
Independent and Combined Effects of Prenatal Alcohol Exposure and Prenatal Stress on Fetal HPA Axis Development

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Sample Characteristics
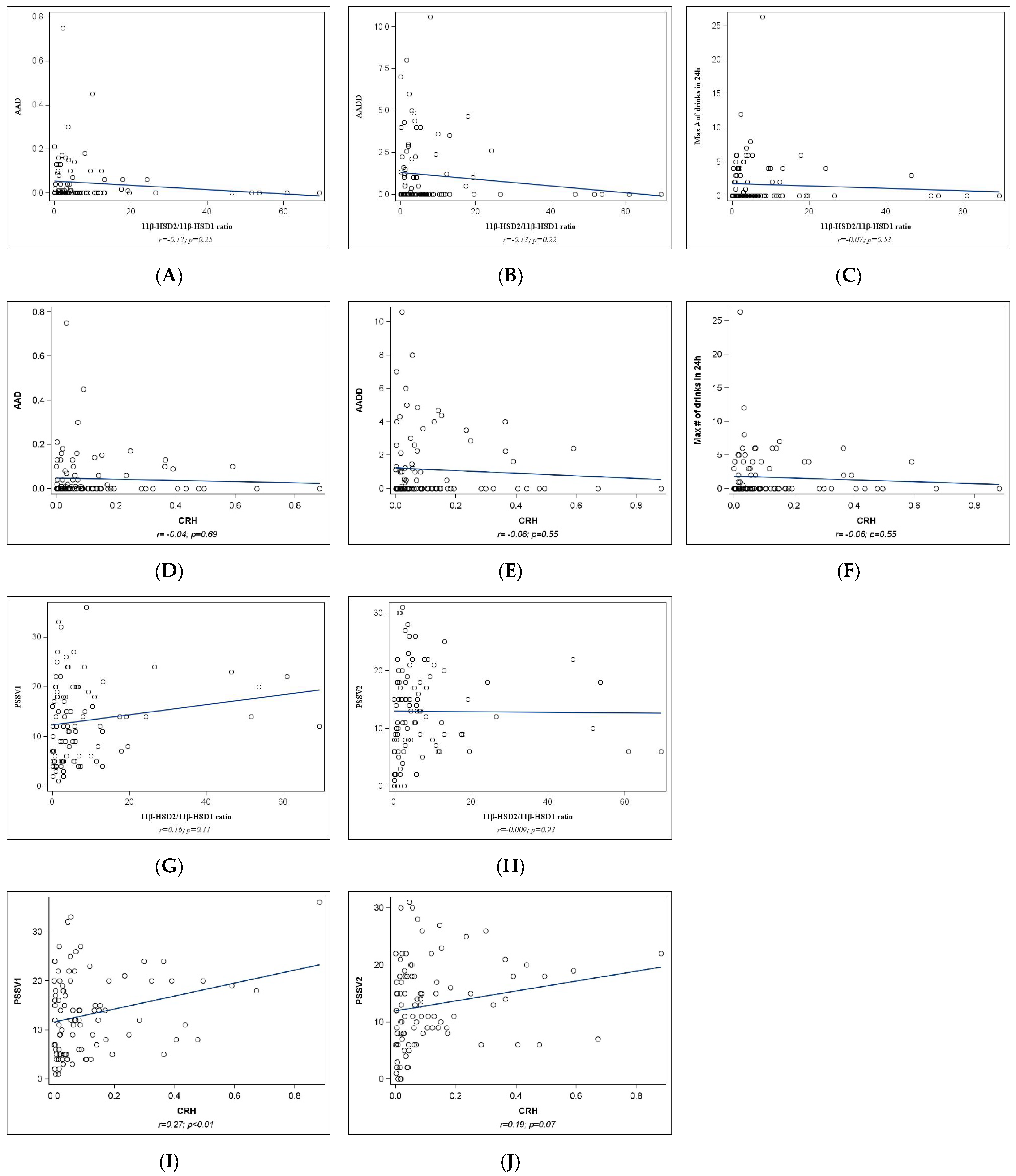
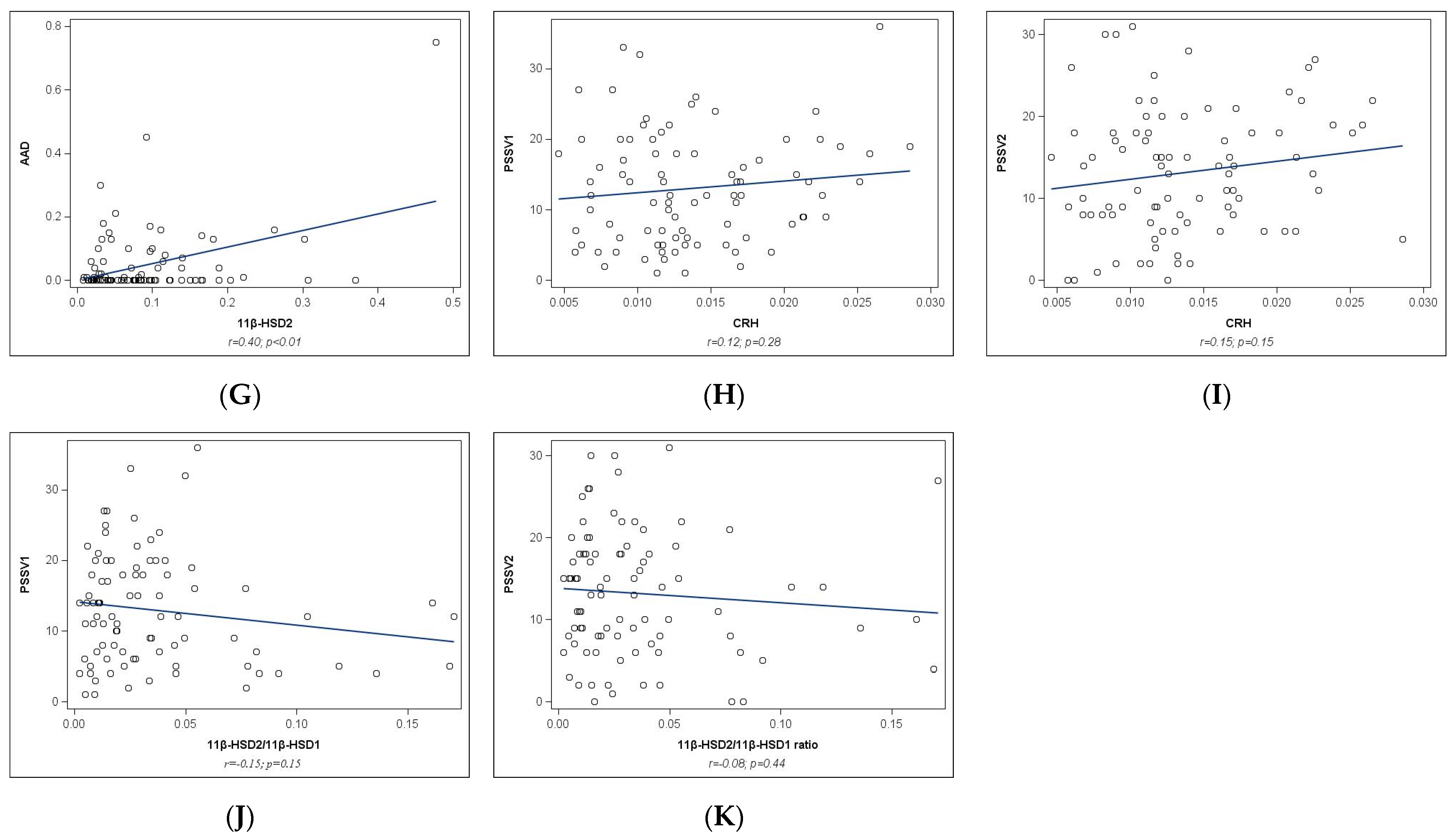
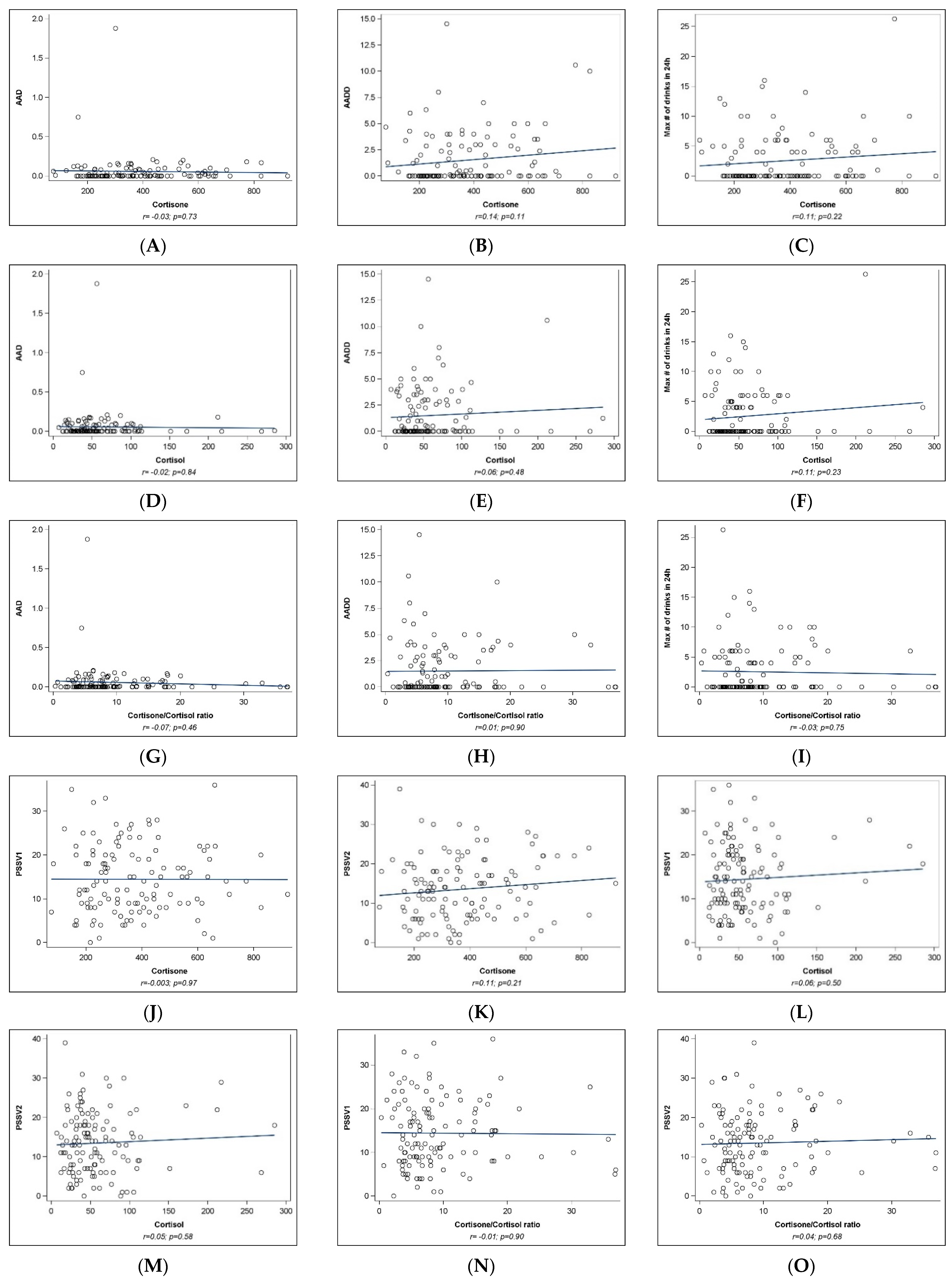
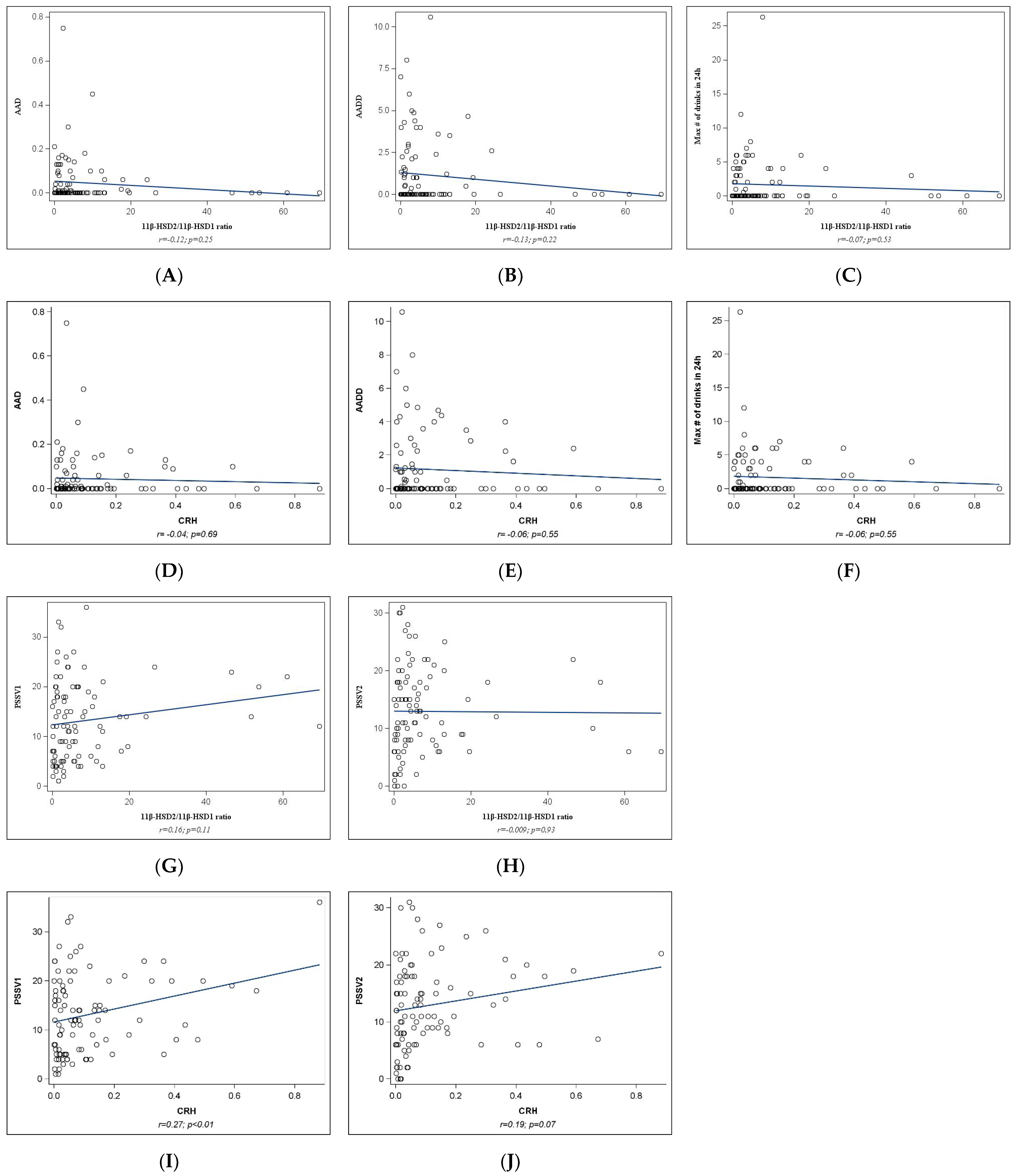
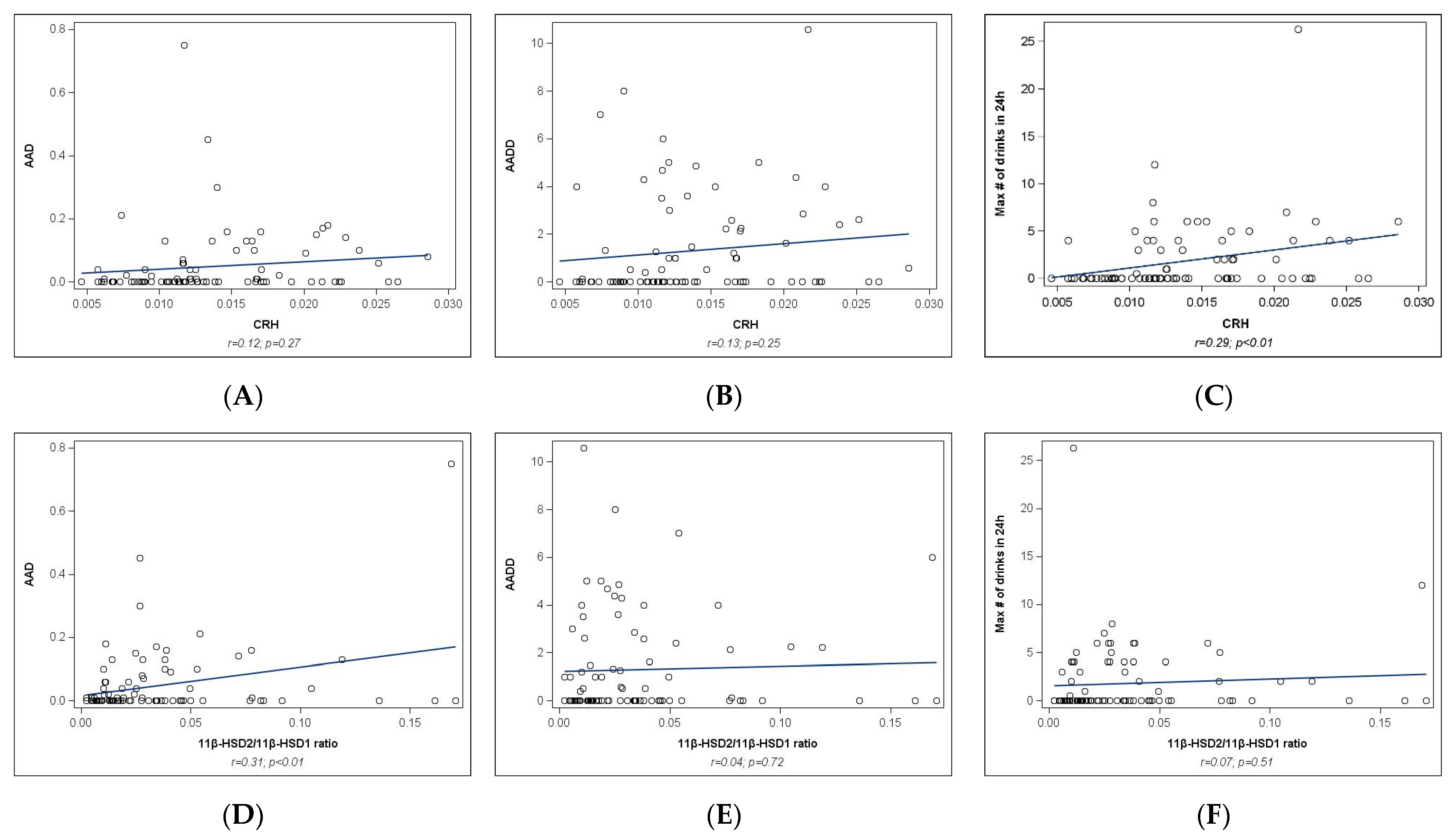
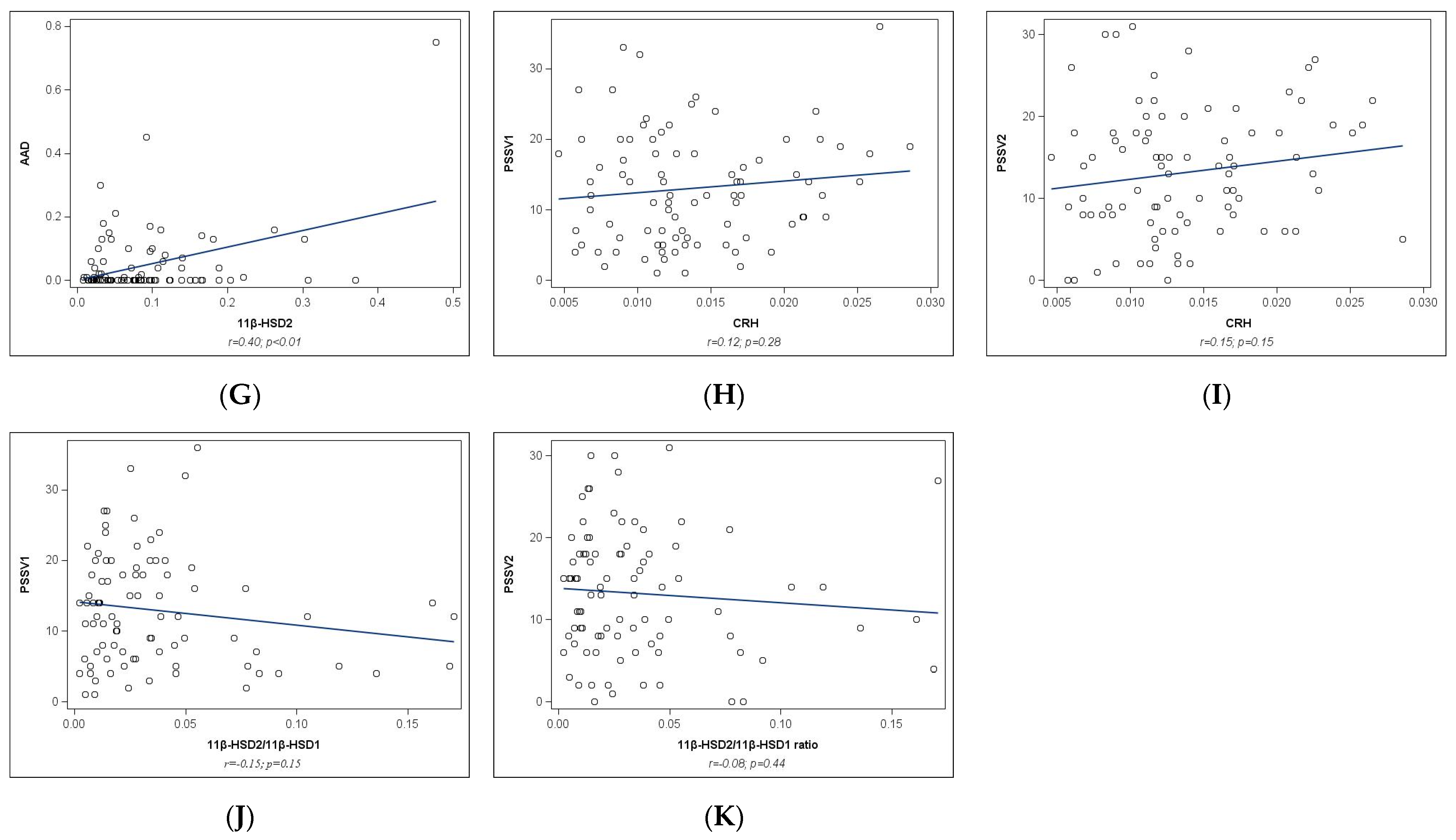
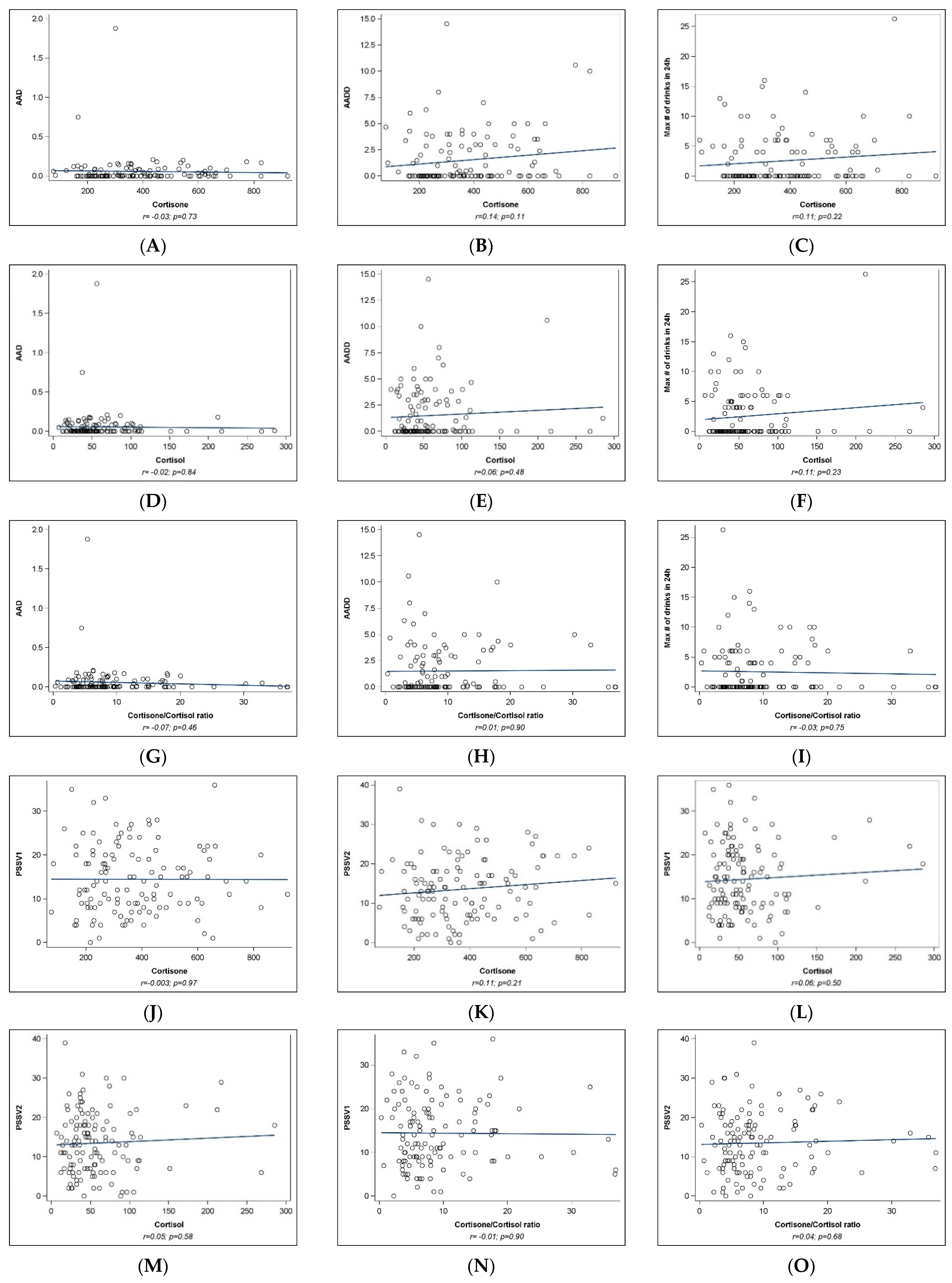
2.2. Univariate Analyses
2.3. Multivariable Analyses
2.3.1. pCRH Expression
2.3.2. Placental 11β-HSD2 Expression
3. Discussion
4. Methods and Materials
4.1. Study Design and Population
4.2. Assessment of PAE and PS
4.3. Socio-Demographic Characteristics, Prenatal Environment, and Other Substances
4.4. Placental Tissue and Cord Blood Collection
4.5. mRNA and Protein Initial Homogenization
4.6. Cytosolic and Nuclear Protein Fractionation
4.7. Gene Expression qPCR Analysis
4.8. Placenta Protein Analysis and Umbilical Cord Hormones
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ethen, M.K.; Ramadhani, T.A.; Scheuerle, A.E.; Canfield, M.A.; Wyszynski, D.F.; Druschel, C.M.; Romitti, P.A. National Birth Defects Prevention Study Alcohol Consumption by Women before and during Pregnancy. Matern. Child Health J. 2009, 13, 274–285. [Google Scholar] [CrossRef]
- Tan, C.H.; Denny, C.H.; Cheal, N.E.; Sniezek, J.E.; Kanny, D. Alcohol Use and Binge Drinking among Women of Childbearing Age—United States, 2011–2013. MMWR Morb. Mortal Wkly. Rep. 2015, 64, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Chambers, C.D.; Kalberg, W.O.; Zellner, J.; Feldman, H.; Buckley, D.; Kopald, D.; Hasken, J.M.; Xu, R.; Honerkamp-Smith, G.; et al. Prevalence of Fetal Alcohol Spectrum Disorders in 4 US Communities. JAMA 2018, 319, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Hoyme, H.E.; Kalberg, W.O.; Elliott, A.J.; Blankenship, J.; Buckley, D.; Marais, A.-S.; Manning, M.A.; Robinson, L.K.; Adam, M.P.; Abdul-Rahman, O.; et al. Updated Clinical Guidelines for Diagnosing Fetal Alcohol Spectrum Disorders. Pediatrics 2016, 138, e20154256. [Google Scholar] [CrossRef]
- Madigan, S.; Oatley, H.; Racine, N.; Fearon, R.M.P.; Schumacher, L.; Akbari, E.; Cooke, J.E.; Tarabulsy, G.M. A Meta-Analysis of Maternal Prenatal Depression and Anxiety on Child Socioemotional Development. J. Am. Acad. Child Adolesc. Psychiatry 2018, 57, 645–657.e8. [Google Scholar] [CrossRef]
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Räikkönen, K.; King, S.; et al. Prenatal Developmental Origins of Behavior and Mental Health: The Influence of Maternal Stress in Pregnancy. Neurosci. Biobehav. Rev. 2020, 117, 26–64. [Google Scholar] [CrossRef]
- Tarabulsy, G.M.; Pearson, J.; Vaillancourt-Morel, M.-P.; Bussières, E.-L.; Madigan, S.; Lemelin, J.-P.; Duchesneau, A.-A.; Hatier, D.-E.; Royer, F. Meta-Analytic Findings of the Relation between Maternal Prenatal Stress and Anxiety and Child Cognitive Outcome. J. Dev. Behav. Pediatr. 2014, 35, 38–43. [Google Scholar] [CrossRef]
- Manzari, N.; Matvienko-Sikar, K.; Baldoni, F.; O’Keeffe, G.W.; Khashan, A.S. Prenatal Maternal Stress and Risk of Neurodevelopmental Disorders in the Offspring: A Systematic Review and Meta-Analysis. Soc. Psychiatry Psychiatr. Epidemiol. 2019, 54, 1299–1309. [Google Scholar] [CrossRef]
- Kingston, D.; McDonald, S.; Austin, M.-P.; Tough, S. Association between Prenatal and Postnatal Psychological Distress and Toddler Cognitive Development: A Systematic Review. PLoS ONE 2015, 10, e0126929. [Google Scholar] [CrossRef]
- Gray, S.A.O.; Jones, C.W.; Theall, K.P.; Glackin, E.; Drury, S.S. Thinking Across Generations: Unique Contributions of Maternal Early Life and Prenatal Stress to Infant Physiology. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Dipietro, J.A. Maternal Stress in Pregnancy: Considerations for Fetal Development. J. Adolesc. Health 2012, 51, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Selye, H. A Syndrome Produced by Diverse Nocuous Agents. Nature 1936, 138, 32. [Google Scholar] [CrossRef]
