
Potential Rheumatoid Arthritis-Associated Interstitial Lung Disease Treatment and Computational Approach for Future Drug Development


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Anti-Fibrotic Agents for ILD
2.1. Nintedanib
2.2. Pirfenidone
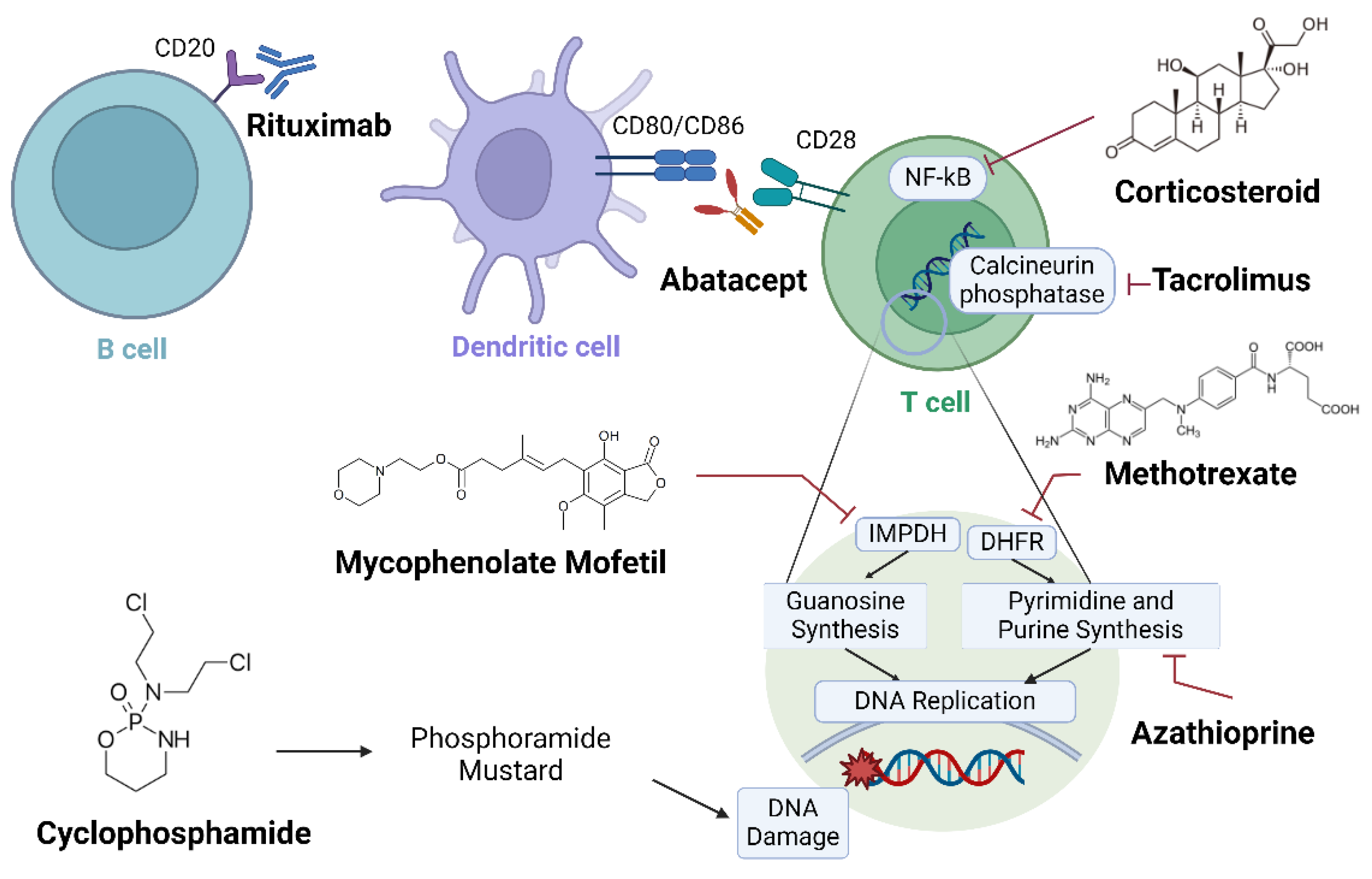
3. Conventional Synthetic DMARDs for RA-ILD
3.1. Methotrexate (MTX)
3.2. Cyclophosphamide (Cyc)
3.3. Mycophenolate Mofetil (MMF)
3.4. Azathioprine (AZA)
3.5. Tacrolimus (TAC)
4. Biologic DMARDs
4.1. Rituximab (RTX)
4.2. TNF Inhibitors
4.3. Abatacept (ABA)
4.4. Tocilizumab (TCZ)
5. Corticosteroid (Glucocorticoid)
6. Targeted Synthetic DMARDs (JAK Inhibitors)
6.1. Tofacitinib
6.2. Ruxolitinib
6.3. Baricitinib
6.4. Upadacitinib
7. Computational Methods for Further RA-ILD Drug Development
7.1. Molecular Targets Suggested for RA-ILD Fibrosis
7.2. Computational Approach for Drug Repurposing
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABA | Abatacept |
| AZA | Azathioprine |
| bDMARD | Biological disease-modifying antirheumatic drug |
| CSF1R | Colony-stimulating factor 1 receptor |
| CTD-ILD | Connective tissue disease-interstitial lung disease |
| CTLA4 | Cytotoxic T-lymphocyte-associated antigen 4 |
| CYC | Cyclophosphamide |
| DL | Deep learning |
| DLCO | Diffusing capacity of lungs for carbon monoxide |
| DMARD | Disease-modifying antirheumatic drug |
| DNN | Deep neural network |
| DTI | Drug–target interaction |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| EndoMT | Endothelial–mesenchymal transition |
| FDA | Food and Drug Administration |
| FGF | Fibroblast growth factor |
| FVC | Forced vital capacity |
| HRCT | High-resolution computed tomography |
| ID1 | Inhibitor of DNA binding protein 1 |
| IFN-γ | Interferon gamma |
| IGFBP2 | Insulin-like growth factor binding protein 2 |
| ILD | Interstitial lung disease |
| IPF | Idiopathic pulmonary fibrosis |
| JAK | Janus kinase |
| Lck | Lymphocyte-specific tyrosine protein kinase |
| MMF | Mycophenolate mofetil |
| MTX | Methotrexate |
| NSIP | Nonspecific interstitial pneumonia |
| OP | Organizing pneumonia |
| PDGF | Platelet-derived growth factor |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| QSAR | Quantitative structure–activity relationship |
| RA | Rheumatoid arthritis |
| RA-ILD | Rheumatoid arthritis-associated interstitial lung disease |
| RTX | Rituximab |
| SSc | Systemic sclerosis |
| SSc-ILD | Systemic sclerosis-associated interstitial lung disease |
| TAC | Tacrolimus |
| TCZ | Tocilizumab |
| TGF-β | Tumor growth factor beta |
| TKI | Tyrosine kinase inhibitor |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor alpha |
| UIP | Usual interstitial pneumonia |
| VEGFR | Vascular growth factor receptor |
References
- Liang, M.; Matteson, E.L.; Abril, A.; Distler, J.H.W. The role of antifibrotics in the treatment of rheumatoid arthritis–associated interstitial lung disease. Ther. Adv. Musculoskelet. Dis. 2022, 14, 1759720X2210744. [Google Scholar] [CrossRef]
- Norton, S.; Koduri, G.; Nikiphorou, E.; Dixey, J.; Williams, P.; Young, A. A study of baseline prevalence and cumulative incidence of comorbidity and extra-articular manifestations in RA and their impact on outcome. Rheumatology 2013, 52, 99–110. [Google Scholar] [CrossRef]
- Wilsher, M.; Voight, L.; Milne, D.; Teh, M.; Good, N.; Kolbe, J.; Williams, M.; Pui, K.; Merriman, T.; Sidhu, K.; et al. Prevalence of airway and parenchymal abnormalities in newly diagnosed rheumatoid arthritis. Respir. Med. 2012, 106, 1441–1446. [Google Scholar] [CrossRef]
- Huang, S.; Kronzer, V.L.; Dellaripa, P.F.; Deane, K.D.; Bolster, M.B.; Nagaraja, V.; Khanna, D.; Doyle, T.J.; Sparks, J.A. Rheumatoid arthritis-associated interstitial lung disease: Current update on prevalence, risk factors, and pharmacologic treatment. Curr. Treat. Options Rheumatol. 2020, 6, 337–353. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Sato, S.; Nishizawa, T.; Kawabe, R.; Oba, T.; Kato, A.; Horikoshi, M.; Akasaka, K.; Amano, M.; Sasaki, H.; et al. Impact of radiological honeycombing in rheumatoid arthritis-associated interstitial lung disease. BMC Pulm. Med. 2020, 20, 25. [Google Scholar] [CrossRef] [PubMed]
