

Novel Perspectives in Chronic Kidney Disease-Specific Cardiovascular Disease
 , ,
, , 

Abstract
1. Introduction
2. Clinical Manifestations of CRS
2.1. Coronary Artery Disease
2.2. Left Ventricular Hypertrophy (LVH)
2.3. Valvular Heart Disease
2.4. Sudden Cardiac Death
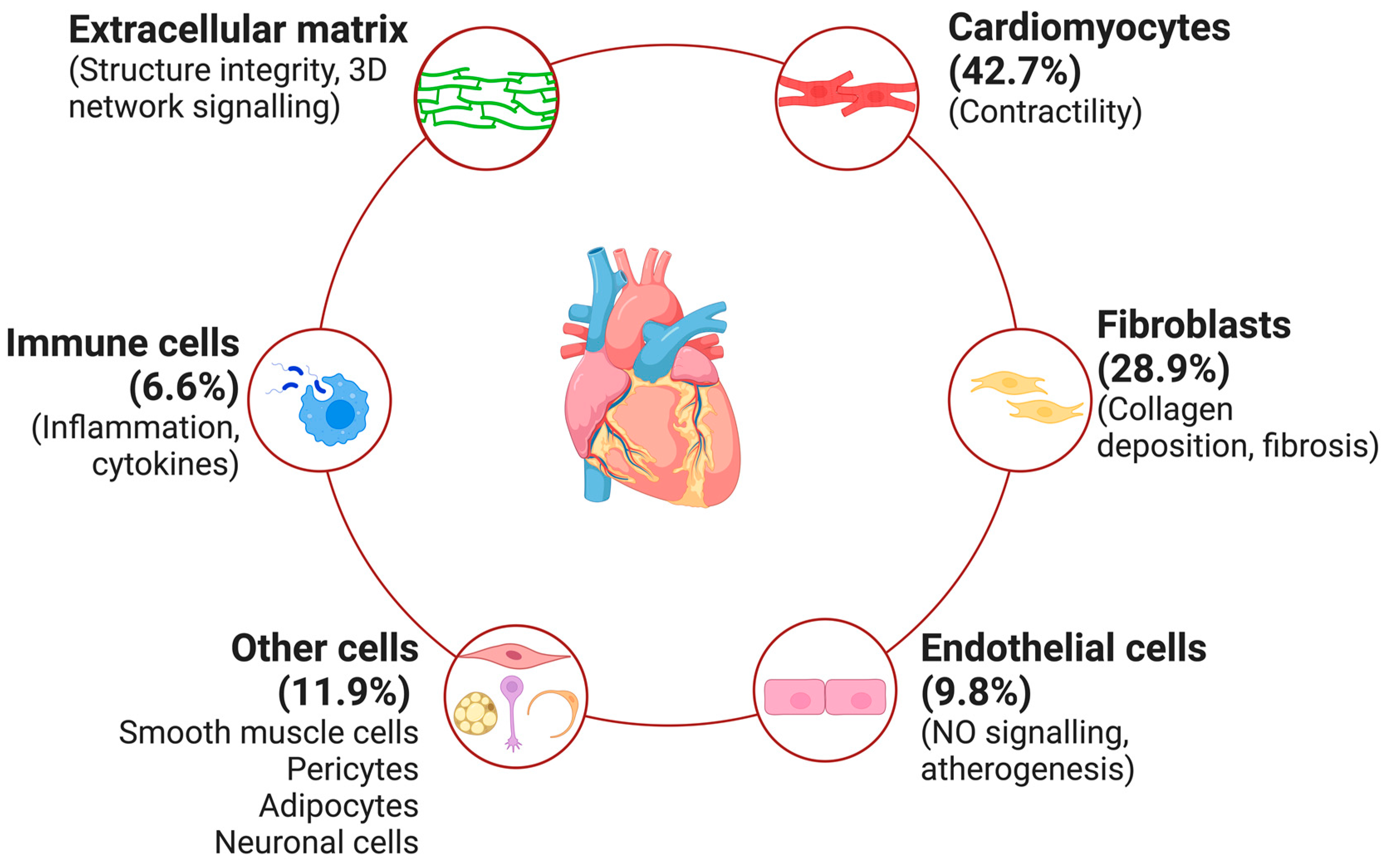
3. Molecular Mechanisms Underlying CRS
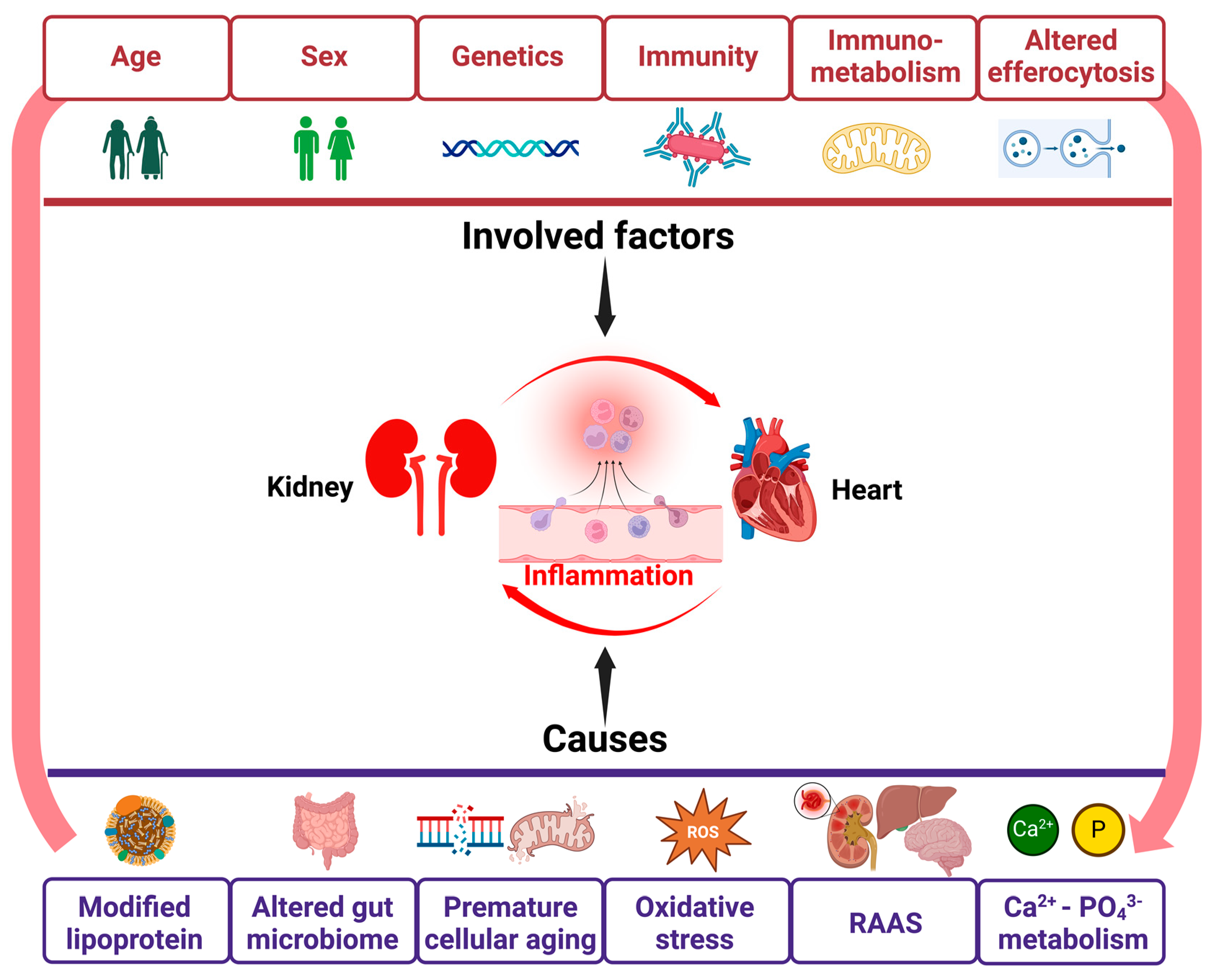
4. Inflammation in CRS (Figure 2)

4.1. Genetics of Inflammation
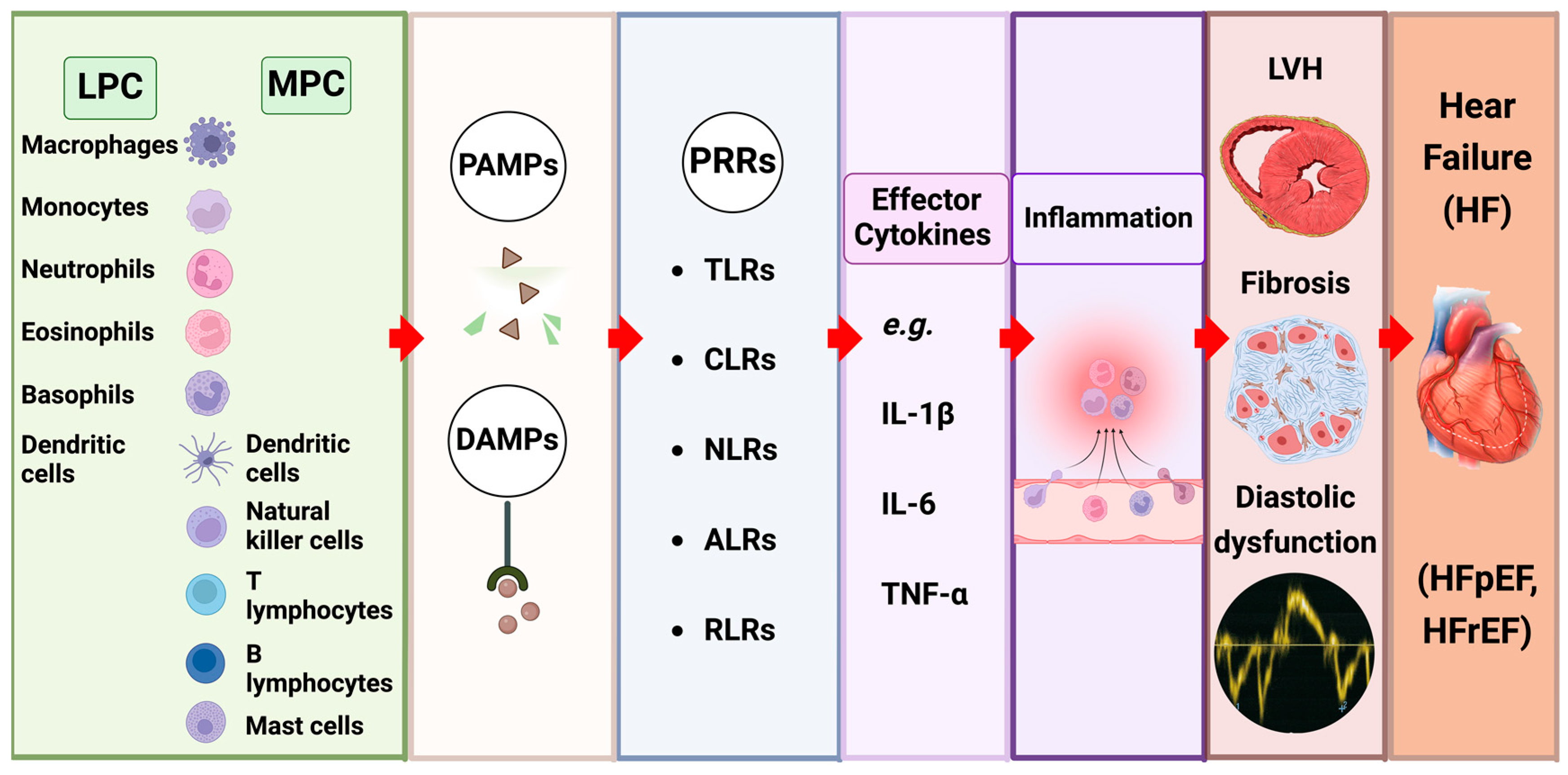
4.2. The Impact of Innate Immune Signalling (Figure 3)

