020, 12, e12062. [Google Scholar] [CrossRef] [PubMed]
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna, M.; Pegueroles, J.; Montal, V.; et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997. [Google Scholar] [CrossRef]
- Roberts, B.R.; Lind, M.; Wagen, A.Z.; Rembach, A.; Frugier, T.; Li, Q.X.; Ryan, T.M.; McLean, C.A.; Doecke, J.D.; Rowe, C.C.; et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer’s disease: Correlation with amyloid PET. Brain 2017, 140, 1486–1498. [Google Scholar] [CrossRef]
- Abushakra, S.; Hey, J.; Blennow, K.; Scheltens, P.; Reiman, E.M.; Hort, J.; Sheardova, K.; Rutgers, S.M.; Prins, N.D.; Dautzenberg, P.; et al. Effects of oral ALZ-801, an amyloid oligomer inhibitor, on plasma biomarkers in APOE4 carriers with Early Alzheimer’s disease: Results of six-month interim analysis from a phase 2 biomarker study. Alzheimers Dement. 2022, 18, e069141. Available online: https://alz-journals.onlinelibrary.wiley.com/doi/epdf/10.1002/alz.069141 (accessed on 14 September 2023). [CrossRef]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A randomized, double-blind phase 2b proof-of-concept clinical trial in Early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Abushakra, S.; Porsteinsson, A.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Hendrix, S.; Wang, P.; Shen, L.; et al. Clinical benefits of tramiprosate in Alzheimer’s disease are associated with higher number of APOE4 alleles: The “APOE4 gene-dose effect”. J. Prev. Alzheimers Dis. 2016, 3, 219–228. [Google Scholar] [CrossRef]
- Gaspar, R.C.; Villarreal, S.A.; Bowles, N.; Hepler, R.W.; Joyce, J.G.; Shughrue, P.J. Oligomers of beta-amyloid are sequestered into and seed new plaques in the brains of an AD mouse model. Exp. Neurol. 2010, 223, 394–400. [Google Scholar] [CrossRef]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Sehlin, D.; Englund, H.; Simu, B.; Karlsson, M.; Ingelsson, M.; Nikolajeff, F.; Lannfelt, L.; Pettersson, F.E. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS ONE 2012, 7, e32014. [Google Scholar] [CrossRef]
- Esparza, T.J.; Zhao, H.; Cirrito, J.R.; Cairns, N.J.; Bateman, R.J.; Holtzman, D.M.; Brody, D.L. Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 2013, 73, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion channel formation by amyloid-β42 oligomers but not amyloid-β40 in cellular membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef] [PubMed]
- Madhu, P.; Mukhopadhyay, S. Distinct types of amyloid-β oligomers displaying diverse neurotoxicity mechanisms in Alzheimer’s disease. J. Cell Biochem. 2021, 122, 1594–1608. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and identification of an endogenous metabolite of tramiprosate and its prodrug ALZ-801 that inhibits beta amyloid oligomer formation in the human brain. CNS Drugs 2018, 32, 849–861. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.N.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.; Weller, R.O.; Carare, R.O. Lymphatic clearance of the brain: Perivascular, paravascular and significance for neurodegenerative diseases. Cell. Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef]
- Taoka, T.; Masutani, Y.; Kawai, H.; Nakane, T.; Matsuoka, K.; Yasuno, F.; Kishimoto, T.; Naganawa, S. Evaluation of glymphatic system activity with the diffusion MR technique: Diffusion tensor image analysis along the perivascular space (DTI-ALPS) in Alzheimer’s disease cases. Jpn. J. Radiol. 2017, 35, 172–178. [Google Scholar] [CrossRef]
- Kamagata, K.; Andica, C.; Takabayashi, K.; Saito, Y.; Taoka, T.; Nozaki, H.; Kikuta, J.; Fujita, S.; Hagiwara, A.; Kamiya, K.; et al. Association of MRI indices of glymphatic system with amyloid deposition and cognition in mild cognitive impairment and Alzheimer disease. Neurology 2022, 99, e2648–e2660. [Google Scholar] [CrossRef]
- Pujadas, L.; Rossi, D.; Andrés, R.; Teixeira, C.M.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef]
