
Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown

 ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
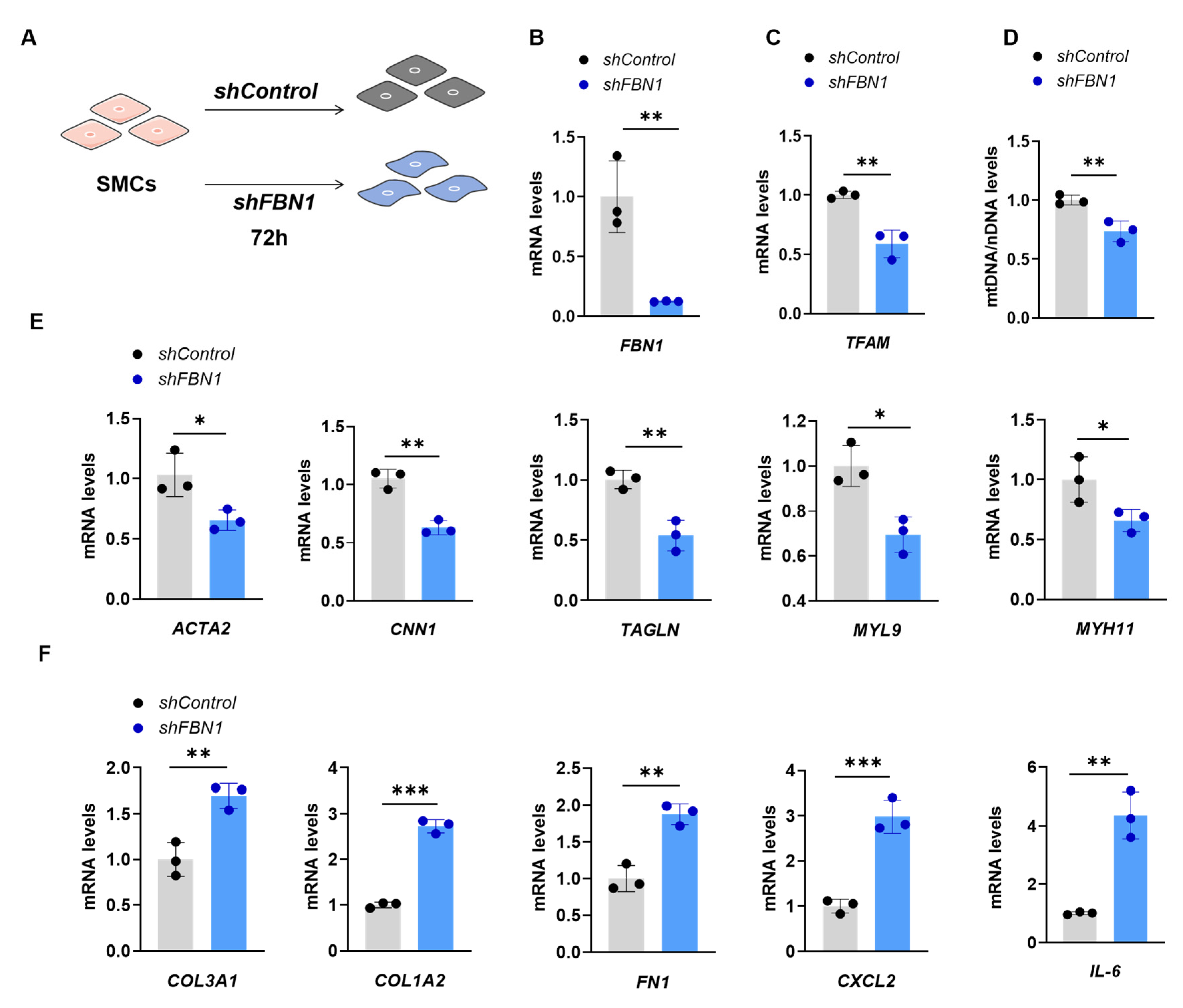
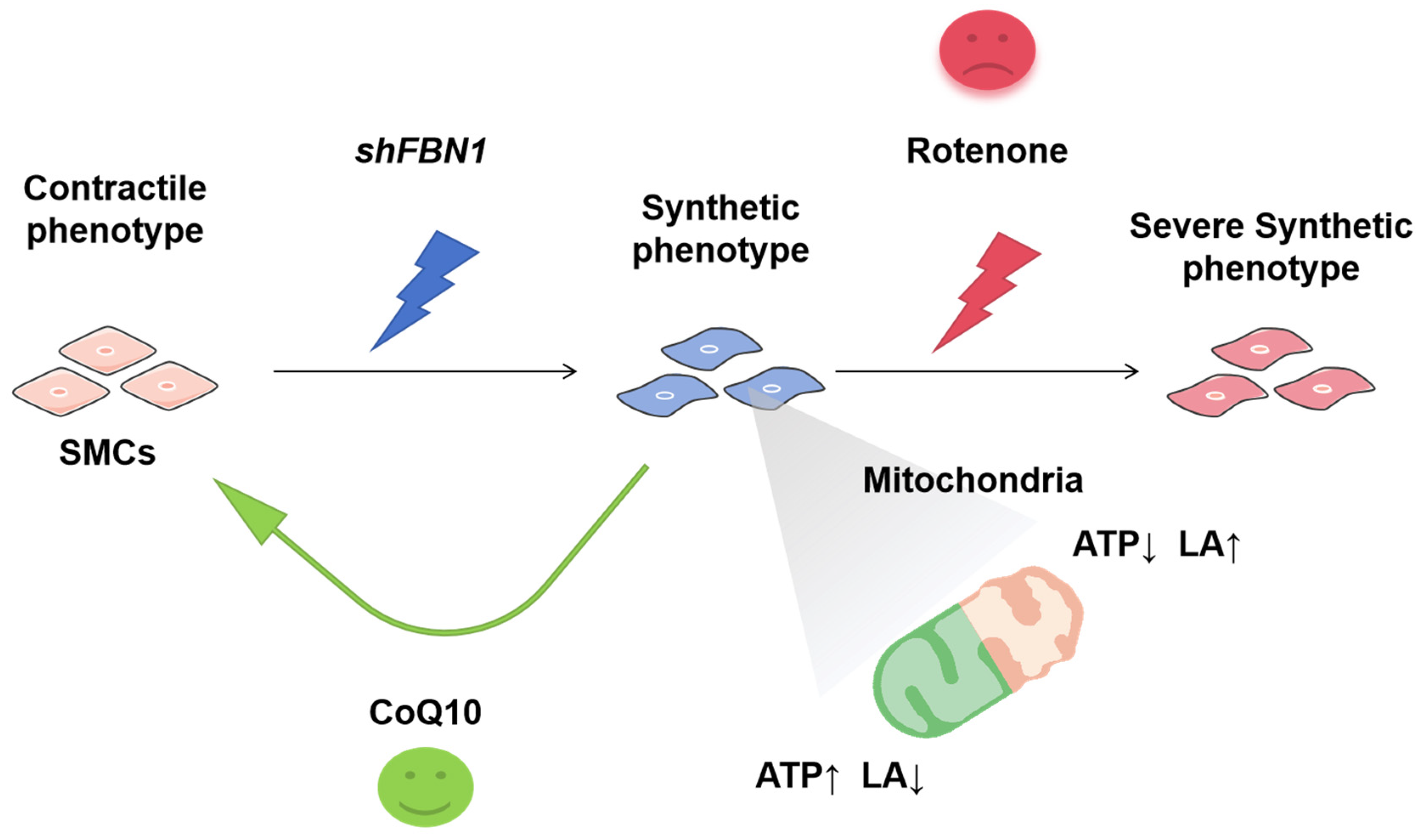
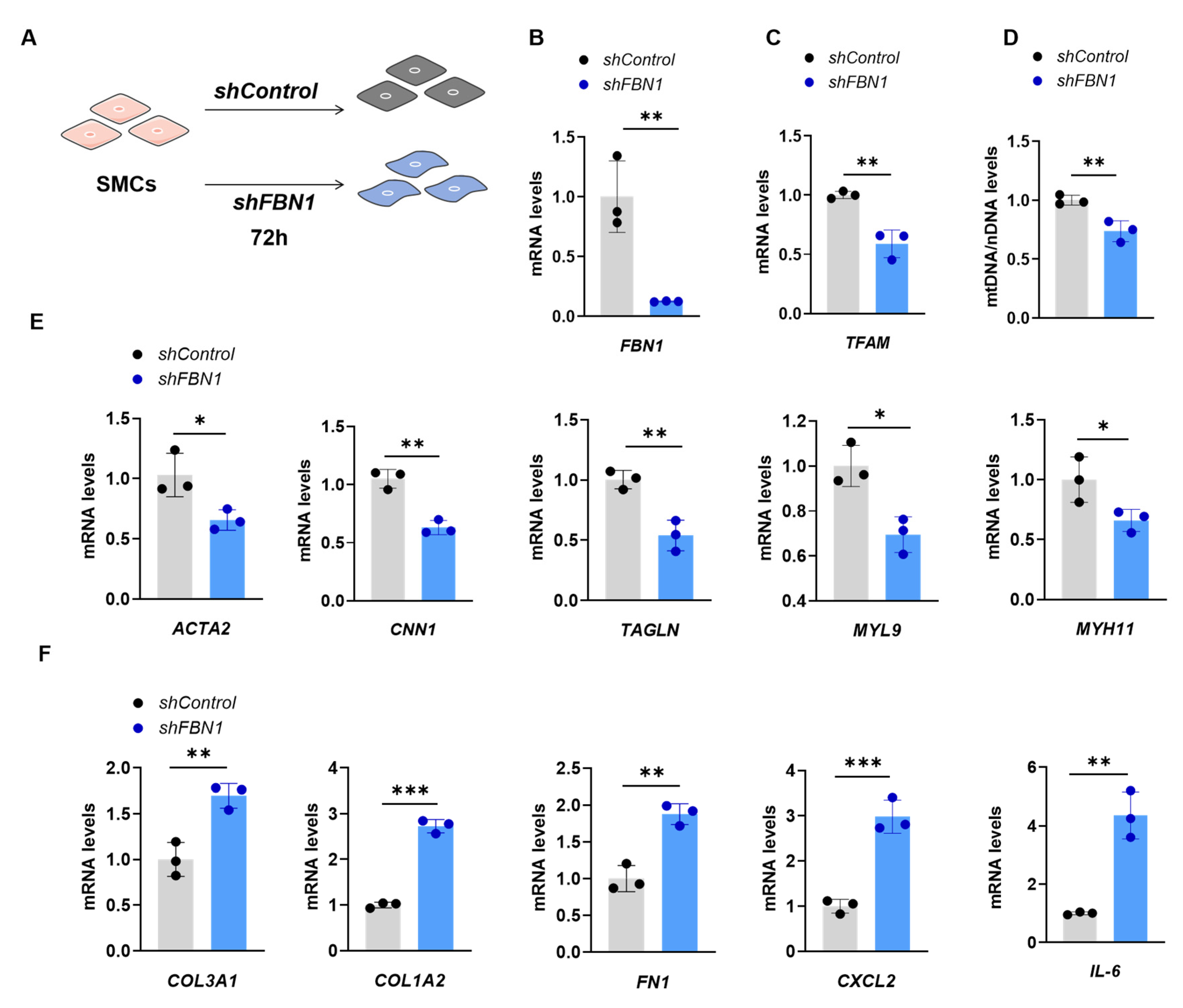
2.1. SMCs with FBN1 Knockdown Present Features of Decreased TFAM Expression, mtDNA Content and Changed Phenotypic Transformation
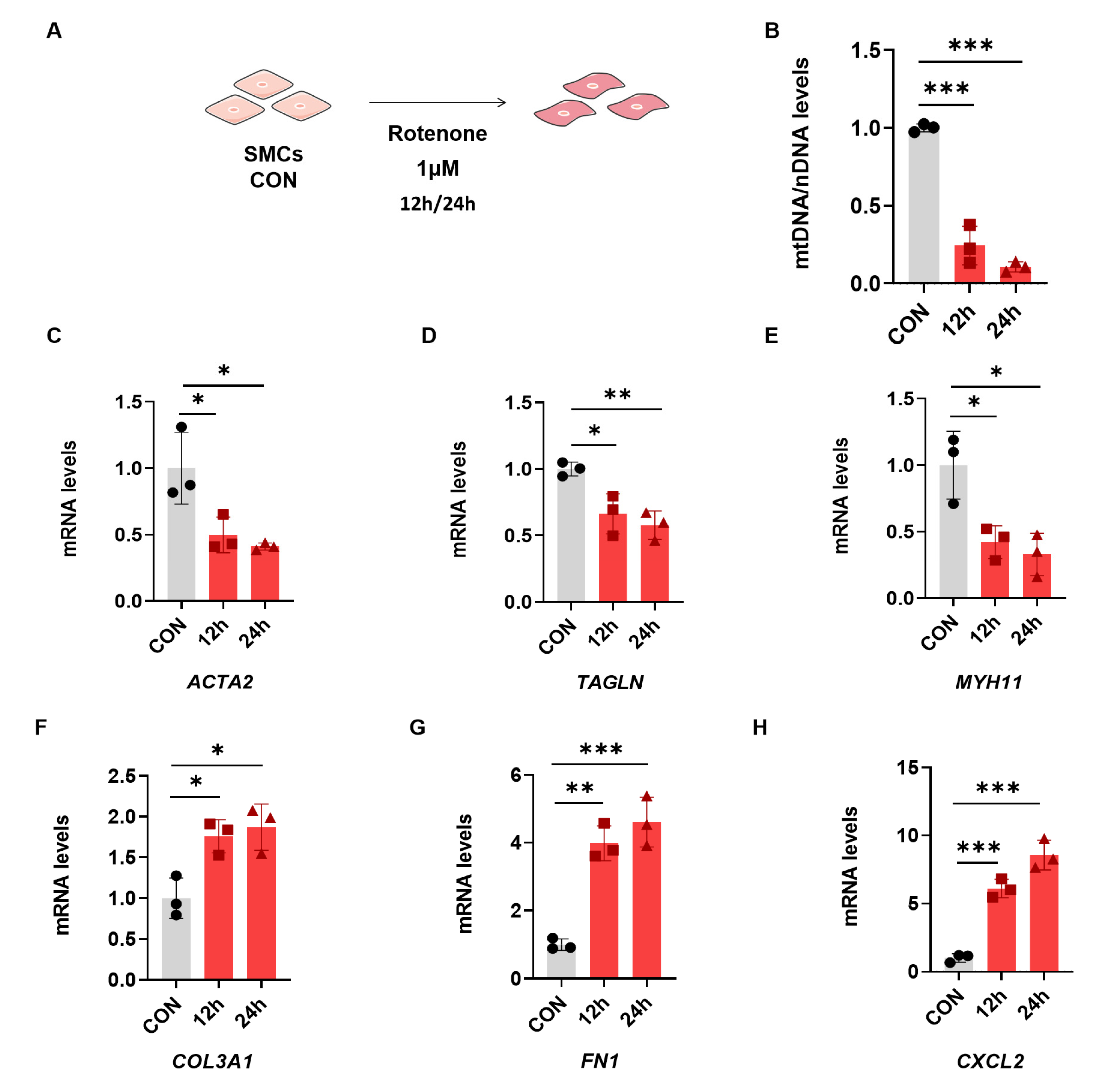
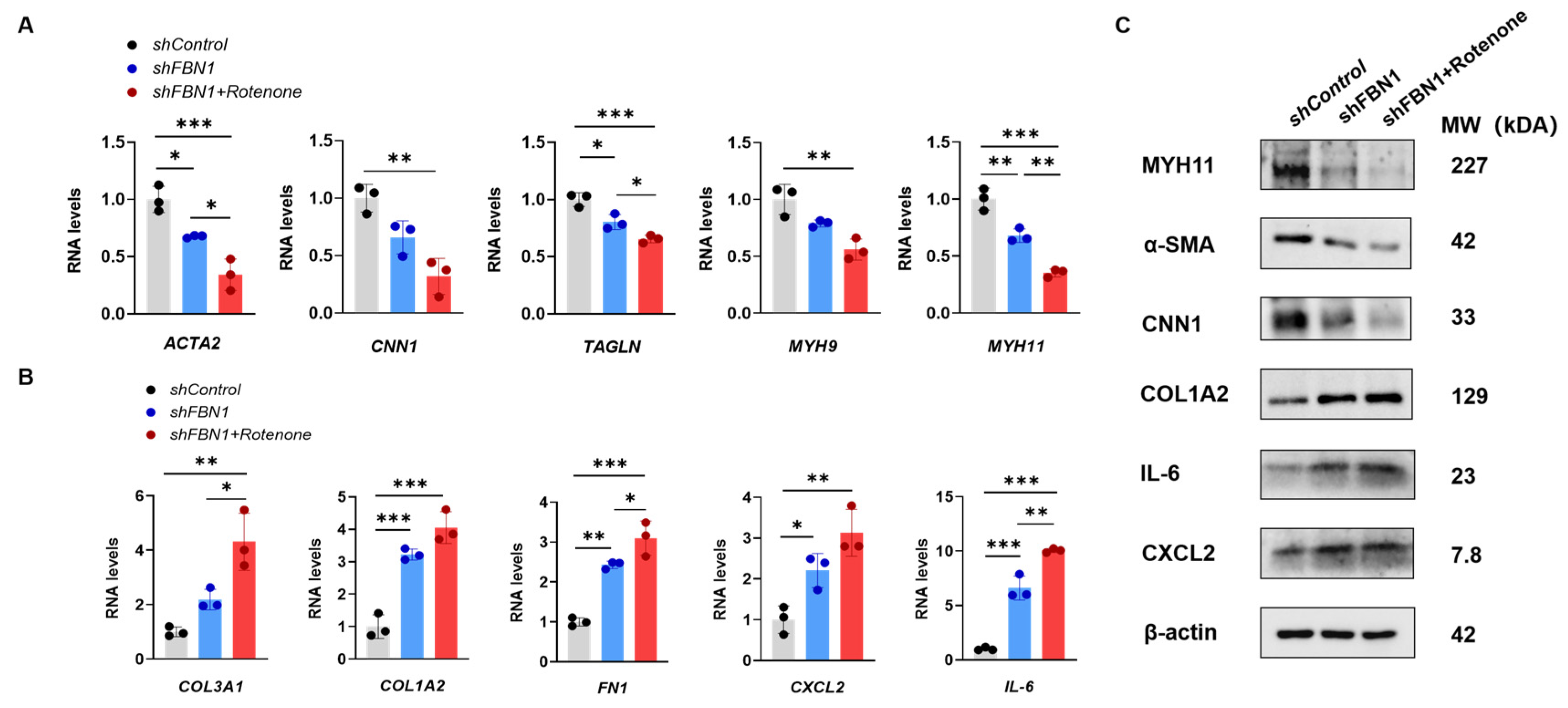
2.2. Rotenone-Induced Mitochondrial Dysfunction Elicits the Phenotypic Transformation of SMCs
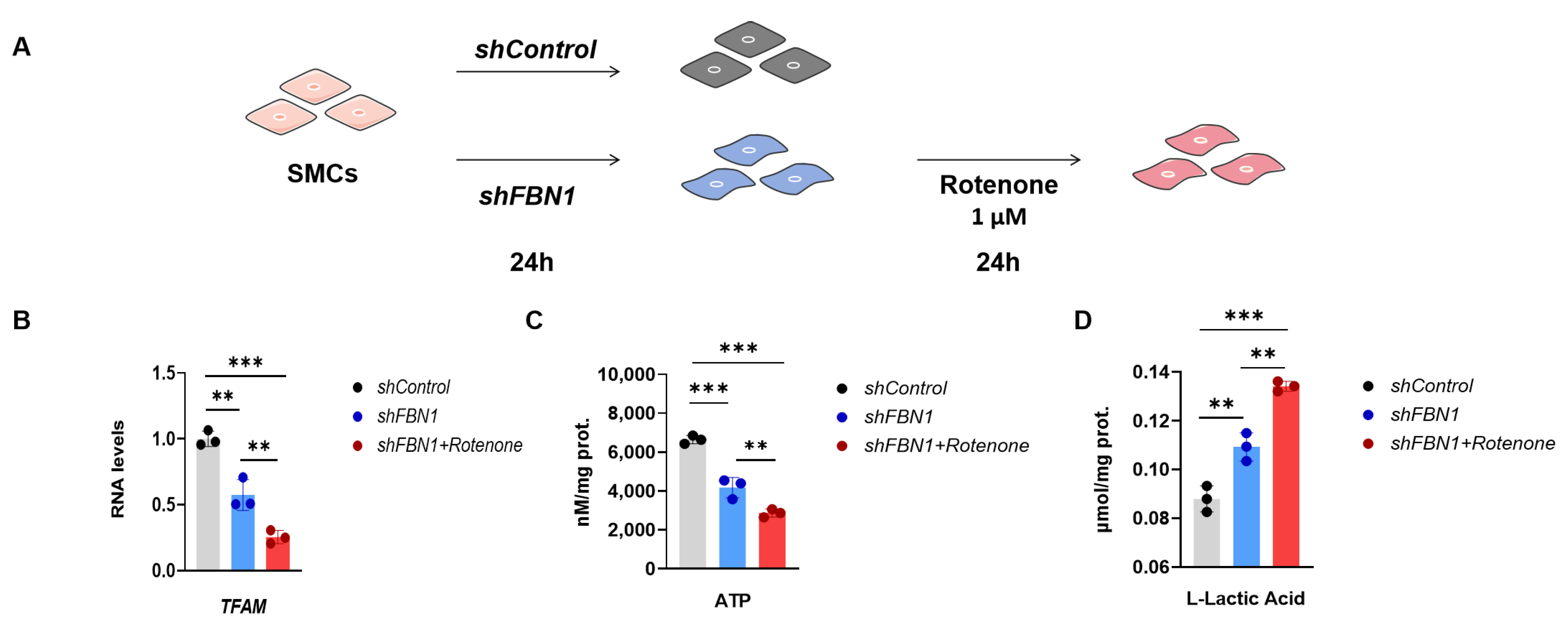
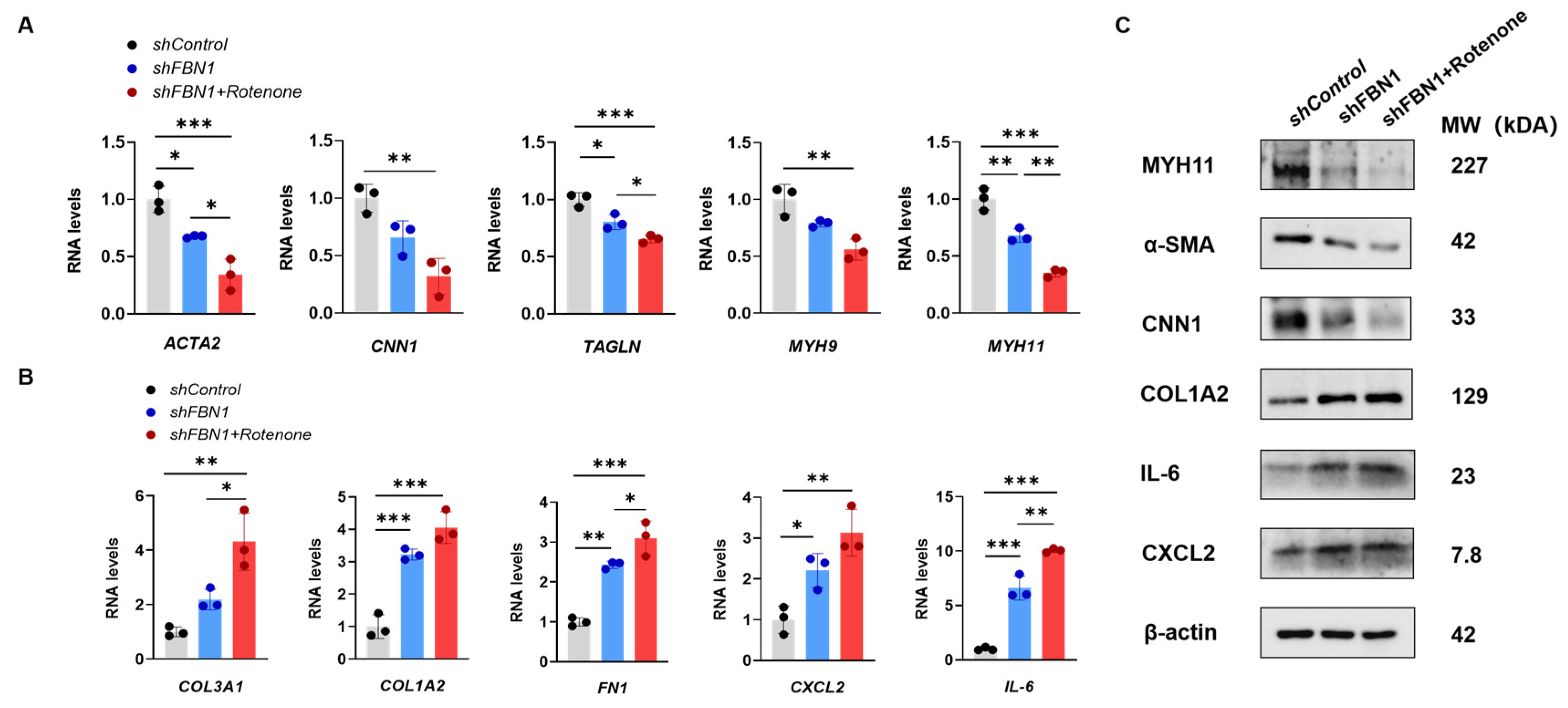
2.3. Mitochondrial Dysfunction Aggravates Synthetic Phenotype in an SMC Model of MFS
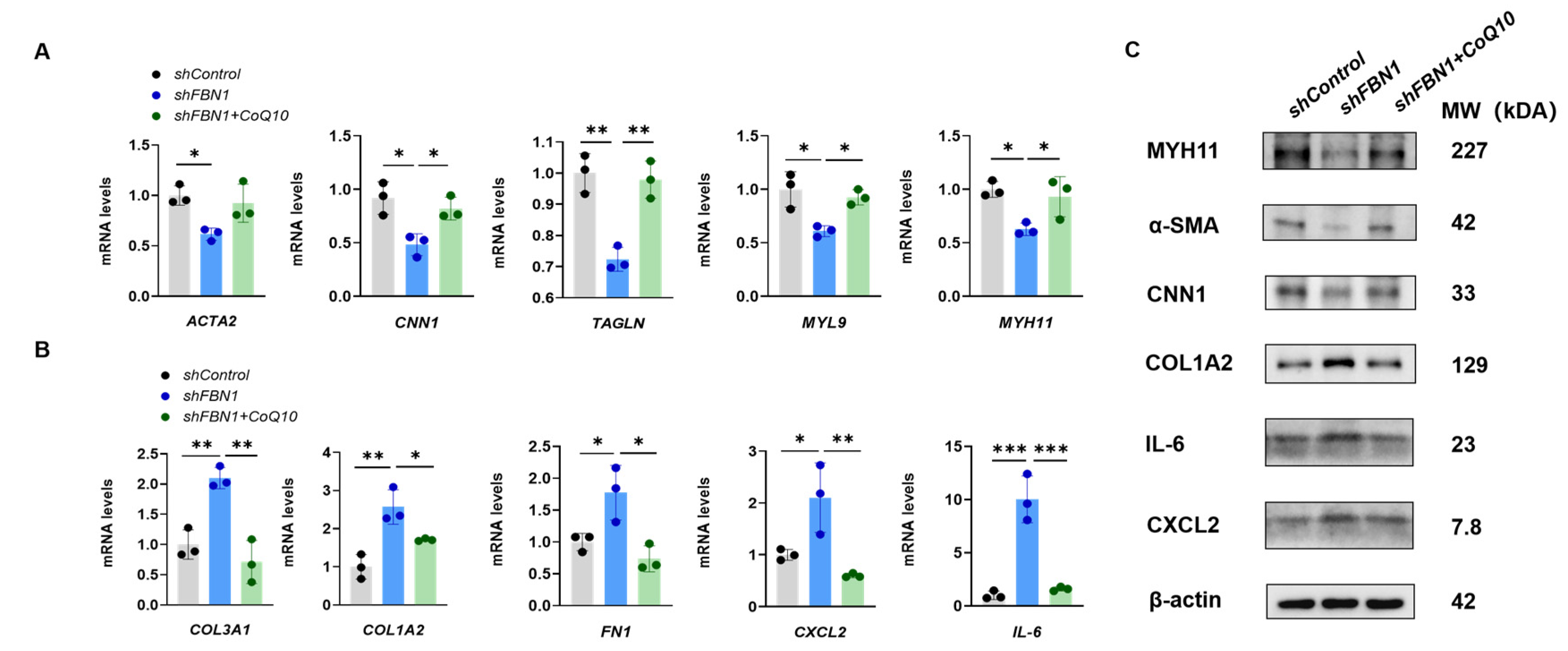
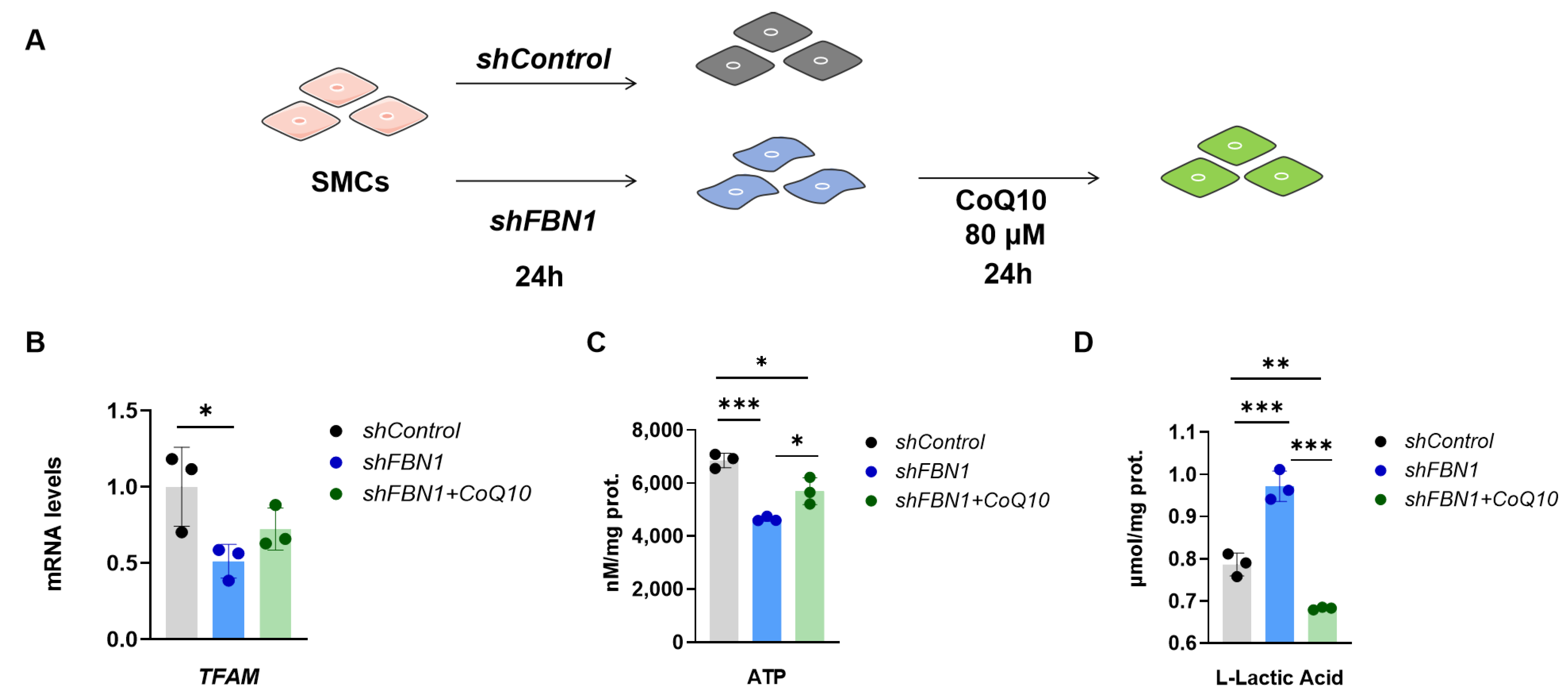
2.4. CoQ10 Reverts Phenotypic Transformation in a Cell Model of MFS
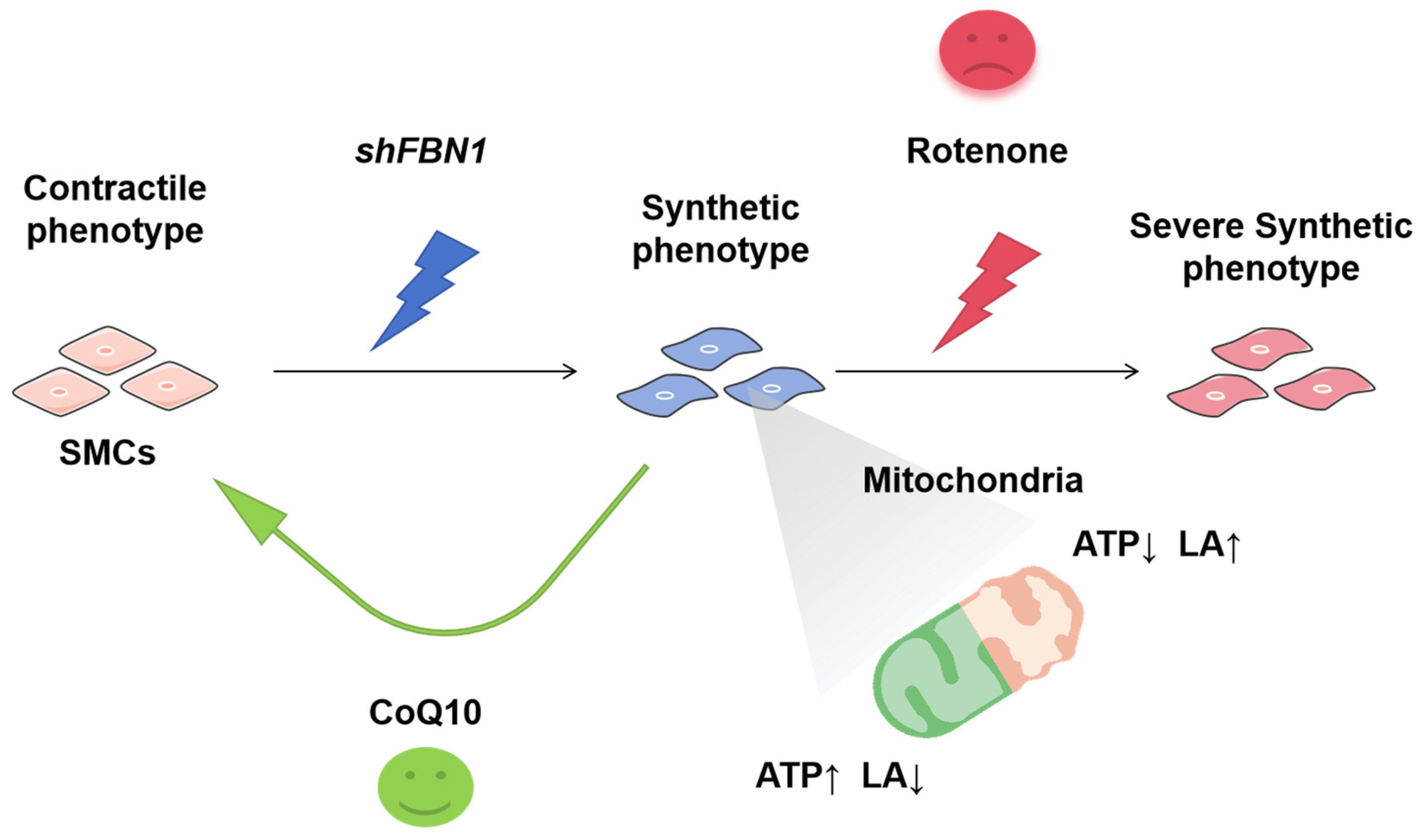
3. Discussion
4. Materials and Methods
4.1. Cell Procedures
4.2. Lentivirus Production and Infection
4.3. RNA and DNA Extraction
4.4. Real-Time PCR
4.5. mtDNA Copy Number Analysis
4.6. Mitochondrial Mass
4.7. Intracellular ATP Assay
4.8. Extracellular L-Lactic Acid Assay
4.9. Immunoblotting