- Pacák, K.; Palkovits, M. Stressor Specificity of Central Neuroendocrine Responses: Implications for Stress-Related Disorders. Endocr. Rev. 2001, 22, 502–548. [Google Scholar] [CrossRef] [PubMed]
- Nast, I.; Bolten, M.; Meinlschmidt, G.; Hellhammer, D.H. How to Measure Prenatal Stress? A Systematic Review of Psychometric Instruments to Assess Psychosocial Stress during Pregnancy. Paediatr. Perinat. Epidemiol. 2013, 27, 313–322. [Google Scholar] [CrossRef]
- Bertilsson, J.; Niehorster, D.C.; Fredriksson, P.J.; Dahl, M.; Granér, S.; Fredriksson, O.; Mårtensson, J.M.; Magnusson, M.; Fransson, P.-A.; Nyström, M. Stress Levels Escalate When Repeatedly Performing Tasks Involving Threats. Front. Psychol. 2019, 10, 1562. [Google Scholar] [CrossRef] [PubMed]
- Ulrich-Lai, Y.M.; Herman, J.P. Neural Regulation of Endocrine and Autonomic Stress Responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef]
- Tsigos, C.; Kyrou, I.; Kassi, E.; Chrousos, G.P. Stress: Endocrine Physiology and Pathophysiology. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Lan, N.; Chiu, M.P.Y.; Ellis, L.; Weinberg, J. Prenatal Alcohol Exposure and Prenatal Stress Differentially Alter Glucocorticoid Signaling in the Placenta and Fetal Brain. Neuroscience 2017, 342, 167–179. [Google Scholar] [CrossRef]
- Breese, G.R.; Sinha, R.; Heilig, M. Chronic Alcohol Neuroadaptation and Stress Contribute to Susceptibility for Alcohol Craving and Relapse. Pharmacol. Ther. 2011, 129, 149–171. [Google Scholar] [CrossRef]
- Wang, J.X.; Roeske, W.R.; Wang, W.; Yamamura, H.I. Himbacine Recognizes a High Affinity Subtype of M2 Muscarinic Cholinergic Receptors in the Rat Cerebral Cortex. Brain Res. 1988, 446, 155–158. [Google Scholar] [CrossRef]
- Keiver, K.; Bertram, C.P.; Orr, A.P.; Clarren, S. Salivary Cortisol Levels Are Elevated in the Afternoon and at Bedtime in Children with Prenatal Alcohol Exposure. Alcohol 2015, 49, 79–87. [Google Scholar] [CrossRef]
- Comeau, W.L.; Lee, K.; Anderson, K.; Weinberg, J. Prenatal Alcohol Exposure and Adolescent Stress Increase Sensitivity to Stress and Gonadal Hormone Influences on Cognition in Adult Female Rats. Physiol. Behav. 2015, 148, 157–165. [Google Scholar] [CrossRef]
- Spanagel, R.; Noori, H.R.; Heilig, M. Stress and Alcohol Interactions: Animal Studies and Clinical Significance. Trends Neurosci. 2014, 37, 219–227. [Google Scholar] [CrossRef]
- Räikkönen, K.; Pesonen, A.-K.; O’Reilly, J.R.; Tuovinen, S.; Lahti, M.; Kajantie, E.; Villa, P.; Laivuori, H.; Hämäläinen, E.; Seckl, J.R.; et al. Maternal Depressive Symptoms during Pregnancy, Placental Expression of Genes Regulating Glucocorticoid and Serotonin Function and Infant Regulatory Behaviors. Psychol. Med. 2015, 45, 3217–3226. [Google Scholar] [CrossRef]
- Bellisario, V.; Panetta, P.; Balsevich, G.; Baumann, V.; Noble, J.; Raggi, C.; Nathan, O.; Berry, A.; Seckl, J.; Schmidt, M.; et al. Maternal High-Fat Diet Acts as a Stressor Increasing Maternal Glucocorticoids’ Signaling to the Fetus and Disrupting Maternal Behavior and Brain Activation in C57BL/6J Mice. Psychoneuroendocrinology 2015, 60, 138–150. [Google Scholar] [CrossRef]
- Hirst, J.J.; Cumberland, A.L.; Shaw, J.C.; Bennett, G.A.; Kelleher, M.A.; Walker, D.W.; Palliser, H.K. Loss of Neurosteroid-Mediated Protection Following Stress during Fetal Life. J. Steroid Biochem. Mol. Biol. 2016, 160, 181–188. [Google Scholar] [CrossRef]
- Quinn, K.E.; Reynolds, L.P.; Grazul-Bilska, A.T.; Borowicz, P.P.; Ashley, R.L. Placental Development during Early Pregnancy: Effects of Embryo Origin on Expression of Chemokine Ligand Twelve (CXCL12). Placenta 2016, 43, 77–80. [Google Scholar] [CrossRef]
- Steinbrekera, B.; Roghair, R. Modeling the Impact of Growth and Leptin Deficits on the Neuronal Regulation of Blood Pressure. J. Endocrinol. 2016, 231, R47–R60. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.E.; Labrecque, M.T.; Solomon, B.R.; Ali, A.; Allan, A.M. Prenatal Arsenic Exposure Alters the Programming of the Glucocorticoid Signaling System during Embryonic Development. Neurotoxicol. Teratol. 2015, 47, 66–79. [Google Scholar] [CrossRef]