- Yu, N.N.; Tan, M.S.; Yu, J.T.; Xie, A.M.; Tan, L. The role of Reelin signaling in Alzheimer’s disease. Mol. Neurobiol. 2016, 53, 5692–5700. [Google Scholar] [CrossRef]
- Marck, A.T.; Fritschle, K.E.; Calvier, L.; Herz, J. Reelin changes hippocampal learning in aging and Alzheimer’s disease. Behav. Brain Res. 2021, 414, 113482. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benvenist, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Fluid biomarker-based molecular phenotyping of Alzheimer’s disease patients in research and clinical settings. Prog. Mol. Biol. Transl. Sci. 2019, 168, 3–23. [Google Scholar] [CrossRef]
- Janelidze, S.; Zetterberg, H.; Mattsson, N.; Palmqvist, S.; Vanderstichele, H.; Lindberg, O.; van Westen, D.; Stomrud, E.; Minthon, L.; Blennow, K.; et al. CSF Aβ42/Aβ40 and Aβ42/Aβ38 ratios: Better diagnostic markers of Alzheimer disease. Ann. Clin. Transl. Neurol. 2016, 3, 154–165. [Google Scholar] [CrossRef]
- Karisetty, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Zhang, H.; Elefant, F. Amyloid-β peptide impact on synaptic function and neuroepigenetic gene control reveal new therapeutic strategies for Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 577622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Z.; Hu, H.; Zhao, M.; Sun, L. Microglia in Alzheimer’s Disease: A target for therapeutic intervention. Front. Cell. Neurosci. 2021, 15, 749587. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Editorial: A is for amyloid. J. Prev. Alzheimers Dis. 2020, 7, 140–141. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Selkoe, D.J. Systematic analysis of time-dependent neural effects of soluble amyloid β oligomers in culture and in vivo: Prevention by scyllo-inositol. Neurobiol. Dis. 2015, 82, 152–163. [Google Scholar] [CrossRef]
- Savage, M.J.; Kalinina, J.; Wolfe, A.; Tugusheva, K.; Korn, R.; Cash-Mason, T.; Maxwell, J.W.; Hatcher, N.G.; Haugabook, S.J.; Wu, G.; et al. A sensitive aβ oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluid. J. Neurosci. 2014, 34, 2884–2897. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Nilson, A.N.; Kaye, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jackson, R.J.; Hong, W.; Taylor, W.M.; Corbett, G.T.; Moreno, A.; Liu, W.; Li, S.; Frosch, M.P.; Slutsky, I.; et al. Human brain-derived Aβ oligomers bind to synapses and disrupt synaptic activity in a manner that requires APP. J. Neurosci. 2017, 37, 11947–11966. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Wang, Z.; Liu, W.; O’Malley, T.T.; Jin, M.; Willem, M.; Haass, C.; Frosch, M.P.; Walsh, D.M. Diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in Alzheimer’s disease brain. Acta Neuropathol. 2018, 136, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Serrano-Pozo, A.; Hori, Y.; Adams, K.W.; Takeda, S.; Banerji, A.O.; Mitani, A.; Joyner, D.; Thyssen, D.H.; Bacskai, B.J.; et al. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide. J. Neurosci. 2012, 32, 15181–15192. [Google Scholar] [CrossRef]
- Tai, L.M.; Bilousova, T.; Jungbauer, L.; Roeske, S.K.; Youmans, K.L.; Yu, C.; Poon, W.W.; Cornwell, L.B.; Miller, C.A.; Vinters, H.V.; et al. Levels of soluble apolipoprotein E/amyloid-β (Aβ) complex are reduced and oligomeric Aβ increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J. Biol. Chem. 2013, 288, 5914–5926. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.H.; Abushakra, S.; Blennow, K.; Scheltens, P.; Hort, J.; Sheardova, K.; Prins, N.D.; Rutgers, M.S.; Dautzenberg, P.L.; Pazdert, L.; et al. Effects of ALZ-801, an oral amyloid oligomer inhibitor, on biomarkers of Alzheimer’s disease (AD): 12-month results of phase 2 biomarker study in early AD (S26.0007). In Proceedings of the American Academy of Neurology Conference, Boston, MA, USA, 22–27 April 2023; Available online: https://www.aan.com/MSA/Public/Events/AbstractDetails/52624 (accessed on 14 September 2023).