- Bongartz, T.; Nannini, C.; Medina-Velasquez, Y.F.; Achenbach, S.J.; Crowson, C.S.; Ryu, J.H.; Vassallo, R.; Gabriel, S.E.; Matteson, E.L. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: A population-based study. Arthritis Rheum. 2010, 62, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Fragoulis, G.E.; Nikiphorou, E.; Larsen, J.; Korsten, P.; Conway, R. Methotrexate-Associated Pneumonitis and Rheumatoid Arthritis-Interstitial Lung Disease: Current Concepts for the Diagnosis and Treatment. Front. Med. 2019, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, M.S.; Khaychuk, V.; Wittstock, K.; Han, X.; Crocket, G.; Lin, M.; Hill, S.L.; Ferri, L. Rheumatoid arthritis-associated interstitial lung disease: Epidemiology, risk/prognostic factors, and treatment landscape. Clin. Exp. Rheumatol. 2021, 39, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Kelly, C. Treatment of rheumatoid arthritis-associated interstitial lung disease: A perspective review. Ther. Adv. Musculoskelet. Dis. 2015, 7, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Oka, S.; Higuchi, T.; Shimada, K.; Hashimoto, A.; Matsui, T.; Tohma, S. Biomarkers for interstitial lung disease and acute-onset diffuse interstitial lung disease in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2021, 13, 1759720X211022506. [Google Scholar] [CrossRef] [PubMed]
- Matson, S.; Lee, J.; Eickelberg, O. Two sides of the same coin? A review of the similarities and differences between idiopathic pulmonary fibrosis and rheumatoid arthritis-associated interstitial lung disease. Eur. Respir. J. 2021, 57, 2002533. [Google Scholar] [CrossRef]
- Kass, D.J.; Nouraie, M.; Glassberg, M.K.; Ramreddy, N.; Fernandez, K.; Harlow, L.; Zhang, Y.; Chen, J.; Kerr, G.S.; Reimold, A.M.; et al. Comparative Profiling of Serum Protein Biomarkers in Rheumatoid Arthritis-Associated Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis. Arthritis Rheumatol. 2020, 72, 409–419. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Papiris, S.A.; Kagouridis, K.; Papadaki, G.; Kolilekas, L.; Manali, E.D. Treating CTDs related fibrotic ILDs by immunosuppressants: “facts and faults”. Lung 2014, 192, 221–223. [Google Scholar] [CrossRef]
- Cipriani, N.A.; Strek, M.; Noth, I.; Gordon, I.O.; Charbeneau, J.; Krishnan, J.A.; Krausz, T.; Husain, A.N. Pathologic quantification of connective tissue disease-associated versus idiopathic usual interstitial pneumonia. Arch. Pathol. Lab. Med. 2012, 136, 1253–1258. [Google Scholar] [CrossRef]
- Cassone, G.; Manfredi, A.; Vacchi, C.; Luppi, F.; Coppi, F.; Salvarani, C.; Sebastiani, M. Treatment of Rheumatoid Arthritis-Associated Interstitial Lung Disease: Lights and Shadows. J. Clin. Med. 2020, 9, 1082. [Google Scholar] [CrossRef]
- Kim, E.J.; Elicker, B.M.; Maldonado, F.; Webb, W.R.; Ryu, J.H.; Van Uden, J.H.; Lee, J.S.; King, T.E.; Collard, H.R. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur. Respir. J. 2010, 35, 1322–1328. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Wuyts, W.; Wijsenbeek, M.; Wells, A.U. Medical Therapy in Idiopathic Pulmonary Fibrosis. Semin. Respir. Crit. Care Med. 2016, 37, 368–377. [Google Scholar] [CrossRef]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef]
- Wollin, L.; Distler, J.H.W.; Redente, E.F.; Riches, D.W.H.; Stowasser, S.; Schlenker-Herceg, R.; Maher, T.M.; Kolb, M. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019, 54, 1900161. [Google Scholar] [CrossRef]
- Wollin, L.; Ostermann, A.; Williams, C. Nintedanib inhibits pro-fibrotic mediators from T cells with relevance to connective tissue disease-associated interstitial lung disease. Eur. Respir. J. 2017, 50, PA903. [Google Scholar] [CrossRef]
- Sato, S.; Shinohara, S.; Hayashi, S.; Morizumi, S.; Abe, S.; Okazaki, H.; Chen, Y.; Goto, H.; Aono, Y.; Ogawa, H.; et al. Anti-fibrotic efficacy of nintedanib in pulmonary fibrosis via the inhibition of fibrocyte activity. Respir. Res. 2017, 18, 172. [Google Scholar] [CrossRef]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages During the Fibrotic Process: M2 as Friend and Foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef]
- Tandon, K.; Herrmann, F.; Ayaub, E.; Parthasarathy, P.; Ackermann, M.; Inman, M.D.; Kolb, M.R.J.; Wollin, L.; Ask, K. Nintedanib Attenuates the Polarization of Profibrotic Macrophages Through the Inhibition of Tyrosine Phosphorylation on CSF1 Receptor. In A72. MECHANISMS DRIVING FIBROSIS; American Thoracic Society International Conference Abstracts; American Thoracic Society: New York, NY, USA, 2017; p. A2397. [Google Scholar]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Matteson, E.L.; Aringer, M.; Burmester, G.R.; Mueller, H.; Moros, L.; Kolb, M. Effect of nintedanib in patients with progressive pulmonary fibrosis associated with rheumatoid arthritis: Data from the INBUILD trial. Clin. Rheumatol. 2023, 42, 2311–2319. [Google Scholar] [CrossRef]