- Wyrwoll, C.; Keith, M.; Noble, J.; Stevenson, P.L.; Bombail, V.; Crombie, S.; Evans, L.C.; Bailey, M.A.; Wood, E.; Seckl, J.R.; et al. Fetal Brain 11β-Hydroxysteroid Dehydrogenase Type 2 Selectively Determines Programming of Adult Depressive-like Behaviors and Cognitive Function, but Not Anxiety Behaviors in Male Mice. Psychoneuroendocrinology 2015, 59, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A.S. Stress-Induced Perinatal and Transgenerational Epigenetic Programming of Brain Development and Mental Health. Neurosci. Biobehav. Rev. 2015, 48, 70–91. [Google Scholar] [CrossRef] [PubMed]
- Welberg, L.A.; Seckl, J.R.; Holmes, M.C. Inhibition of 11beta-Hydroxysteroid Dehydrogenase, the Foeto-Placental Barrier to Maternal Glucocorticoids, Permanently Programs Amygdala GR mRNA Expression and Anxiety-like Behaviour in the Offspring. Eur. J. Neurosci. 2000, 12, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.J.; Kotelevtsev, Y.; Mullins, J.J.; Seckl, J.R.; Holmes, M.C. Intracellular Regeneration of Glucocorticoids by 11beta-Hydroxysteroid Dehydrogenase (11beta-HSD)-1 Plays a Key Role in Regulation of the Hypothalamic-Pituitary-Adrenal Axis: Analysis of 11beta-HSD-1-Deficient Mice. Endocrinology 2001, 142, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Clifton, V.L.; Cuffe, J.; Moritz, K.M.; Cole, T.J.; Fuller, P.J.; Lu, N.Z.; Kumar, S.; Chong, S.; Saif, Z. Review: The Role of Multiple Placental Glucocorticoid Receptor Isoforms in Adapting to the Maternal Environment and Regulating Fetal Growth. Placenta 2017, 54, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Saif, Z.; Hodyl, N.A.; Stark, M.J.; Fuller, P.J.; Cole, T.; Lu, N.; Clifton, V.L. Expression of Eight Glucocorticoid Receptor Isoforms in the Human Preterm Placenta Vary with Fetal Sex and Birthweight. Placenta 2015, 36, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Bronson, S.L.; Bale, T.L. The Placenta as a Mediator of Stress Effects on Neurodevelopmental Reprogramming. Neuropsychopharmacology 2016, 41, 207–218. [Google Scholar] [CrossRef]
- McGowan, P.O.; Matthews, S.G. Prenatal Stress, Glucocorticoids, and Developmental Programming of the Stress Response. Endocrinology 2018, 159, 69–82. [Google Scholar] [CrossRef]
- Jensen Peña, C.; Monk, C.; Champagne, F.A. Epigenetic Effects of Prenatal Stress on 11β-Hydroxysteroid Dehydrogenase-2 in the Placenta and Fetal Brain. PLoS ONE 2012, 7, e39791. [Google Scholar] [CrossRef]
- Haley, D.W.; Handmaker, N.S.; Lowe, J. Infant Stress Reactivity and Prenatal Alcohol Exposure. Alcohol Clin. Exp. Res. 2006, 30, 2055–2064. [Google Scholar] [CrossRef]
- Allan, A.M.; Goggin, S.L.; Caldwell, K.K. Prenatal Alcohol Exposure Modifies Glucocorticoid Receptor Subcellular Distribution in the Medial Prefrontal Cortex and Impairs Frontal Cortex-Dependent Learning. PLoS ONE 2014, 9, e96200. [Google Scholar] [CrossRef]
- Harris, A.; Seckl, J. Glucocorticoids, Prenatal Stress and the Programming of Disease. Horm. Behav. 2011, 59, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.J.; Wolff, C.R.; El-Emawy, A.; Staples, M.C.; Perrone-Bizzozero, N.I.; Savage, D.D. Effects of Moderate Drinking during Pregnancy on Placental Gene Expression. Alcohol 2010, 44, 673–690. [Google Scholar] [CrossRef]
- Gitau, R.; Cameron, A.; Fisk, N.M.; Glover, V. Fetal Exposure to Maternal Cortisol. Lancet 1998, 352, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Saleh, A.I.; Al Olabi, R.; Al Shehabi, T.S.; Eid, A.H. Glucocorticoid-Induced Fetal Origins of Adult Hypertension: Association with Epigenetic Events. Vasc. Pharmacol. 2016, 82, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Murphy, V.E.; Clifton, V.L. Alterations in Human Placental 11beta-Hydroxysteroid Dehydrogenase Type 1 and 2 with Gestational Age and Labour. Placenta 2003, 24, 739–744. [Google Scholar] [CrossRef]
- Chapman, K.; Holmes, M.; Seckl, J. 11β-Hydroxysteroid Dehydrogenases: Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed]