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Oller, J.; Gabande-Rodriguez, E.; Ruiz-Rodriguez, M.J.; Desdin-Mico, G.; Aranda, J.F.; Rodrigues-Diez, R.; Ballesteros-Martinez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuna, P.; et al. Extracellular tuning of mitochondrial respiration leads to aortic aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- Dietz, H.C.; Cutting, G.R.; Pyeritz, R.E.; Maslen, C.L.; Sakai, L.Y.; Corson, G.M.; Puffenberger, E.G.; Hamosh, A.; Nanthakumar, E.J.; Curristin, S.M.; et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991, 352, 337–339. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cao, H.; Hu, G.; Wu, Y.; Xu, Y.; Cui, H.; Lu, H.S.; Zheng, L. The mechanism and therapy of aortic aneurysms. Signal Transduct. Target. Ther. 2023, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Lacolley, P.; Regnault, V.; Avolio, A.P. Smooth muscle cell and arterial aging: Basic and clinical aspects. Cardiovasc. Res. 2018, 114, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, X.; An, P.; Luo, J.; Luo, Y. Mitochondrial homeostasis in VSMCs as a central hub in vascular remodeling. Int. J. Mol. Sci. 2023, 24, 3483. [Google Scholar] [CrossRef]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.G.; Wu, X.C.