- Silva, A.J.S.G.; Martins, A.; Ferreira, C.C.; Oliveira, C.P.; Guimarães, F.; Gago, L.; Duarte, A.C.; Costa, C.; Salvador, M.J.; Teixeira, V.; et al. Pos1307 Nintedanib in Rheumatic Disease-Associated Interstitial Lung Disease—A Multicentre Nationwide Cohort Study. Ann. Rheum. Dis. 2023, 82 (Suppl. S1), 1001–1002. [Google Scholar] [CrossRef]
- Redente, E.F.; Aguilar, M.A.; Black, B.P.; Edelman, B.L.; Bahadur, A.N.; Humphries, S.M.; Lynch, D.A.; Wollin, L.; Riches, D.W.H. Nintedanib reduces pulmonary fibrosis in a model of rheumatoid arthritis-associated interstitial lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L998–L1009. [Google Scholar] [CrossRef]
- Conte, E.; Gili, E.; Fagone, E.; Fruciano, M.; Iemmolo, M.; Vancheri, C. Effect of pirfenidone on proliferation, TGF-β-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2014, 58, 13–19. [Google Scholar] [CrossRef]
- Nakazato, H.; Oku, H.; Yamane, S.; Tsuruta, Y.; Suzuki, R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-alpha at the translational level. Eur. J. Pharmacol. 2002, 446, 177–185. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Misra, H.P.; Rabideau, C. Pirfenidone inhibits NADPH-dependent microsomal lipid peroxidation and scavenges hydroxyl radicals. Mol. Cell. Biochem. 2000, 204, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Gan, D.; Cheng, W.; Ke, L.; Sun, A.R.; Jia, Q.; Chen, J.; Lin, J.; Li, J.; Xu, Z.; Zhang, P. Repurposing of Pirfenidone (Anti-Pulmonary Fibrosis Drug) for Treatment of Rheumatoid Arthritis. Front. Pharmacol. 2021, 12, 631891. [Google Scholar] [CrossRef]
- Solomon, J.J.; Danoff, S.K.; Woodhead, F.A.; Hurwitz, S.; Maurer, R.; Glaspole, I.; Dellaripa, P.F.; Gooptu, B.; Vassallo, R.; Cox, P.G.; et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: A randomised, double-blind, placebo-controlled, phase 2 study. Lancet Respir. Med. 2023, 11, 87–96. [Google Scholar] [CrossRef]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Qi, X.; Sun, Z.; Zhang, T.; Cui, Y.; Shu, Q. The Efficacy and Safety of Pirfenidone Combined With Immunosuppressant Therapy in Connective Tissue Disease-Associated Interstitial Lung Disease: A 24-Week Prospective Controlled Cohort Study. Front. Med. 2022, 9, 871861. [Google Scholar] [CrossRef]
- Maher, T.M.; Corte, T.J.; Fischer, A.; Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Axmann, J.; Kirchgaessler, K.-U.; Samara, K.; Gilberg, F.; et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir. Med. 2020, 8, 147–157. [Google Scholar] [CrossRef]
- Shu, Q. Efficacy, Safety, Immune Function of Pirfenidone in the Treatment of Connetive Tissue Disease-Related Interstitial Lung Disease(CTD-LID). Clinical Trial Registration, 2022. Available online: https://clinicaltrials.gov (accessed on 2 January 2024).
- Fragoulis, G.E.; Conway, R.; Nikiphorou, E. Methotrexate and interstitial lung disease: Controversies and questions. A narrative review of the literature. Rheumatology 2019, 58, 1900–1906. [Google Scholar] [CrossRef]
- Kiely, P.; Busby, A.D.; Nikiphorou, E.; Sullivan, K.; Walsh, D.A.; Creamer, P.; Dixey, J.; Young, A. Is incident rheumatoid arthritis interstitial lung disease associated with methotrexate treatment? Results from a multivariate analysis in the ERAS and ERAN inception cohorts. BMJ Open 2019, 9, e028466. [Google Scholar] [CrossRef]
- Juge, P.-A.; Lee, J.S.; Lau, J.; Kawano-Dourado, L.; Rojas Serrano, J.; Sebastiani, M.; Koduri, G.; Matteson, E.; Bonfiglioli, K.; Sawamura, M.; et al. Methotrexate and rheumatoid arthritis associated interstitial lung disease. Eur. Respir. J. 2021, 57, 2000337. [Google Scholar] [CrossRef]
- Kim, K.; Woo, A.; Park, Y.; Yong, S.H.; Lee, S.H.; Lee, S.H.; Leem, A.Y.; Kim, S.Y.; Chung, K.S.; Kim, E.Y.; et al. Protective effect of methotrexate on lung function and mortality in rheumatoid arthritis-related interstitial lung disease: A retrospective cohort study. Ther. Adv. Respir. Dis. 2022, 16, 17534666221135314. [Google Scholar] [CrossRef] [PubMed]
- Vastra Gotaland Region. Effects of Tofacitinib vs Methotrexate on Clinical and Molecular Disease Activity Markers in Joints and Lungs in Early Rheumatoid Arthritis (PULMORA)—A Randomized, Controlled, Open-label, Assessor-blinded, Phase IV Trial. Clinical Trial Registration, 2022. Available online: https://clinicaltrials.gov (accessed on 2 January 2024).