- Heussner, K.; Ruebner, M.; Huebner, H.; Rascher, W.; Menendez-Castro, C.; Hartner, A.; Fahlbusch, F.B.; Rauh, M. Species Differences of 11beta-Hydroxysteroid Dehydrogenase Type 2 Function in Human and Rat Term Placenta Determined via LC-MS/MS. Placenta 2016, 37, 79–84. [Google Scholar] [CrossRef]
- Challis, J.R.; Sloboda, D.; Matthews, S.G.; Holloway, A.; Alfaidy, N.; Patel, F.A.; Whittle, W.; Fraser, M.; Moss, T.J.; Newnham, J. The Fetal Placental Hypothalamic-Pituitary-Adrenal (HPA) Axis, Parturition and Post Natal Health. Mol. Cell Endocrinol. 2001, 185, 135–144. [Google Scholar] [CrossRef]
- Wadhwa, P.D.; Garite, T.J.; Porto, M.; Glynn, L.; Chicz-DeMet, A.; Dunkel-Schetter, C.; Sandman, C.A. Placental Corticotropin-Releasing Hormone (CRH), Spontaneous Preterm Birth, and Fetal Growth Restriction: A Prospective Investigation. Am. J. Obstet. Gynecol. 2004, 191, 1063–1069. [Google Scholar] [CrossRef]
- Wadhwa, P.D.; Sandman, C.A.; CHICZ-DeMET, A.; Porto, M. Placental CRH Modulates Maternal Pituitary-Adrenal Function in Human Pregnancy. Ann. N. Y. Acad. Sci. 1997, 814, 276–281. [Google Scholar] [CrossRef]
- Bakhireva, L.N.; Leeman, L.; Roberts, M.; Rodriguez, D.E.; Jacobson, S.W. You Didn’t Drink During Pregnancy, Did You? Alcohol Clin. Exp. Res. 2021, 45, 543–547. [Google Scholar] [CrossRef]
- Ruffaner-Hanson, C.; Noor, S.; Sun, M.S.; Solomon, E.; Marquez, L.E.; Rodriguez, D.E.; Allan, A.M.; Caldwell, K.K.; Bakhireva, L.N.; Milligan, E.D. The Maternal-Placental-Fetal Interface: Adaptations of the HPA Axis and Immune Mediators Following Maternal Stress and Prenatal Alcohol Exposure. Exp. Neurol. 2022, 355, 114121. [Google Scholar] [CrossRef]
- Michael, A.E.; Papageorghiou, A.T. Potential Significance of Physiological and Pharmacological Glucocorticoids in Early Pregnancy. Hum. Reprod. Update 2008, 14, 497–517. [Google Scholar] [CrossRef]
- Burton, P.J.; Waddell, B.J. Dual Function of 11beta-Hydroxysteroid Dehydrogenase in Placenta: Modulating Placental Glucocorticoid Passage and Local Steroid Action. Biol. Reprod. 1999, 60, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Howland, M.A.; Sandman, C.A.; Glynn, L.M. Developmental Origins of the Human Hypothalamic-Pituitary-Adrenal Axis. Expert Rev. Endocrinol. Metab. 2017, 12, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, M.; Mnif, M.F.; Charfi, N.; Kacem, F.H.; Naceur, B.B.; Mnif, F.; Dammak, M.; Rekik, N.; Abid, M. Adrenal Diseases during Pregnancy: Pathophysiology, Diagnosis and Management Strategies. Am. J. Med. Sci. 2014, 347, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Auchus, R.J.; Rainey, W.E. Adrenal Disorders in Pregnancy. Nat. Rev. Endocrinol. 2012, 8, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Gangestad, S.W.; Caldwell Hooper, A.E.; Eaton, M.A. On the Function of Placental Corticotropin-Releasing Hormone: A Role in Maternal-Fetal Conflicts over Blood Glucose Concentrations. Biol. Rev. Camb. Philos. Soc. 2012, 87, 856–873. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Xu, D.; Yu, P.; Fang, M.; Guo, J.; Xu, D.; Qiao, Y.; Chen, S.; Zhang, Y.; Wang, H. P-Gp Expression Inhibition Mediates Placental Glucocorticoid Barrier Opening and Fetal Weight Loss. BMC Med. 2021, 19, 311. [Google Scholar] [CrossRef]
- Taggi, V.; Riera Romo, M.; Piquette-Miller, M.; Meyer Zu Schwabedissen, H.E.; Neuhoff, S. Transporter Regulation in Critical Protective Barriers: Focus on Brain and Placenta. Pharmaceutics 2022, 14, 1376. [Google Scholar] [CrossRef]
- Weinberg, J.; Sliwowska, J.H.; Lan, N.; Hellemans, K.G.C. Prenatal Alcohol Exposure: Foetal Programming, the Hypothalamic-Pituitary-Adrenal Axis and Sex Differences in Outcome. J. Neuroendocrinol. 2008, 20, 470–488. [Google Scholar] [CrossRef]
- Liang, G.; Chen, M.; Pan, X.; Zheng, J.; Wang, H. Ethanol-Induced Inhibition of Fetal Hypothalamic-Pituitary-Adrenal Axis Due to Prenatal Overexposure to Maternal Glucocorticoid in Mice. Exp. Toxicol. Pathol. 2011, 63, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, D.; Luo, H.; Lu, J.; Liu, L.; Ping, J.; Wang, H. Prenatal Xenobiotic Exposure and Intrauterine Hypothalamus-Pituitary-Adrenal Axis Programming Alteration. Toxicology 2014, 325, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Przybycien-Szymanska, M.M.; Mott, N.N.; Pak, T.R. Alcohol Dysregulates Corticotropin-Releasing-Hormone (CRH) Promoter Activity by Interfering with the Negative Glucocorticoid Response Element (nGRE). PLoS ONE 2011, 6, e26647. [Google Scholar] [CrossRef] [PubMed]