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A. Elucidating the Aβ42 anti-aggregation mechanism of action of tramiprosate in Alzheimer’s disease: Integrating molecular analytical methods, pharmacokinetic and clinical data. CNS Drugs 2017, 31, 495–509. [Google Scholar] [CrossRef]
- Uhlmann, R.E.; Rother, C.; Rasmussen, J.; Schelle, J.; Bergmann, C.; Ullrich Gavilanes, E.M.; Fritschi, S.K.; Buehler, A.; Baumann, F.; Skodras, A.; et al. Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat. Neurosci. 2020, 23, 1580–1588. [Google Scholar] [CrossRef]
- Söderberg, L.; Johannesson, M.; Nygren, P.; Laudon, H.; Eriksson, F.; Osswald, G.; Möller, C.; Lannfelt, L. Lecanemab, aducanumab, and gantenerumab—Binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurotherapeutics 2023, 20, 195–206. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirer, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Roche Press Release. Roche Provides Update on Phase III GRADUATE Programme Evaluating Gantenerumab in Alzheimer’s Disease. 14 November 2022. Available online: https://www.globenewswire.com/news-release/2022/11/14/2554515/0/en/Ad-hoc-announcement-pursuant-to-Art-53-LR-Roche-provides-update-on-Phase-III-GRADUATE-programme-evaluating-gantenerumab-in-early-Alzheimer-s-disease.html (accessed on 14 September 2023).
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Saddiki, H.; Fayosse, A.; Cognat, E.; Sabia, S.; Engelborghs, S.; Wallon, D.; Alexopoulos, P.; Blennow, K.; Zetterberg, H.; Parnetti, L.; et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: A cerebrospinal fluid biomarker-based case-control study. PLoS Med. 2020, 17, e1003289. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. 2023, 19, 13–14. [Google Scholar]
- Ward, A.; Crean, S.; Mercaldi, C.J.; Collins, J.M.; Boyd, D.; Cook, M.N.; Arrighi, H.M. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: A systematic review and meta-analysis. Neuroepidemiology 2012, 38, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Schmechel, D.E.; Saunders, A.M.; Strittmatter, W.J.; Crain, B.J.; Hulette, C.M.; Joo, S.H.; Pericak-Vance, M.A.; Goldgaber, D.; Roses, A.D. Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 9649–9653. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, K.; Wilcock, G.K.; Love, S. APOE ε4 influences the pathological phenotype of Alzheimer’s disease by favouring cerebrovascular over parenchymal accumulation of Aβ protein. Neuropathol. Appl. Neurobiol. 2003, 29, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Jack, C.R., Jr.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef]
- Withington, C.G.; Turner, R.S. Amyloid-related imaging abnormalities with anti-amyloid antibodies for the treatment of dementia due to Alzheimer’s disease. Front. Neurol. 2022, 13, 862369. [Google Scholar] [CrossRef]
- Leqembi (lecanemab-irmb) Injection for Intravenous Use [Prescribing Information]; Eisai Inc.: Nutley, NJ, USA, 2023; Available online: https://www.leqembi.com/ (accessed on 14 September 2023).
- Abushakra, S.; Mandelbaum, R.; Barakos, J.; Scheltens, P.; Porsteinsson, A.P.; Watson, D.; MacSweeney, E.; Sabbagh, M.; Liang, E.; Kesslak, P.; et al. Prevalence of amyloid-related imaging abnormalities in APOE4/4 homozygotes with early Alzheimer’s disease (AD): Baseline findings from ongoing clinical trials of the oral anti-amyloid agent ALZ-801 (valiltramiprosate) (P5-6.003). In Proceedings of the American Academy of Neurology Conference, Boston, MA, USA, 22–27 April 2023. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Dutysheva, E.A.; Kanunikov, I.E.; Guzhova, I.V.; Margulis, B.A. Protein interactome of amyloid-β as a therapeutic target. Pharmaceuticals 2023, 16, 312. [Google Scholar] [CrossRef]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. J. Mol. Biol. 2019, 431, 2248–2265. [Google Scholar] [CrossRef] [PubMed]
- Parhizkar, S.; Holtzman, D.M. APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol. 2022, 59, 101594. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xiong, M.; Gratuze, M.; Bao, X.; Shi, Y.; Andhey, P.S.; Manis, M.; Schroeder, C.; Yin, Z.; Madore, C.; et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 2021, 109, 1657–1674.e7. [Google Scholar] [CrossRef]