- Ahmed, A.R.; Hombal, S.M. Cyclophosphamide (Cytoxan). A review on relevant pharmacology and clinical uses. J. Am. Acad. Dermatol. 1984, 11, 1115–1126. [Google Scholar] [CrossRef]
- Ahlmann, M.; Hempel, G. The effect of cyclophosphamide on the immune system: Implications for clinical cancer therapy. Cancer Chemother. Pharmacol. 2016, 78, 661–671. [Google Scholar] [CrossRef]
- Schupp, J.C.; Köhler, T.; Müller-Quernheim, J. Usefulness of Cyclophosphamide Pulse Therapy in Interstitial Lung Diseases. Respir. Int. Rev. Thorac. Dis. 2016, 91, 296–301. [Google Scholar] [CrossRef]
- Barnes, H.; Holland, A.E.; Westall, G.P.; Goh, N.S.; Glaspole, I.N. Cyclophosphamide for connective tissue disease-associated interstitial lung disease. Cochrane Database Syst. Rev. 2018, 1, CD010908. [Google Scholar] [CrossRef]
- Maher, T.M.; Tudor, V.A.; Saunders, P.; Gibbons, M.A.; Fletcher, S.V.; Denton, C.P.; Hoyles, R.K.; Parfrey, H.; Renzoni, E.A.; Kokosi, M.; et al. Rituximab versus intravenous cyclophosphamide in patients with connective tissue disease-associated interstitial lung disease in the UK (RECITAL): A double-blind, double-dummy, randomised, controlled, phase 2b trial. Lancet Respir. Med. 2023, 11, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Almazor, M.E.; Belseck, E.; Shea, B.; Wells, G.; Tugwell, P. Cyclophosphamide for treating rheumatoid arthritis. Cochrane Database Syst. Rev. 2000, 2000, CD001157. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Ohbe, H.; Ikeda, K.; Uda, K.; Furuya, H.; Furuta, S.; Nakajima, M.; Sasabuchi, Y.; Matsui, H.; Fushimi, K.; et al. Intravenous cyclophosphamide in acute exacerbation of rheumatoid arthritis-related interstitial lung disease: A propensity-matched analysis using a nationwide inpatient database. Semin. Arthritis Rheum. 2021, 51, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Ritter, M.L.; Pirofski, L. Mycophenolate mofetil: Effects on cellular immune subsets, infectious complications, and antimicrobial activity. Transpl. Infect. Dis. Off. J. Transplant. Soc. 2009, 11, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Morath, C.; Schwenger, V.; Beimler, J.; Mehrabi, A.; Schmidt, J.; Zeier, M.; Muranyi, W. Antifibrotic actions of mycophenolic acid. Clin. Transplant. 2006, 20 (Suppl. S17), 25–29. [Google Scholar] [CrossRef]
- Saketkoo, L.A.; Espinoza, L.R. Rheumatoid Arthritis Interstitial Lung Disease: Mycophenolate Mofetil as an Antifibrotic and Disease-Modifying Antirheumatic Drug. Arch. Intern. Med. 2008, 168, 1718–1719. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Brown, K.K.; Du Bois, R.M.; Frankel, S.K.; Cosgrove, G.P.; Fernandez-Perez, E.R.; Huie, T.J.; Krishnamoorthy, M.; Meehan, R.T.; Olson, A.L.; et al. Mycophenolate mofetil improves lung function in connective tissue disease-associated interstitial lung disease. J. Rheumatol. 2013, 40, 640–646. [Google Scholar] [CrossRef]
- Omair, M.A.; Alhamad, E.H. Mycophenolate mofetil is an effective therapy for connective tissue disease-associated interstitial lung disease. Int. J. Clin. Rheumatol. 2017, 12, 67. [Google Scholar]
- Cassone, G.; Sebastiani, M.; Vacchi, C.; Erre, G.L.; Salvarani, C.; Manfredi, A. Efficacy and safety of mycophenolate mofetil in the treatment of rheumatic disease-related interstitial lung disease: A narrative review. Drugs Context 2021, 10, 2020-8-8. [Google Scholar] [CrossRef]
- Schiff, M.; Beaulieu, A.; Scott, D.L.; Rashford, M. Mycophenolate mofetil in the treatment of adults with advanced rheumatoid arthritis: Three 24-week, randomized, double-blind, placebo- or ciclosporin-controlled trials. Clin. Drug Investig. 2010, 30, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Maltzman, J.S.; Koretzky, G.A. Azathioprine: Old drug, new actions. J. Clin. Investig. 2003, 111, 1122–1124. [Google Scholar] [CrossRef]
- Bérezné, A.; Ranque, B.; Valeyre, D.; Brauner, M.; Allanore, Y.; Launay, D.; Le Guern, V.; Kahn, J.-E.; Couderc, L.-J.; Constans, J.; et al. Therapeutic strategy combining intravenous cyclophosphamide followed by oral azathioprine to treat worsening interstitial lung disease associated with systemic sclerosis: A retrospective multicenter open-label study. J. Rheumatol. 2008, 35, 1064–1072. [Google Scholar]
- Vorselaars, A.D.M.; Wuyts, W.A.; Vorselaars, V.M.M.; Zanen, P.; Deneer, V.H.M.; Veltkamp, M.; Thomeer, M.; van Moorsel, C.H.M.; Grutters, J.C. Methotrexate vs azathioprine in second-line therapy of sarcoidosis. Chest 2013, 144, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Terras Alexandre, A.; Martins, N.; Raimundo, S.; Melo, N.; Catetano Mota, P.; Novais E Bastos, H.; Pereira, J.M.; Cunha, R.; Guimarães, S.; Souto Moura, C.; et al. Impact of Azathioprine use in chronic hypersensitivity pneumonitis patients. Pulm. Pharmacol. Ther. 2020, 60, 101878. [Google Scholar] [CrossRef] [PubMed]
- Matson, S.M.; Baqir, M.; Moua, T.; Marll, M.; Kent, J.; Iannazzo, N.S.; Boente, R.D.; Donatelli, J.M.; Dai, J.; Diaz, F.J.; et al. Treatment Outcomes for Rheumatoid Arthritis-Associated Interstitial Lung Disease: A Real-World, Multisite Study of the Impact of Immunosuppression on Pulmonary Function Trajectory. Chest 2023, 163, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Boerner, E.B.; Cuyas, M.; Theegarten, D.; Ohshimo, S.; Costabel, U.; Bonella, F. Azathioprine for Connective Tissue Disease-Associated Interstitial Lung Disease. Respir. Int. Rev. Thorac. Dis. 2020, 99, 628–636. [Google Scholar] [CrossRef]