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1-Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- He, L.; Zhou, Q.L.; Huang, Z.; Xu, J.; Zhou, H.; Lv, D.G.; Lu, L.Q.; Huang, S.F.; Tang, M.Z.; Zhong, J.C.; et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. J. Cell. Physiol. 2019, 234, 8668–8682. [Google Scholar] [CrossRef]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA Damage Can Promote Atherosclerosis Independently of Reactive Oxygen Species Through Effects on Smooth Muscle Cells and Monocytes and Correlates With Higher-Risk Plaques in Humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Luo, Y.; Luo, J.; An, P.; Zhao, Y.; Zhao, W.; Fang, Z.; Xia, Y.; Zhu, L.; Xu, T.; Zhang, X.; et al. The activator protein-1 complex governs a vascular degenerative transcriptional programme in smooth muscle cells to trigger aortic dissection and rupture. Eur. Heart J. 2023, 45, 287–305. [Google Scholar] [CrossRef]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012, 15, 545–553. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Li, N.Y.; Ragheb, K.; Lawler, G.; Sturgist, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [PubMed]
- Franken, R.; Hibender, S.; den Hartog, A.W.; Radonic, T.; de Vries, C.J.M.; Zwinderman, A.H.; Groenink, M.; Mulder, B.J.M.; de Waard, V. No Beneficial Effect of General and Specific Anti-Inflammatory Therapies on Aortic Dilatation in Marfan Mice. PLoS ONE 2014, 9, e107221. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Lee, S.Y.; Gayraud, B.; Andrikopoulos, K.; Shapiro, S.D.; Bunton, T.; Biery, N.J.; Dietz, H.C.; Sakai, L.Y.; Ramirez, F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-l. Proc. Natl. Acad. Sci. USA 1999, 96, 3819–3823. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Dhillon, P.; Huang, S.Z.; Sheng, X.; Shrestha, R.; Qiu, C.X.; Kaufman, B.A.; Park, J.; Pei, L.M.; Baur, J.; et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. 2019, 30, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease—A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010, 121, E266–E369. [Google Scholar] [CrossRef] [PubMed]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.S.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef]
- Mullen, M.; Jin, X.Y.; Child, A.; Stuart, A.G.; Dodd, M.; Aragon-Martin, J.A.; Gaze, D.; Kiotsekoglou, A.; Yuan, L.; Hu, J.T.; et al. Irbesartan in Marfan syndrome (AIMS): A double-blind, placebo-controlled randomised trial. Lancet 2019, 394, 2263–2270. [Google Scholar] [CrossRef]