- Gianoulakis, C. Alcohol-Seeking Behavior: The Roles of the Hypothalamic-Pituitary-Adrenal Axis and the Endogenous Opioid System. Alcohol Health Res. World 1998, 22, 202–210. [Google Scholar]
- Zhang, X.; Sliwowska, J.H.; Weinberg, J. Prenatal Alcohol Exposure and Fetal Programming: Effects on Neuroendocrine and Immune Function. Exp. Biol. Med. 2005, 230, 376–388. [Google Scholar] [CrossRef]
- Hellemans, K.G.C.; Verma, P.; Yoon, E.; Yu, W.; Weinberg, J. Prenatal Alcohol Exposure Increases Vulnerability to Stress and Anxiety-like Disorders in Adulthood. Ann. N. Y. Acad. Sci. 2008, 1144, 154–175. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.W.; Mletzko, T.C.; Alagbe, O.; Musselman, D.L.; Nemeroff, C.B.; Miller, A.H.; Heim, C.M. Increased Stress-Induced Inflammatory Responses in Male Patients with Major Depression and Increased Early Life Stress. Am. J. Psychiatry 2006, 163, 1630–1633. [Google Scholar] [CrossRef]
- Lian, S.; Guo, J.; Wang, L.; Li, W.; Wang, J.; Ji, H.; Kong, F.; Xu, B.; Li, S.; Yang, H. Impact of Prenatal Cold Stress on Placental Physiology, Inflammatory Response, and Apoptosis in Rats. Oncotarget 2017, 8, 115304–115314. [Google Scholar] [CrossRef]
- Coles, C.D.; Grant, T.M.; Kable, J.A.; Stoner, S.A.; Perez, A. Collaborative Initiative on Fetal Alcohol Spectrum Disorders Prenatal Alcohol Exposure and Mental Health at Midlife: A Preliminary Report on Two Longitudinal Cohorts. Alcohol Clin. Exp. Res. 2022, 46, 232–242. [Google Scholar] [CrossRef]
- Lester, B.M.; Marsit, C.J. Epigenetic Mechanisms in the Placenta Related to Infant Neurodevelopment. Epigenomics 2018, 10, 321–333. [Google Scholar] [CrossRef]
- Lowe, J.R.; DiDomenico, J.; Stephen, J.M.; Roberts, M.H.; Rodriguez, D.E.; Bakhireva, L.N. Early Developmental Trajectory of Children with Prenatal Alcohol and Opioid Exposure. Pediatr. Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; DiDomenico, J.; Roberts, M.H.; Marquez, L.E.; Rai, R.; Weinberg, J.; Jacobson, S.W.; Stephen, J.; Bakhireva, L.N. Impact of Low-Level Prenatal Alcohol Exposure and Maternal Stress on Autonomic Regulation. Pediatr. Res. 2023, 95, 350–358. [Google Scholar] [CrossRef]
- Christiaens, I.; Hegadoren, K.; Olson, D.M. Adverse Childhood Experiences Are Associated with Spontaneous Preterm Birth: A Case-Control Study. BMC Med. 2015, 13, 124. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.L.; Rosenblum, K.L.; Muzik, M.; Jester, J.; Freeman, S.; Huth-Bocks, A.; Waddell, R.; Alfafara, E.; Miller, A.; Lawler, J.; et al. Infant Mental Health Home Visiting Mitigates Impact of Maternal Adverse Childhood Experiences on Toddler Language Competence: A Randomized Controlled Trial. J. Dev. Behav. Pediatr. 2022, 43, e227–e236. [Google Scholar] [CrossRef] [PubMed]
- Appleton, A.A.; Kiley, K.; Holdsworth, E.A.; Schell, L.M. Social Support During Pregnancy Modifies the Association Between Maternal Adverse Childhood Experiences and Infant Birth Size. Matern. Child Health J. 2019, 23, 408–415. [Google Scholar] [CrossRef]
- Murphy, S.K.; Fineberg, A.M.; Maxwell, S.D.; Alloy, L.B.; Zimmermann, L.; Krigbaum, N.Y.; Cohn, B.A.; Drabick, D.A.G.; Ellman, L.M. Maternal Infection and Stress during Pregnancy and Depressive Symptoms in Adolescent Offspring. Psychiatry Res. 2017, 257, 102–110. [Google Scholar] [CrossRef]
- Catalano, P.M.; Shankar, K. Obesity and Pregnancy: Mechanisms of Short Term and Long Term Adverse Consequences for Mother and Child. BMJ 2017, 356, j1. [Google Scholar] [CrossRef]
- Walsh, K.; McCormack, C.A.; Webster, R.; Pinto, A.; Lee, S.; Feng, T.; Krakovsky, H.S.; O’Grady, S.M.; Tycko, B.; Champagne, F.A.; et al. Maternal Prenatal Stress Phenotypes Associate with Fetal Neurodevelopment and Birth Outcomes. Proc. Natl. Acad. Sci. USA 2019, 116, 23996–24005. [Google Scholar] [CrossRef]