- Gratuze, M.; Schlachetzki, J.C.M.; D’Oliveira Albanus, R.; Jain, N.; Novotny, B.; Brase, L.; Rodriguez, L.; Mansel, C.; Kipnis, M.; O’Brien, S.; et al. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron 2023, 111, 202–219.e7. [Google Scholar] [CrossRef]
- Jolly-Amado, A.; Kulkarni, N.; Nash, K.R. Reelin signaling in neurodevelopmental disorders and neurodegenerative diseases. Brain Sci. 2023, 13, 1479. [Google Scholar] [CrossRef]
- Lane-Donovan, C.; Philips, G.T.; Wasser, C.R.; Durakoglugil, M.S.; Masiulis, I.; Upadhaya, A.; Pohlkamp, T.; Coskun, C.; Kotti, T.; Steller, L.; et al. Reelin protects against amyloid β toxicity. Sci. Signal. 2015, 8, ra67. [Google Scholar] [CrossRef]
- Bufill, E.; Roura-Poch, P.; Sala-Matavera, I.; Antón, S.; Lleó, A.; Sánchez-Saudinós, B.; Tomàs-Abadal, L.; Puig, T.; Abós, J.; Bernades, S.; et al. Reelin signaling pathway genotypes and Alzheimer disease in a Spanish population. Alzheimer Dis. Assoc. Disord. 2015, 29, 169–172. [Google Scholar] [CrossRef]
- Bracher-Smith, M.; Leonenko, G.; Baker, E.; Crawford, K.; Graham, A.C.; Salih, D.A.; Howell, B.W.; Hardy, J.; Escott-Price, V. Whole genome analysis in APOE4 homozygotes identifies the DAB1-RELN pathway in Alzheimer’s disease pathogenesis. Neurobiol. Aging 2022, 119, 67–76. [Google Scholar] [CrossRef]
- Lopera, F.; Marino, C.; Chandrahas, A.S.; O’Hare, M.; Villalba-Moreno, N.D.; Aguillon, D.; Baena, A.; Sanchez, J.S.; Vila-Castelar, C.; Ramirez Gomez, L.; et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat. Med. 2023, 29, 1243–1252. [Google Scholar] [CrossRef]
- Degenhardt, E.K.; Witte, M.M.; Case, M.G.; Yu, P.; Henley, D.B.; Hochstetler, H.M.; D’Souza, D.N.; Trzepacz, P.T. Florbetapir F18 PET amyloid neuroimaging and characteristics in patients with mild and moderate Alzheimer dementia. Psychosomatics 2016, 57, 208–216. [Google Scholar] [CrossRef]
- Karikari, T.K.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018, 14, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Mattsson-Calgren, N.; Andersson, E.; Janelidze, S.; Ossenkoppele, R.; Insel, P.; Strandberg, O.; Zetterberg, H.; Rosen, H.J.; Rabinovici, G.; Chai, X.; et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive tau PET in Alzheimer’s disease. Sci. Adv. 2020, 6, eaaz2387. [Google Scholar] [CrossRef] [PubMed]
- Milà-Alomà, M.; Ashton, N.J.; Shekari, M.; Salvadó, G.; Ortiz-Romero, P.; Montoliu-Gaya, L.; Benedet, A.L.; Karikari, T.K.; Lantero-Rodriguez, J.; Vanmechelen, E.; et al. Plasma p-tau231 and p-tau217 as state markers of amyloid-β pathology in preclinical Alzheimer’s disease. Nat. Med. 2022, 28, 1797–1801. [Google Scholar] [CrossRef] [PubMed]
- Verberk, I.M.W.; Thijssen, E.; Koelewijn, J.; Mauroo, K.; Vanbrabant, J.; de Wilde, A.; Zwan, M.D.; Verfaillie, S.C.J.; Ossenkoppele, R.; Barkhof, F.; et al. Combination of plasma amyloid beta (1–42/1–40) and glial fibrillary acidic protein strongly associates with cerebral amyloid pathology. Alzheimers Res. Ther. 2020, 12, 118. [Google Scholar] [CrossRef] [PubMed]
- Salvadó, G.; Ossenkoppele, R.; Ashton, N.J.; Beach, T.G.; Serrano, G.E.; Reiman, E.M.; Zetterberg, H.; Mattsson-Carlgren, N.; Janelidze, S.; Blennow, K.; et al. Specific associations between plasma biomarkers and postmortem amyloid plaque and tau tangle loads. EMBO Mol. Med. 2023, 15, e17123. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvo, M.J.; Lu, M.; Burnham, S.C.; Schade, A.E.; Dage, J.L.; Shcherbinin, S.; Collins, E.C.; Sims, J.R.; Mintun, M.A. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: A secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 2022, 79, 1250–1259. [Google Scholar] [CrossRef]
- Bittner, T.; Blennow, K.; Scelsi, M.; Palermo, G.; Kollmorgen, G.; Smith, J.; Zetterberg, H.; Doody, R.S. GRADUATE I and II Results. Effect of Subcutaneous Gantenerumab on Fluid Biomarkers of AD Pathology and Neurodegeneration with Insights on Plasma Biomarkers and New CSF Biomarkers. Available online: https://medically.gene.com/global/en/unrestricted/neuroscience/adpd-2023/adpd-2023-presentation-bittner-graduate-i-and-ii-result.html (accessed on 14 September 2023).