- Groenink, M.; den Hartog, A.W.; Franken, R.; Radonic, T.; de Waard, V.; Timmermans, J.; Scholte, A.J.; van den Berg, M.P.; Spijkerboer, A.M.; Marquering, H.A.; et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: A randomized controlled trial. Eur. Heart J. 2013, 34, 3491–3500. [Google Scholar] [CrossRef]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Tierney, E.S.S.; Levine, J.C.; Atz, A.M.; et al. Atenolol versus Losartan in Children and Young Adults with Marfan’s Syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.H.; Wu, M.H.; Wang, J.K.; Lu, C.W.; Chiu, S.N.; Chen, C.A.; Lin, M.T.; Hu, F.C. Losartan Added to β-Blockade Therapy for Aortic Root Dilation in Marfan Syndrome: A Randomized, Open-Label Pilot Study. Mayo Clin. Proc. 2013, 88, 271–276. [Google Scholar] [CrossRef]
- Milleron, O.; Arnoult, F.; Ropers, J.; Aegerter, P.; Detaint, D.; Delorme, G.; Attias, D.; Tubach, F.; Dupuis-Girod, S.; Plauchu, H.; et al. Marfan Sartan: A randomized, double-blind, placebo-controlled trial. Eur. Heart J. 2015, 36, 2160–2166. [Google Scholar] [CrossRef]