- Oldham, J.M.; Lee, C.; Valenzi, E.; Witt, L.J.; Adegunsoye, A.; Hsu, S.; Chen, L.; Montner, S.; Chung, J.H.; Noth, I.; et al. Azathioprine response in patients with fibrotic connective tissue disease-associated interstitial lung disease. Respir. Med. 2016, 121, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Broen, J.C.A.; van Laar, J.M. Mycophenolate mofetil, azathioprine and tacrolimus: Mechanisms in rheumatology. Nat. Rev. Rheumatol. 2020, 16, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Kurita, T.; Yasuda, S.; Oba, K.; Odani, T.; Kono, M.; Otomo, K.; Fujieda, Y.; Oku, K.; Bohgaki, T.; Amengual, O.; et al. The efficacy of tacrolimus in patients with interstitial lung diseases complicated with polymyositis or dermatomyositis. Rheumatology 2015, 54, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Labirua-Iturburu, A.; Selva-O’Callaghan, A.; Martínez-Gómez, X.; Trallero-Araguás, E.; Labrador-Horrillo, M.; Vilardell-Tarrés, M. Calcineurin inhibitors in a cohort of patients with antisynthetase-associated interstitial lung disease. Clin. Exp. Rheumatol. 2013, 31, 436–439. [Google Scholar] [PubMed]
- Yamano, Y.; Taniguchi, H.; Kondoh, Y.; Ando, M.; Kataoka, K.; Furukawa, T.; Johkoh, T.; Fukuoka, J.; Sakamoto, K.; Hasegawa, Y. Multidimensional improvement in connective tissue disease-associated interstitial lung disease: Two courses of pulse dose methylprednisolone followed by low-dose prednisone and tacrolimus. Respirology 2018, 23, 1041–1048. [Google Scholar] [CrossRef]
- Kim, J.-W.; Chung, S.W.; Pyo, J.Y.; Chang, S.H.; Kim, M.U.; Park, C.H.; Lee, J.S.; Lee, J.S.; Ha, Y.-J.; Kang, E.H.; et al. Methotrexate, leflunomide and tacrolimus use and the progression of rheumatoid arthritis-associated interstitial lung disease. Rheumatology 2023, 62, 2377–2385. [Google Scholar] [CrossRef]
- Pescovitz, M.D. Rituximab, an anti-cd20 monoclonal antibody: History and mechanism of action. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2006, 6, 859–866. [Google Scholar] [CrossRef]
- Smith, M.R. Rituximab (monoclonal anti-CD20 antibody): Mechanisms of action and resistance. Oncogene 2003, 22, 7359–7368. [Google Scholar] [CrossRef]
- Ansari, M.A.; Nadeem, A.; Attia, S.M.; Bakheet, S.A.; Alasmari, A.F.; Alomar, H.A.; Al-Mazroua, H.A.; Alhamed, A.S.; Shahid, M.; Alqinyah, M.; et al. Rituximab exerts its anti-arthritic effects via inhibiting NF-κB/GM-CSF/iNOS signaling in B cells in a mouse model of collagen-induced arthritis. Heliyon 2023, 9, e16673. [Google Scholar] [CrossRef]
- Cohen, S.B.; Emery, P.; Greenwald, M.W.; Dougados, M.; Furie, R.A.; Genovese, M.C.; Keystone, E.C.; Loveless, J.E.; Burmester, G.-R.; Cravets, M.W.; et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006, 54, 2793–2806. [Google Scholar] [CrossRef]
- Emery, P.; Fleischmann, R.; Filipowicz-Sosnowska, A.; Schechtman, J.; Szczepanski, L.; Kavanaugh, A.; Racewicz, A.J.; van Vollenhoven, R.F.; Li, N.F.; Agarwal, S.; et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: Results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006, 54, 1390–1400. [Google Scholar] [CrossRef]
- Fui, A.; Bergantini, L.; Selvi, E.; Mazzei, M.A.; Bennett, D.; Pieroni, M.G.; Rottoli, P.; Bargagli, E. Rituximab therapy in interstitial lung disease associated with rheumatoid arthritis. Intern. Med. J. 2020, 50, 330–336. [Google Scholar] [CrossRef]
- Vadillo, C.; Nieto, M.A.; Romero-Bueno, F.; Leon, L.; Sanchez-Pernaute, O.; Rodriguez-Nieto, M.J.; Freites, D.; Jover, J.A.; Álvarez-Sala, J.L.; Abasolo, L. Efficacy of rituximab in slowing down progression of rheumatoid arthritis-related interstitial lung disease: Data from the NEREA Registry. Rheumatology 2020, 59, 2099–2108. [Google Scholar] [CrossRef]
- Md Yusof, M.Y.; Kabia, A.; Darby, M.; Lettieri, G.; Beirne, P.; Vital, E.M.; Dass, S.; Emery, P. Effect of rituximab on the progression of rheumatoid arthritis-related interstitial lung disease: 10 years’ experience at a single centre. Rheumatology 2017, 56, 1348–1357. [Google Scholar] [CrossRef]
- Vassallo, R.; Matteson, E.; Thomas, C.F. Clinical response of rheumatoid arthritis-associated pulmonary fibrosis to tumor necrosis factor-alpha inhibition. Chest 2002, 122, 1093–1096. [Google Scholar] [CrossRef]
- Koo, B.S.; Hong, S.; Kim, Y.J.; Kim, Y.-G.; Lee, C.-K.; Yoo, B. Mortality in patients with rheumatoid arthritis-associated interstitial lung disease treated with an anti-tumor necrosis factor agent. Korean J. Intern. Med. 2015, 30, 104–109. [Google Scholar] [CrossRef]
- Hagiwara, K.; Sato, T.; Takagi-Kobayashi, S.; Hasegawa, S.; Shigihara, N.; Akiyama, O. Acute exacerbation of preexisting interstitial lung disease after administration of etanercept for rheumatoid arthritis. J. Rheumatol. 2007, 34, 1151–1154. [Google Scholar]
- Korhonen, R.; Moilanen, E. Abatacept, a novel CD80/86-CD28 T cell co-stimulation modulator, in the treatment of rheumatoid arthritis. Basic Clin. Pharmacol. Toxicol. 2009, 104, 276–284. [Google Scholar] [CrossRef]
- Israël-Assayag, E.; Fournier, M.; Cormier, Y. Blockade of T cell costimulation by CTLA4-Ig inhibits lung inflammation in murine hypersensitivity pneumonitis. J. Immunol. Baltim. Md 1950 1999, 163, 6794–6799. [Google Scholar] [CrossRef]
- Boleto, G.; Guignabert, C.; Pezet, S.; Cauvet, A.; Sadoine, J.; Tu, L.; Nicco, C.; Gobeaux, C.; Batteux, F.; Allanore, Y.; et al. T-cell costimulation blockade is effective in experimental digestive and lung tissue fibrosis. Arthritis Res. Ther. 2018, 20, 197. [Google Scholar] [CrossRef]
- Fernández-Díaz, C.; Castañeda, S.; Melero-González, R.B.; Ortiz-Sanjuán, F.; Juan-Mas, A.; Carrasco-Cubero, C.; Casafont-Solé, I.; Olivé, A.; Rodríguez-Muguruza, S.; Almodóvar-González, R.; et al. Abatacept in interstitial lung disease associated with rheumatoid arthritis: National multicenter study of 263 patients. Rheumatology 2020, 59, 3906–3916. [Google Scholar] [CrossRef]
- Tardella, M.; Di Carlo, M.; Carotti, M.; Giovagnoni, A.; Salaffi, F. Abatacept in rheumatoid arthritis-associated interstitial lung disease: Short-term outcomes and predictors of progression. Clin. Rheumatol. 2021, 40, 4861–4867. [Google Scholar] [CrossRef]
- Vicente-Rabaneda, E.F.; Atienza-Mateo, B.; Blanco, R.; Cavagna, L.; Ancochea, J.; Castañeda, S.; González-Gay, M.Á. Efficacy and safety of abatacept in interstitial lung disease of rheumatoid arthritis: A systematic literature review. Autoimmun. Rev. 2021, 20, 102830. [Google Scholar] [CrossRef]