- Bittner, T.; Scelsi, M.; Kollmorgen, G.; Jethwa, A.; Kerchner, G.A.; Fontoura, P.; Baudler, M.; Doody, R.S. Gantenerumab treatment increases plasma beta-amyloid(1–42) and decreases plasma pTau. Alzheimers Dement. 2022, 18, e065684. Available online: https://www.researchgate.net/publication/366456921_Gantenerumab_treatment_increases_plasma_beta-amyloid1-42_and_decreases_plasma_pTau (accessed on 14 September 2023). [CrossRef]
- Biogen, Inc.; Eisai, Inc. Press Release. FDA Grants Traditional Approval for LEQEMBI® (lecanemab-irmb) for the Treatment of Alzheimer’s Disease. 6 July 2023. Available online: https://media-us.eisai.com/2023-07-06-FDA-Grants-Traditional-Approval-for-LEQEMBI-R-lecanemab-irmb-for-the-Treatment-of-Alzheimers-Disease (accessed on 14 September 2023).
- U.S. Food and Drug Administration News Release. FDA Converts Novel Alzheimer’s Disease Treatment to Traditional Approval. Action Follows Confirmatory Trial to Verify Clinical Benefit. 6 July 2023. Available online: https://www.fda.gov/news-events/press-announcements/fda-converts-novel-alzheimers-disease-treatment-traditional-approval (accessed on 14 September 2023).
- Solopova, E.; Romero-Frenandez, W.; Harmsen, H.; Ventura-Antunes, L.; Wang, E.; Shostak, A.; Maldonado, J.; Donahue, M.; Schultz, D.; Coyne, T.M.; et al. Fatal Iatrogenic Cerebral Amyloid-Related Encephalitis in a Patient Treated with Lecanemab for Alzheimer’s Disease: Neuroimaging and Neuropathology. 2023. Available online: https://www.medrxiv.org/content/10.1101/2023.04.26.23289061v1 (accessed on 14 September 2023). [CrossRef]
- Reish, N.J.; Jamshidi, P.; Stamm, B.; Flanagan, M.E.; Sugg, E.; Tang, M.; Donohue, K.L.; McCord, M.; Krumpelman, C.; Mesulam, M.M.; et al. Multiple cerebral hemorrhages in a patient receiving lecanemab and treated with t-PA for stroke. N. Eng. J. Med. 2023, 388, 478–479. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; Ehrlich, M.E. Moving the needle on Alzheimer’s disease with an anti-oligomer antibody. N. Eng. J. Med. 2023, 388, 80–81. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarker | Amyloid (A) | Tau (T) | Neuronal Injury (N) |
|---|---|---|---|
| Imaging Biomarker | Amyloid PET | Tau PET | Volumetric MRI (hippocampal volume or cortical thickness) |
| CSF or Plasma Biomarker | Aβ42/Aβ40 ratio | P-tau181 P-tau217 | Total tau or NfL (axonal injury) Neurogranin (synaptic injury) |
| Drug | Clinical Trials | Plasma P-tau181 % Reduction Annualized | CDR-SB % Clinical Benefit |
|---|---|---|---|
| Gantenerumab 1200 mg subcutaneously every 4 weeks | Phase 3 | 2–4% | 6–8% |
| Aducanumab 10 mg/kg IV infusion monthly | Phase 3 | 9–11% | 22% |
| Lecanemab 10 mg/kg IV infusion twice monthly | Phase 2, Phase 3 | 18–20% | 26–27% |
| ALZ-801/valiltramiprosate 265 mg oral tablet twice daily | Phase 2 | 41% | Phase 3 ongoing |