- Traylor, C.S.; Johnson, J.D.; Kimmel, M.C.; Manuck, T.A. Effects of Psychological Stress on Adverse Pregnancy Outcomes and Nonpharmacologic Approaches for Reduction: An Expert Review. Am. J. Obstet. Gynecol. MFM 2020, 2, 100229. [Google Scholar] [CrossRef]
- Treatment and Management of Mental Health Conditions During Pregnancy and Postpartum: ACOG Clinical Practice Guideline No. 5. Obstet. Gynecol. 2023, 141, 1262–1288. [CrossRef]
- Green, C.; George, N.; Park, Y.; Denny, C.H.; Weber, M.K.; Meaney-Delman, D.; Kim, S.Y. Screening and Brief Interventions for Alcohol Use During Pregnancy: Practices Among US Primary Care Clinicians, DocStyles 2019. Prev. Chronic Dis. 2023, 20, E25. [Google Scholar] [CrossRef]
- Blevins, C.E.; Marsh, E.L.; Stein, M.D.; Schatten, H.T.; Abrantes, A.M. Project CHOICE: Choosing Healthy Options in Coping with Emotions, an EMA/EMI plus in-Person Intervention for Alcohol Use. Subst. Abuse 2021, 42, 569–576. [Google Scholar] [CrossRef]
- Velasquez, M.M.; von Sternberg, K.L.; Floyd, R.L.; Parrish, D.; Kowalchuk, A.; Stephens, N.S.; Ostermeyer, B.; Green, C.; Seale, J.P.; Mullen, P.D. Preventing Alcohol and Tobacco Exposed Pregnancies: CHOICES Plus in Primary Care. Am. J. Prev. Med. 2017, 53, 85–95. [Google Scholar] [CrossRef]
- Shu, Q.; Li, W.; Li, J.; Wang, W.; Liu, C.; Sun, K. Cross-Talk between cAMP and MAPK Pathways in HSD11B2 Induction by hCG in Placental Trophoblasts. PLoS ONE 2014, 9, e107938. [Google Scholar] [CrossRef]
- Togher, K.L.; Togher, K.L.; O’Keeffe, M.M.; O’Keeffe, M.M.; Khashan, A.S.; Khashan, A.S.; Gutierrez, H.; Gutierrez, H.; Kenny, L.C.; Kenny, L.C.; et al. Epigenetic Regulation of the Placental HSD11B2 Barrier and Its Role as a Critical Regulator of Fetal Development. Epigenetics 2014, 9, 816–822. [Google Scholar] [CrossRef]
- Yue, Y.; Xu, F.; Zhang, J.; Zhao, M.; Zhou, F. Sufentanil Alleviates Pre-Eclampsia via Silencing microRNA-24-3p to Target 11β-Hydroxysteroid Dehydrogenase Type 2. Bioengineered 2022, 13, 11456–11470. [Google Scholar] [CrossRef]
- Kable, J.A.; Mukherjee, R.A.S. Neurodevelopmental Disorder Associated with Prenatal Exposure to Alcohol (ND-PAE): A Proposed Diagnostic Method of Capturing the Neurocognitive Phenotype of FASD. Eur. J. Med. Genet. 2017, 60, 49–54. [Google Scholar] [CrossRef]
- Jacobson, S.W.; Chiodo, L.M.; Sokol, R.J.; Jacobson, J.L. Validity of Maternal Report of Prenatal Alcohol, Cocaine, and Smoking in Relation to Neurobehavioral Outcome. Pediatrics 2002, 109, 815–825. [Google Scholar] [CrossRef]
- Bakhireva, L.N.; Savage, D.D. Focus on: Biomarkers of Fetal Alcohol Exposure and Fetal Alcohol Effects. Alcohol Res. Health 2011, 34, 56–63. [Google Scholar]
- Cohen, S.; Kamarck, T.; Mermelstein, R. A Global Measure of Perceived Stress. J. Health Soc. Behav. 1983, 24, 385. [Google Scholar] [CrossRef]
- Nielsen, M.G.; Ørnbøl, E.; Vestergaard, M.; Bech, P.; Larsen, F.B.; Lasgaard, M.; Christensen, K.S. The Construct Validity of the Perceived Stress Scale. J. Psychosom. Res. 2016, 84, 22–30. [Google Scholar] [CrossRef]
- Yılmaz Koğar, E.; Koğar, H. A Systematic Review and Meta-Analytic Confirmatory Factor Analysis of the Perceived Stress Scale (PSS-10 and PSS-14). Stress Health 2023, 40, e3285. [Google Scholar] [CrossRef]
- Holbrook, B.D.; Davies, S.; Cano, S.; Shrestha, S.; Jantzie, L.L.; Rayburn, W.F.; Bakhireva, L.N.; Savage, D.D. The Association between Prenatal Alcohol Exposure and Protein Expression in Human Placenta. Birth Defects Res. 2019, 111, 749–759. [Google Scholar] [CrossRef]
- Mushahary, D.; Gautam, P.; Sundaram, C.S.; Sirdeshmukh, R. Expanded Protein Expression Profile of Human Placenta Using Two-Dimensional Gel Electrophoresis. Placenta 2013, 34, 193–196. [Google Scholar] [CrossRef]
- Mine, K.; Katayama, A.; Matsumura, T.; Nishino, T.; Kuwabara, Y.; Ishikawa, G.; Murata, T.; Sawa, R.; Otsubo, Y.; Shin, S.; et al. Proteome Analysis of Human Placentae: Pre-Eclampsia Versus Normal Pregnancy. Placenta 2007, 28, 676–687. [Google Scholar] [CrossRef]