- Milewicz, D.M.; Braverman, A.C.; De Backer, J.; Morris, S.A.; Boileau, C.; Maumenee, I.H.; Jondeau, G.; Evangelista, A.; Pyeritz, R.E.; European Reference Network Rare, M.; et al. Marfan syndrome. Nat. Rev. Dis. Primers 2021, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445. [Google Scholar] [CrossRef]
- Chen, C.; Chen, Y.; Guan, M.-X. A peep into mitochondrial disorder: Multifaceted from mitochondrial DNA mutations to nuclear gene modulation. Protein Cell 2015, 6, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.-X.; Yan, Q.; Li, X.; Bykhovskaya, Y.; Gallo-Teran, J.; Hajek, P.; Umeda, N.; Zhao, H.; Garrido, G.; Mengesha, E.; et al. Mutation in TRMU related to transfer RNA modification modulates the phenotypic expression of the deafness-associated mitochondrial 12S ribosomal RNA mutations. Am. J. Hum. Genet. 2006, 79, 291–302. [Google Scholar] [CrossRef]
- Sifuentes-Franco, S.; Carolina Sanchez-Macias, D.; Carrillo-Ibarra, S.; Jose Rivera-Valdes, J.; Zuniga, L.Y.; Aleyda Sanchez-Lopez, V. Antioxidant and anti-Inflammatory effects of coenzyme Q10 supplementation on infectious diseases. Healthcare 2022, 10, 487. [Google Scholar] [CrossRef]
- Fiorillo, C.; Becatti, M.; Attanasio, M.; Lucarini, L.; Nassi, N.; Evangelisti, L.; Porciani, M.C.; Nassi, P.; Gensini, G.F.; Abbate, R.; et al. Evidence for oxidative stress in plasma of patients with Marfan syndrome. Int. J. Cardiol. 2010, 145, 544–546. [Google Scholar] [CrossRef]