| Oligomer Selectivity and Pharmaceutical Profile | Biogen Aducanumab IV Infusion 10 mg/kg Monthly | Eli Lilly Donanemab IV Infusion 1400 mg Monthly | Eisai Lecanemab IV Infusion 10 mg/kg Twice Monthly | Alzheon ALZ-801/Tramiprosate Oral Tablet 265 mg Twice Daily |
|---|---|---|---|---|
| Relative Selectivity for Soluble Amyloid Oligomers | + | + | ++ | +++ Fully blocks formation of neurotoxic oligomers |
| Brain Penetration Ability to Cross Blood–Brain Barrier | Low 1.5% | Low 0.1% | Low 0.3% | High 40% |
| Study Population | Early AD All genotypes | Early AD All genotypes | Early AD All genotypes | Mild AD APOE4/4 subgroup |
| Cognitive Benefit ADAS-cog (% vs. placebo) | 27% | 19% | 26% | 125% |
| Functional Benefit CDR-SB (% vs. placebo) | 22% | 29% | 27% | 81% |
| ARIA-E Brain Edema (% in active arm) | 35% 64% in APOE4/4 | 24% 41% in APOE4/4 | 13% 33% in APOE4/4 | 0% |
| Outcome Measure | Aducanumab ENGAGE * Phase 3 N~550/arm | Aducanumab EMERGE Phase 3 N~550/arm | Donanemab Phase 3 † N~850/arm | Lecanemab Phase 3 ‡ N~900/arm |
|---|---|---|---|---|
| CDR-SB | −2% (27%) p = NS | 22% p = 0.012 | 29% p < 0.0001 | 27% p < 0.0001 |
| ADAS-cog | 11% p = NS | 27% p = 0.010 | 19% p < 0.001 | 26% p < 0.001 |
| ADCS—iADL | 18% p = NS | 40% p < 0.001 | 28% p < 0.001 | 37% p < 0.0001 |
| ARIA | Aducanumab ENGAGE Phase 3 N~550/arm | Aducanumab EMERGE Phase 3 N~550/arm | Donanemab Phase 3 N~850/arm | Lecanemab Phase 3 N~900/arm |
|---|---|---|---|---|
| ARIA-E Incidence | 36% 64% in homozygotes * | 35% 64% in homozygotes * | 24% 41% in homozygotes | 15% 33% in homozygotes |
| ARIA-E Symptomatic | 6% | 6% | 6% | 3% |
| ARIA-H Incidence | 19% | 20% | 31% | 17% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolar, M.; Hey, J.A.; Power, A.; Abushakra, S. The Single Toxin Origin of Alzheimer’s Disease and Other Neurodegenerative Disorders Enables Targeted Approach to Treatment and Prevention. Int. J. Mol. Sci. 2024, 25, 2727. https://doi.org/10.3390/ijms25052727
Tolar M, Hey JA, Power A, Abushakra S. The Single Toxin Origin of Alzheimer’s Disease and Other Neurodegenerative Disorders Enables Targeted Approach to Treatment and Prevention. International Journal of Molecular Sciences. 2024; 25(5):2727. https://doi.org/10.3390/ijms25052727
Chicago/Turabian StyleTolar, Martin, John A. Hey, Aidan Power, and Susan Abushakra. 2024. "The Single Toxin Origin of Alzheimer’s Disease and Other Neurodegenerative Disorders Enables Targeted Approach to Treatment and Prevention" International Journal of Molecular Sciences 25, no. 5: 2727. https://doi.org/10.3390/ijms25052727
APA StyleTolar, M., Hey, J. A., Power, A., & Abushakra, S. (2024). The Single Toxin Origin of Alzheimer’s Disease and Other Neurodegenerative Disorders Enables Targeted Approach to Treatment and Prevention. International Journal of Molecular Sciences, 25(5), 2727. https://doi.org/10.3390/ijms25052727