- Pasternak, Y.; Ohana, M.; Biron-Shental, T.; Cohen-Hagai, K.; Benchetrit, S.; Zitman-Gal, T. Thioredoxin, Thioredoxin Interacting Protein and Transducer and Activator of Transcription 3 in Gestational Diabetes. Mol. Biol. Rep. 2020, 47, 1199–1206. [Google Scholar] [CrossRef]
- Baghirova, S.; Hughes, B.G.; Hendzel, M.J.; Schulz, R. Sequential Fractionation and Isolation of Subcellular Proteins from Tissue or Cultured Cells. MethodsX 2015, 2, 440–445. [Google Scholar] [CrossRef]
- Li, L.; Schust, D.J. Isolation, Purification and in Vitro Differentiation of Cytotrophoblast Cells from Human Term Placenta. Reprod. Biol. Endocrinol. 2015, 13, 71. [Google Scholar] [CrossRef]
- Balducci, E.; Emanuelli, M.; Magni, G.; Raffaelli, N.; Ruggieri, S.; Vita, A.; Natalini, P. Nuclear Matrix-Associated NMN Adenylyltransferase Activity in Human Placenta. Biochem. Biophys. Res. Commun. 1992, 189, 1275–1279. [Google Scholar] [CrossRef]
- Bustin, S. Absolute Quantification of mRNA Using Real-Time Reverse Transcription Polymerase Chain Reaction Assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef]
- Hromadnikova, I.; Kotlabova, K.; Doucha, J.; Dlouha, K.; Krofta, L. Absolute and Relative Quantification of Placenta-Specific MicroRNAs in Maternal Circulation with Placental Insufficiency–Related Complications. J. Mol. Diagn. 2012, 14, 160–167. [Google Scholar] [CrossRef]
- Mina, T.H.; Räikkönen, K.; Riley, S.C.; Norman, J.E.; Reynolds, R.M. Maternal Distress Associates with Placental Genes Regulating Fetal Glucocorticoid Exposure and IGF2: Role of Obesity and Sex. Psychoneuroendocrinology 2015, 59, 112–122. [Google Scholar] [CrossRef]
- Cleal, J.K.; Day, P.; Hanson, M.A.; Lewis, R.M. Measurement of Housekeeping Genes in Human Placenta. Placenta 2009, 30, 1002–1003. [Google Scholar] [CrossRef]
- Meller, M.; Vadachkoria, S.; Luthy, D.A.; Williams, M.A. Evaluation of Housekeeping Genes in Placental Comparative Expression Studies. Placenta 2005, 26, 601–607. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control (n = 76) | PAE (n = 48) | p | |
|---|---|---|---|
| Maternal characteristics | |||
| Mean ± SD | Mean ± SD | ||
| Maternal age at enrollment (years) | 29.7 ± 5.7 | 29.2 ± 5.9 | 0.63 1 |
| Years of education (years) | 14.7 ± 3.5 | 14.8 ± 3.5 | 0.97 1 |
| N (%) | N (%) | ||
| Marital status: | |||
| Single/separated/divorced | 15 (19.7) | 15 (31.3) | 0.14 2 |
| Married/cohabitating | 61 (80.3) | 33 (68.8) | |
| Ethnicity (Hispanic/Latinx) | 47 (61.8) | 33 (68.8) | 0.43 2 |
| Race: | |||
| White | 51 (67.1) | 37 (77.1) | 0.82 3 |
| Black or African American | 3 (4.0) | 1 (2.1) | |
| American Indian or Alaskan Native | 5 (6.6) | 3 (6.3) | |
| Multi-racial/other | 13 (17.1) | 5 (10.4) | |
| Prefer not to report | 4 (5.3) | 2 (4.2) | |
| Education: | |||
| High school or less | 28 (36.8) | 16 (33.3) | 0.48 2 |
| Some college or vocational school | 15 (19.7) | 14 (29.2) | |
| College degree or higher | 33 (43.4) | 18 (37.5) | |
| Family income *: | |||
| Under $30,000 | 25 (32.9) | 17 (35.4) | 0.57 2 |
| $30,000–49,000 | 13 (17.1) | 13 (27.1) | |
| $50,000–69,000 | 13 (17.1) | 5 (10.4) | |
| $70,000 or over | 24 (31.6) | 12 (25.0) | |
| Currently employed | 46 (60.5) | 30 (62.5) | 0.83 2 |
| Health insurance: | 0.90 2 | ||
| Employer-based insurance | 30 (39.5) | 18 (37.5) | |
| Medicaid | 27 (35.5) | 19 (39.6) | |
| Other ** | 19 (25.0) | 11 (22.9) | |
| Maternal prenatal stress: | |||
| PSS-V1 (2nd trimester) | 12.8 ± 7.8 | 17.0 ± 7.2 | <0.01 1 |
| PSS-V2 (3rd trimester) | 11.8 ± 7.8 | 16.2 ±7.2 | <0.01 1 |
| Perinatal and infant outcomes | |||
| Mean + SD | Mean + SD | ||
| Gestational age at birth (weeks) | 39.0 ± 1.3 | 39.1 ± 1.3 | 0.58 1 |
| Birth weight percentile | 48.1 ± 28.8 | 50.8 ± 27.2 | 0.60 1 |
| Birth height percentile | 61.4 ± 28.7 | 63.9 ± 29.5 | 0.53 1 |
| OFC percentile | 55.5 ± 29.1 | 57.3 ± 29.4 | 0.81 1 |
| Control (n = 76) | PAE (n = 48) | |
|---|---|---|
| Periconceptional period | ||
| AAD, Mean ± SD | 0.01 ± 0.02 | 0.58 ± 1.11 |
| AADD, Mean ± SD | 0.09 ± 0.27 | 2.19 ± 1.38 |
| Maximum number of drinks in 24 h, Mean ± SD | 0.07 ± 0.30 | 6.46 ± 4.73 |
| Any binge drinking episodes, Mean ± SD | 0.00 ± 0.00 | 3.71 ± 5.75 |