4.3. Inflammatory Cytokines as Effector Molecules in CRS
4.4. Innate Immune Cells in CRS
5. Contributors to Inflammation in CRS
5.1. Modified Lipoproteins
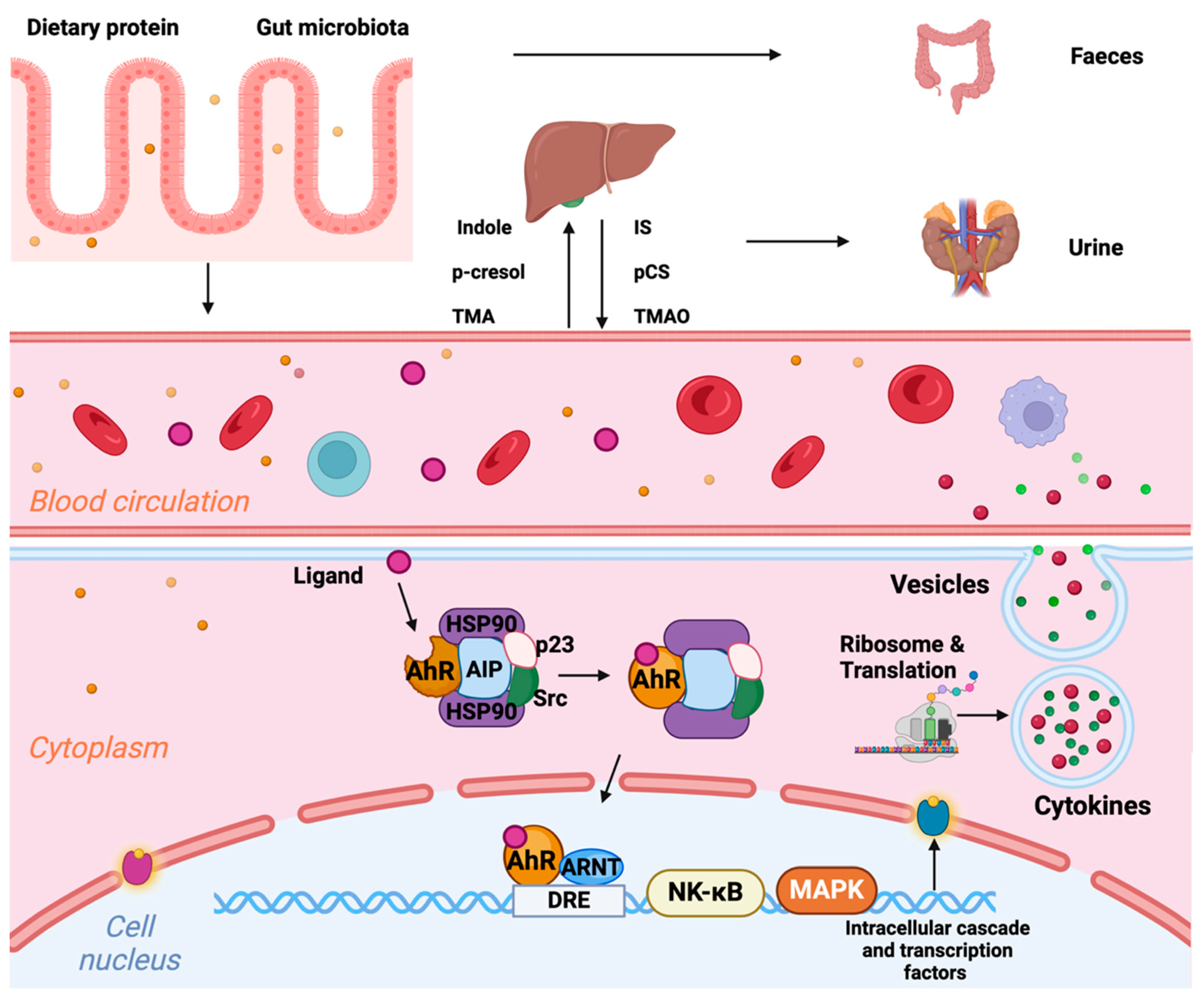
5.2. Altered Gut Microbiome and Disturbed Intestinal Barrier Function (Figure 4)

5.3. Premature Cellular Aging
5.4. Altered Bone Mineral Metabolism
6. Biomarkers in CRS (Table 1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Marker | Pathophysiological Significance | Healthy Adult Plasma Range | Relevant Pharmacological Agents (Clinical or Experimental) | References |