- Jiménez-Altayó, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; del Blanco, D.G.; López-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barberà, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef]
- Zhang, P.; Samuels, D.C.; Lehmann, B.; Stricker, T.; Pietenpol, J.; Shyr, Y.; Guo, Y. Mitochondria sequence mapping strategies and practicability of mitochondria variant detection from exome and RNA sequencing data. Brief. Bioinform. 2016, 17, 224–232. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef]
- Yi, Z.; Zhang, X.; Tang, W.; Yu, Y.; Wei, X.; Zhang, X.; Wei, W. Strand-selective base editing of human mitochondrial DNA using mitoBEs. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Cho, S.-I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.-S. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 2022, 185, 1764–1776. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; Kotrys, A.V.; Raguram, A.; Huang, T.P.; Mootha, V.K.; Liu, D.R. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol. 2022, 40, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Czajka, A.; Cunningham, P. Accurate quantification of mouse mitochondrial DNA without co-amplification of nuclear mitochondrial insertion sequences. Mitochondrion 2016, 29, 59–64. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhang, Z.; Wan, S.; Qi, J.; Hao, Y.; An, P.; Luo, Y.; Luo, J. Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown. Int. J. Mol. Sci. 2024, 25, 2662. https://doi.org/10.3390/ijms25052662
Zhang X, Zhang Z, Wan S, Qi J, Hao Y, An P, Luo Y, Luo J. Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown. International Journal of Molecular Sciences. 2024; 25(5):2662. https://doi.org/10.3390/ijms25052662
Chicago/Turabian StyleZhang, Xu, Zhengyang Zhang, Sitong Wan, Jingyi Qi, Yanling Hao, Peng An, Yongting Luo, and Junjie Luo. 2024. "Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown" International Journal of Molecular Sciences 25, no. 5: 2662. https://doi.org/10.3390/ijms25052662
APA StyleZhang, X., Zhang, Z., Wan, S., Qi, J., Hao, Y., An, P., Luo, Y., & Luo, J. (2024). Ameliorative Effect of Coenzyme Q10 on Phenotypic Transformation in Human Smooth Muscle Cells with FBN1 Knockdown. International Journal of Molecular Sciences, 25(5), 2662. https://doi.org/10.3390/ijms25052662