| Any binge drinking episodes, n (%) | 0 (0.0) | 40 (83.3) |
| ≥2 binge episodes, n (%) | 0 (0.0) | 34 (70.8) |
| ≥2 positive biomarkers, n (%) | 0 (0.0) | 2 (4.2) |
| During pregnancy | ||
| AAD, Mean ± SD | 0.00 ± 0.00 | 0.001 ± 0.01 |
| AADD, Mean ± SD | 0.00 ± 0.00 | 0.03 ± 0.10 |
| Maximum number of drinks in 24 h, Mean ± SD | 0.00 ± 0.00 | 0.16 ± 0.41 |
| Any binge drinking episodes, n (%) | 0 (0.0) | 43 (89.6) |
| ≥2 positive biomarkers, n (%) | 0 (0.0) | 1 (2.1) |
| Across pregnancy and periconceptional period | ||
| Mean AAD, Mean ± SD | 0.00 ± 0.01 | 0.15 ± 0.28 |
| Mean AADD, Mean ± SD | 0.17 ± 0.53 | 3.66 ± 2.83 |
| Maximum number of drinks in 24 h, Mean ± SD | 0.07 ± 0.30 | 6.46 ± 4.73 |
| Any binge drinking episodes, n (%) | 0 (0.0) | 44 (91.7) |
| ≥2 binge episodes, n (%) | 0 (0.0) | 34 (70.8) |
| ≥2 positive biomarkers, n (%) | 0 (0.0) | 7 (14.6) |
| Other substances during pregnancy | ||
| Marijuana, n (%) | 8 (10.5) | 17 (35.4) |
| Tobacco, n (%) | 1 (1.3) | 8 (16.7) |
| Control | PAE | p 1 | |
|---|---|---|---|
| Mean ± SD | Mean ± SD | ||
| Placenta (n = 95 for gene expression, n = 87 for protein expression) * | |||
| Gene pCRH | 0.11 ± 0.17 | 0.12 ± 0.15 | 0.62 |
| Protein pCRH | 0.01 ± 0.01 | 0.02 ± 0.01 | <0.001 |
| Gene 11β-HSD2 | 0.07 ± 0.09 | 0.06 ± 0.07 | 0.84 |
| Protein 11β-HSD2 * | 0.08 ± 0.07 | 0.11 ± 0.10 | 0.058 |
| Umbilical cord blood (n = 124) | |||
| (n = 76) | (n = 48) | ||
| Cortisone | 366.9 ± 161.9 | 384.7 ± 183.3 | 0.62 |
| Cortisol | 58.4 ± 44.8 | 59.0 ± 47.5 | 0.70 |
| Cortisone/Cortisol ratio | 9.2 ± 7.4 | 9.5 ±6.9 | 0.67 |
| PSS-V1 | AAD | Tobacco | Marijuana | |
|---|---|---|---|---|
| β (SE) | β (SE) | β (SE) | β (SE) | |
| Gene pCRH (n = 95) | ||||
| Model 1 | 0.005 (0.002) ** | −0.05 (0.15) | -- | -- |
| Model 2 | 0.006 (0.002) ** | 0.01 (0.16) | −0.05 (0.08) | −0.04 (0.04) |
| Model 3 | 0.006 (0.002) ** | 0.004 (0.17) | −0.04 (0.09) | −0.03 (0.04) |
| Protein pCRH (n = 87) | ||||
| PSS-V1 | Max number of drinks in 24 h | Tobacco | Marijuana | |
| β (SE) | β (SE) | β (SE) | β (SE) | |
| Model 1 | 0.0001 (0.0001) | 0.0004 (0.0002) ** | -- | -- |
| Model 2 | 0.0001 (0.0001) | 0.0003 (0.0002) * | −0.002 (0.003) | 0.002 (0.002) |
| Model 3 | 0.0001 (0.0001) | 0.0004 (0.0002) * | −0.001 (0.003) | 0.002 (0.002) |
| PSS-V1 | AAD | PSSV1 *AAD | Tobacco | Marijuana | |
|---|---|---|---|---|---|
| β (SE) | β (SE) | β (SE) | β (SE) | β (SE) | |
| Protein 11β-HSD2 (N = 87) | |||||
| Model 1 | 0.0002(0.001) | 0.58(0.12) ** | −0.03(0.01) ** | -- | -- |
| Model 2 | 0.0003(0.001) | 0.59(0.11) ** | −0.02(0.01) * | −0.08(0.04) * | −0.02(0.02) |
| Model 3 | 0.0004(0.001) | 0.53(0.11) ** | −0.02(0.01) | −0.08(0.04) * | −0.02(0.02) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakhireva, L.N.; Solomon, E.; Roberts, M.H.; Ma, X.; Rai, R.; Wiesel, A.; Jacobson, S.W.; Weinberg, J.; Milligan, E.D. Independent and Combined Effects of Prenatal Alcohol Exposure and Prenatal Stress on Fetal HPA Axis Development. Int. J. Mol. Sci. 2024, 25, 2690. https://doi.org/10.3390/ijms25052690
Bakhireva LN, Solomon E, Roberts MH, Ma X, Rai R, Wiesel A, Jacobson SW, Weinberg J, Milligan ED. Independent and Combined Effects of Prenatal Alcohol Exposure and Prenatal Stress on Fetal HPA Axis Development. International Journal of Molecular Sciences. 2024; 25(5):2690. https://doi.org/10.3390/ijms25052690
Chicago/Turabian StyleBakhireva, Ludmila N., Elizabeth Solomon, Melissa H. Roberts, Xingya Ma, Rajani Rai, Alexandria Wiesel, Sandra W. Jacobson, Joanne Weinberg, and Erin D. Milligan. 2024. "Independent and Combined Effects of Prenatal Alcohol Exposure and Prenatal Stress on Fetal HPA Axis Development" International Journal of Molecular Sciences 25, no. 5: 2690. https://doi.org/10.3390/ijms25052690
APA StyleBakhireva, L. N., Solomon, E., Roberts, M. H., Ma, X., Rai, R., Wiesel, A., Jacobson, S. W., Weinberg, J., & Milligan, E. D. (2024). Independent and Combined Effects of Prenatal Alcohol Exposure and Prenatal Stress on Fetal HPA Axis Development. International Journal of Molecular Sciences, 25(5), 2690. https://doi.org/10.3390/ijms25052690