- Mena-Vázquez, N.; Rojas-Gimenez, M.; Fuego-Varela, C.; García-Studer, A.; Perez-Gómez, N.; Romero-Barco, C.M.; Godoy-Navarrete, F.J.; Manrique-Arija, S.; Gandía-Martínez, M.; Calvo-Gutiérrez, J.; et al. Safety and Effectiveness of Abatacept in a Prospective Cohort of Patients with Rheumatoid Arthritis-Associated Interstitial Lung Disease. Biomedicines 2022, 10, 1480. [Google Scholar] [CrossRef]
- Gudu, T.; Stober, C.; Fifield, M.; Babar, J.; Ostor, A.; Parfrey, H.; Hall, F. P147 Safety of AbatacePt in Rheumatoid arthritis associated Interstitial Lung disease (APRIL). Rheumatology 2021, 60, keab247.143. [Google Scholar] [CrossRef]
- Khanna, D.; Lin, C.J.F.; Furst, D.E.; Goldin, J.; Kim, G.; Kuwana, M.; Allanore, Y.; Matucci-Cerinic, M.; Distler, O.; Shima, Y.; et al. Tocilizumab in systemic sclerosis: A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2020, 8, 963–974. [Google Scholar] [CrossRef]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet Lond. Engl. 2016, 387, 2630–2640. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, A.; Cassone, G.; Furini, F.; Gremese, E.; Venerito, V.; Atzeni, F.; Arrigoni, E.; Della Casa, G.; Cerri, S.; Govoni, M.; et al. Tocilizumab therapy in rheumatoid arthritis with interstitial lung disease: A multicentre retrospective study. Intern. Med. J. 2020, 50, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, N.; Sugiyama, K.; Arifuku, H.; Nakano, K.; Watanabe, H.; Wakayama, T.; Koyama, K.; Hirata, H.; Fukushima, Y. Effect of tocilizumab on interstitial lung disease (ILD) in patients with rheumatoid arthritis (RA). Eur. Respir. J. 2020, 56, 747. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jin, S.-M.; Lee, B.J.; Chung, D.H.; Jang, B.-G.; Park, H.S.; Lee, S.-M.; Yim, J.-J.; Yang, S.-C.; Yoo, C.-G.; et al. Treatment response and long term follow-up results of nonspecific interstitial pneumonia. J. Korean Med. Sci. 2012, 27, 661–667. [Google Scholar] [CrossRef]
- O’Dwyer, D.N.; Armstrong, M.E.; Cooke, G.; Dodd, J.D.; Veale, D.J.; Donnelly, S.C. Rheumatoid Arthritis (RA) associated interstitial lung disease (ILD). Eur. J. Intern. Med. 2013, 24, 597–603. [Google Scholar] [CrossRef]
- Song, J.W.; Lee, H.-K.; Lee, C.K.; Chae, E.J.; Jang, S.J.; Colby, T.V.; Kim, D.S. Clinical course and outcome of rheumatoid arthritis-related usual interstitial pneumonia. Sarcoidosis Vasc. Diffuse Lung Dis. Off. J. WASOG 2013, 30, 103–112. [Google Scholar]
- Bradley, B.; Branley, H.M.; Egan, J.J.; Greaves, M.S.; Hansell, D.M.; Harrison, N.K.; Hirani, N.; Hubbard, R.; Lake, F.; Millar, A.B.; et al. Interstitial lung disease guideline: The British Thoracic Society in collaboration with the Thoracic Society of Australia and New Zealand and the Irish Thoracic Society. Thorax 2008, 63 (Suppl. S5), v1–v58. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez, F.J.; de Andrade, J.A.; Anstrom, K.J.; King, T.E.; Raghu, G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2093–2101. [Google Scholar] [CrossRef]
- Zamora-Legoff, J.A.; Krause, M.L.; Crowson, C.S.; Ryu, J.H.; Matteson, E.L. Risk of serious infection in patients with rheumatoid arthritis-associated interstitial lung disease. Clin. Rheumatol. 2016, 35, 2585–2589. [Google Scholar] [CrossRef]
- Canalis, E.; Mazziotti, G.; Giustina, A.; Bilezikian, J.P. Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporos. Int. 2007, 18, 1319–1328. [Google Scholar] [CrossRef]
- Liu, J.; Wang, F.; Luo, F. The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules 2023, 13, 119. [Google Scholar] [CrossRef]
- Montero, P.; Milara, J.; Roger, I.; Cortijo, J. Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6211. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.L.; Brockwell, N.K.; Parker, B.S. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers 2019, 11, 2002. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, M.; Li, X.; Zhang, J.; Wang, F.; Zhang, C.; Roden, A.; Ryu, J.H.; Warrington, K.J.; Sun, J.; et al. Canonical and noncanonical regulatory roles for JAK2 in the pathogenesis of rheumatoid arthritis-associated interstitial lung disease and idiopathic pulmonary fibrosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22336. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, R.; Chen, C.-W.; Mallano, T.; Dees, C.; Distler, A.; Reich, A.; Bergmann, C.; Ramming, A.; Gelse, K.; et al. JAK1-dependent transphosphorylation of JAK2 limits the antifibrotic effects of selective JAK2 inhibitors on long-term treatment. Ann. Rheum. Dis. 2017, 76, 1467–1475. [Google Scholar] [CrossRef]
- Strand, V.; van Vollenhoven, R.F.; Lee, E.B.; Fleischmann, R.; Zwillich, S.H.; Gruben, D.; Koncz, T.; Wilkinson, B.; Wallenstein, G. Tofacitinib or adalimumab versus placebo: Patient-reported outcomes from a phase 3 study of active rheumatoid arthritis. Rheumatology 2016, 55, 1031–1041. [Google Scholar] [CrossRef]
- Kremer, J.M.; Bloom, B.J.; Breedveld, F.C.; Coombs, J.H.; Fletcher, M.P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R.; Wilkinson, B.; Zerbini, C.A.F.; et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009, 60, 1895–1905. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Tanaka, Y. A review of upadacitinib in rheumatoid arthritis. Mod. Rheumatol. 2020, 30, 779–787. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, D.; Wang, L.; Wang, S.; Roden, A.C.; Zhao, H.; Li, X.; Prakash, Y.S.; Matteson, E.L.; Tschumperlin, D.J.; et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L487–L497. [Google Scholar] [CrossRef]
- Sendo, S.; Saegusa, J.; Yamada, H.; Nishimura, K.; Morinobu, A. Tofacitinib facilitates the expansion of myeloid-derived suppressor cells and ameliorates interstitial lung disease in SKG mice. Arthritis Res. Ther. 2019, 21, 184. [Google Scholar] [CrossRef] [PubMed]
- Citera, G.; Mysler, E.; Madariaga, H.; Cardiel, M.H.; Castañeda, O.; Fischer, A.; Richette, P.; Chartrand, S.; Park, J.K.; Strengholt, S.; et al. Incidence Rates of Interstitial Lung Disease Events in Tofacitinib-Treated Rheumatoid Arthritis Patients: Post Hoc Analysis From 21 Clinical Trials. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2021, 27, e482–e490. [Google Scholar] [CrossRef]
- Fleischmann, R.; Mease, P.J.; Schwartzman, S.; Hwang, L.-J.; Soma, K.; Connell, C.A.; Takiya, L.; Bananis, E. Efficacy of tofacitinib in patients with rheumatoid arthritis stratified by background methotrexate dose group. Clin. Rheumatol. 2017, 36, 15–24. [Google Scholar] [CrossRef]
- Vacchi, C.; Manfredi, A.; Cassone, G.; Cerri, S.; Della Casa, G.; Andrisani, D.; Salvarani, C.; Sebastiani, M. Tofacitinib for the Treatment of Severe Interstitial Lung Disease Related to Rheumatoid Arthritis. Case Rep. Med. 2021, 2021, 6652845. [Google Scholar] [CrossRef]
- Saldarriaga-Rivera, L.M.; López-Villegas, V.J. Janus kinase inhibitors as a therapeutic option in rheumatoid arthritis and associated interstitial lung disease. Report of four cases. Rev. Colomb. Reumatol. Engl. Ed. 2019, 26, 137–139. [Google Scholar] [CrossRef]
- Tardella, M.; Di Carlo, M.; Carotti, M.; Ceccarelli, L.; Giovagnoni, A.; Salaffi, F. A retrospective study of the efficacy of JAK inhibitors or abatacept on rheumatoid arthritis-interstitial lung disease. Inflammopharmacology 2022, 30, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Lescoat, A.; Lelong, M.; Jeljeli, M.; Piquet-Pellorce, C.; Morzadec, C.; Ballerie, A.; Jouneau, S.; Jego, P.; Vernhet, L.; Batteux, F.; et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: Perspectives for scleroderma-associated interstitial lung disease. Biochem. Pharmacol. 2020, 178, 114103. [Google Scholar] [CrossRef] [PubMed]
- Silva-Carmona, M.; Vogel, T.P.; Marchal, S.; Guesmi, M.; Dubus, J.-C.; Leroy, S.; Fabre, A.; Barlogis, V.; Forbes, L.R.; Giovannini-Chami, L. Successful Treatment of Interstitial Lung Disease in STAT3 Gain-of-Function Using JAK Inhibitors. Am. J. Respir. Crit. Care Med. 2020, 202, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Bader-Meunier, B.; Hadchouel, A.; Berteloot, L.; Polivka, L.; Béziat, V.; Casanova, J.-L.; Lévy, R. Effectiveness and safety of ruxolitinib for the treatment of refractory systemic idiopathic juvenile arthritis like associated with interstitial lung disease: A case report. Ann. Rheum. Dis. 2022, 81, e20. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef]
- Salvarani, C.; Sebastiani, M.; Dieude, P.; Garcia, M.; Deberdt, W.; Rogai, V.; de la Torre, I.; Inciarte-Mundo, J.; Balsa, A. Baricitinib and the Risk of Incident Interstitial Lung Disease: A Descriptive Clinical Case Report from Clinical Trials. Rheumatol. Ther. 2021, 8, 1435–1441. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Perillo, F.; Metella Refini, R.; Bergantini, L.; Bellisai, F.; Selvi, E.; Cameli, P.; Manganelli, S.; Conticini, E.; Cantarini, L.; et al. Efficacy of baricitinib in treating rheumatoid arthritis: Modulatory effects on fibrotic and inflammatory biomarkers in a real-life setting. Int. Immunopharmacol. 2020, 86, 106748. [Google Scholar] [CrossRef]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2021, 73, 924–939. [Google Scholar] [CrossRef]
- Canadian Agency for Drugs and Technologies in Health. Pharmacoeconomic Review Report: Upadacitinib (Rinvoq): (AbbVie): Indication: For the Treatment of Adults with Moderately to Severely Active Rheumatoid Arthritis who have had an Inadequate Response or Intolerance to Methotrexate; CADTH Common Drug Reviews; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2020. [Google Scholar]
- Venerito, V.; Manfredi, A.; Carletto, A.; Gentileschi, S.; Atzeni, F.; Guiducci, S.; Lavista, M.; La Corte, L.; Pedrollo, E.; Scardapane, A.; et al. Evolution of Rheumatoid-Arthritis-Associated Interstitial Lung Disease in Patients Treated with JAK Inhibitors: A Retrospective Exploratory Study. J. Clin. Med. 2023, 12, 957. [Google Scholar] [CrossRef]
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Hirota, N.; Ito, T.; Miyazaki, S.; Ebina, M.; Homma, S. Gene expression profiling of lung myofibroblasts reveals the anti-fibrotic effects of cyclosporine. Tohoku J. Exp. Med. 2014, 233, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Wójcik-Pszczoła, K.; Chłoń-Rzepa, G.; Jankowska, A.; Ślusarczyk, M.; Ferdek, P.E.; Kusiak, A.A.; Świerczek, A.; Pociecha, K.; Koczurkiewicz-Adamczyk, P.; Wyska, E.; et al. A Novel, Pan-PDE Inhibitor Exerts Anti-Fibrotic Effects in Human Lung Fibroblasts via Inhibition of TGF-β Signaling and Activation of cAMP/PKA Signaling. Int. J. Mol. Sci. 2020, 21, 4008. [Google Scholar] [CrossRef]
- Xie, X.; Wu, X.; Zhao, D.; Liu, Y.; Du, Q.; Li, Y.; Xu, Y.; Li, Y.; Qiu, Y.; Yang, Y. Fluvoxamine alleviates bleomycin-induced lung fibrosis via regulating the cGAS-STING pathway. Pharmacol. Res. 2023, 187, 106577. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129. [Google Scholar] [CrossRef]
- Li, X.; Zhu, L.; Wang, B.; Yuan, M.; Zhu, R. Drugs and Targets in Fibrosis. Front. Pharmacol. 2017, 8, 855. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C.; Mercer, P.F. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S16–S20. [Google Scholar] [CrossRef]
- Bagnato, G.; Harari, S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2015, 24, 102–114. [Google Scholar] [CrossRef]
- Mahalanobish, S.; Saha, S.; Dutta, S.; Sil, P.C. Matrix metalloproteinase: An upcoming therapeutic approach for idiopathic pulmonary fibrosis. Pharmacol. Res. 2020, 152, 104591. [Google Scholar] [CrossRef]
- Makarev, E.; Izumchenko, E.; Aihara, F.; Wysocki, P.T.; Zhu, Q.; Buzdin, A.; Sidransky, D.; Zhavoronkov, A.; Atala, A. Common pathway signature in lung and liver fibrosis. Cell Cycle Georget. Tex 2016, 15, 1667–1673. [Google Scholar] [CrossRef] [PubMed]
- McEntee, C.P.; Gunaltay, S.; Travis, M.A. Regulation of barrier immunity and homeostasis by integrin-mediated transforming growth factor β activation. Immunology 2020, 160, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Gunaltay, S.; McEntee, C.P.; Shuttleworth, E.E.; Smedley, C.; Houston, S.A.; Fenton, T.M.; Levison, S.; Mann, E.R.; Travis, M.A. Human monocytes and macrophages regulate immune tolerance via integrin αvβ8-mediated TGFβ activation. J. Exp. Med. 2018, 215, 2725–2736. [Google Scholar] [CrossRef] [PubMed]
- Herrera, I.; Cisneros, J.; Maldonado, M.; Ramírez, R.; Ortiz-Quintero, B.; Anso, E.; Chandel, N.S.; Selman, M.; Pardo, A. Matrix metalloproteinase (MMP)-1 induces lung alveolar epithelial cell migration and proliferation, protects from apoptosis, and represses mitochondrial oxygen consumption. J. Biol. Chem. 2013, 288, 25964–25975. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Crampton, S.P.; Hughes, C.C.W. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity 2007, 26, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Pilewski, J.M.; Liu, L.; Henry, A.C.; Knauer, A.V.; Feghali-Bostwick, C.A. Insulin-like growth factor binding proteins 3 and 5 are overexpressed in idiopathic pulmonary fibrosis and contribute to extracellular matrix deposition. Am. J. Pathol. 2005, 166, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-analysis: Diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, W.; Kuca-Warnawin, E.H.; Radzikowska, A.; Maśliński, W. The role of anti-citrullinated protein antibodies (ACPA) in the pathogenesis of rheumatoid arthritis. Cent.-Eur. J. Immunol. 2017, 42, 390–398. [Google Scholar] [CrossRef]
- Correia, C.S.; Briones, M.R.; Guo, R.; Ostrowski, R.A. Elevated anti-cyclic citrullinated peptide antibody titer is associated with increased risk for interstitial lung disease. Clin. Rheumatol. 2019, 38, 1201–1206. [Google Scholar] [CrossRef]
- Shao, T.; Shi, X.; Yang, S.; Zhang, W.; Li, X.; Shu, J.; Alqalyoobi, S.; Zeki, A.A.; Leung, P.S.; Shuai, Z. Interstitial Lung Disease in Connective Tissue Disease: A Common Lesion With Heterogeneous Mechanisms and Treatment Considerations. Front. Immunol. 2021, 12, 684699. [Google Scholar] [CrossRef]
- Mouawad, J.E.; Feghali-Bostwick, C. The Molecular Mechanisms of Systemic Sclerosis-Associated Lung Fibrosis. Int. J. Mol. Sci. 2023, 24, 2963. [Google Scholar] [CrossRef]
- Wu, D.; Gao, W.; Li, X.; Tian, C.; Jiao, N.; Fang, S.; Xiao, J.; Xu, Z.; Zhu, L.; Zhang, G.; et al. Dr AFC: Drug repositioning through anti-fibrosis characteristic. Brief. Bioinform. 2021, 22, bbaa115. [Google Scholar] [CrossRef] [PubMed]
- Mozzicafreddo, M.; Benfaremo, D.; Paolini, C.; Agarbati, S.; Svegliati Baroni, S.; Moroncini, G. Screening and Analysis of Possible Drugs Binding to PDGFRα: A Molecular Modeling Study. Int. J. Mol. Sci. 2023, 24, 9623. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Cuffaro, D.; Nuti, E.; Ojeda-Montes, M.J.; Beltrán-Debón, R.; Mulero, M.; Rossello, A.; Pujadas, G.; Garcia-Vallvé, S. Identification of Broad-Spectrum MMP Inhibitors by Virtual Screening. Molecules 2021, 26, 4553. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Xu, J.; Yan, R.; Liu, H.; Zhao, J.; Xie, Y.; Deng, W.; Liao, W.; Nie, Y. Virtual screening and activity evaluation of multitargeting inhibitors for idiopathic pulmonary fibrosis. Front. Pharmacol. 2022, 13, 998245. [Google Scholar] [CrossRef]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef]
- Zhu, W.; Li, Y.; Zhao, J.; Wang, Y.; Li, Y.; Wang, Y. The mechanism of triptolide in the treatment of connective tissue disease-related interstitial lung disease based on network pharmacology and molecular docking. Ann. Med. 2022, 54, 541–552. [Google Scholar] [CrossRef]
- Lu, Y.; Li, A.; Lai, X.; Jiang, J.; Zhang, L.; Zhong, Z.; Zhao, W.; Tang, P.; Zhao, H.; Ren, X. Identification of differentially expressed genes and signaling pathways using bioinformatics in interstitial lung disease due to tyrosine kinase inhibitors targeting the epidermal growth factor receptor. Investig. New Drugs 2019, 37, 384–400. [Google Scholar] [CrossRef]
- Zhu, H. Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-Learning-Based Drug-Target Interaction Prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Lai, L.; Pei, J. Prediction of Human Cytochrome P450 Inhibition Using a Multitask Deep Autoencoder Neural Network. Mol. Pharm. 2018, 15, 4336–4345. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.L.; Swigris, J.J.; Sprunger, D.B.; Fischer, A.; Fernandez-Perez, E.R.; Solomon, J.; Murphy, J.; Cohen, M.; Raghu, G.; Brown, K.K. Rheumatoid Arthritis–Interstitial Lung Disease–associated Mortality. Am. J. Respir. Crit. Care Med. 2011, 183, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Lee, J.S.; Sverzellati, N.; Rossi, G.; Cottin, V. The Lung in Rheumatoid Arthritis: Focus on Interstitial Lung Disease. Arthritis Rheumatol. 2018, 70, 1544–1554. [Google Scholar] [CrossRef]
- Rangel-Moreno, J.; Hartson, L.; Navarro, C.; Gaxiola, M.; Selman, M.; Randall, T.D. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J. Clin. Investig. 2006, 116, 3183–3194. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, E.; Hong, H.; Lee, Y.-A.; Kim, K.-S. Potential Rheumatoid Arthritis-Associated Interstitial Lung Disease Treatment and Computational Approach for Future Drug Development. Int. J. Mol. Sci. 2024, 25, 2682. https://doi.org/10.3390/ijms25052682
Jeong E, Hong H, Lee Y-A, Kim K-S. Potential Rheumatoid Arthritis-Associated Interstitial Lung Disease Treatment and Computational Approach for Future Drug Development. International Journal of Molecular Sciences. 2024; 25(5):2682. https://doi.org/10.3390/ijms25052682
Chicago/Turabian StyleJeong, Eunji, Hyunseok Hong, Yeon-Ah Lee, and Kyoung-Soo Kim. 2024. "Potential Rheumatoid Arthritis-Associated Interstitial Lung Disease Treatment and Computational Approach for Future Drug Development" International Journal of Molecular Sciences 25, no. 5: 2682. https://doi.org/10.3390/ijms25052682
APA StyleJeong, E., Hong, H., Lee, Y.-A., & Kim, K.-S. (2024). Potential Rheumatoid Arthritis-Associated Interstitial Lung Disease Treatment and Computational Approach for Future Drug Development. International Journal of Molecular Sciences, 25(5), 2682. https://doi.org/10.3390/ijms25052682