|---|---|---|---|---|
| Asymmetric dimethyl arginine (ADMA) |
Reduces the following:
| 0.4–1.0 µmol/L | Antioxidants | [148,153,154] |
| Estrogen | ||||
| Vitamin A | ||||
| ACEi | ||||
| ARBs | ||||
| HMG-CoA reductase inhibitors (statins) | ||||
| Endothelin-1 (ET-1) |
| 0.1–5 pg/mL | ET receptor antagonists (bosentan and ambrisentan)—not beneficial in HF | [155,156,157] |
| SGLT2i | ||||
| Syndecan 4 (SDC4) |
Part of the endothelial glycocalyx
| 5.7–16.05 ng/mL | Not tested in CV literature | [158] |
| N-terminal prohormone of brain natriuretic peptide (NT-proBNP) |
| <125 pg/mL | Nesiritide (recombinant BNP)—not beneficial in HF | [159,160,161] |
| Neprilysin—sacubitril (endopeptidase degrading ANP/BNP)—beneficial with ACEi or ARB | ||||
| High-sensitivity C-reactive protein (hs-CRP) |
| <3 mg/L | No specific drugs target hs-CRP | |
| Interluekin-6 (IL-6) |
Pro-inflammatory, stimulates the following:
| <5 pg/mL | Anti-IL-6 antibody (ziltivekimab) | [69,70,162,163] |
| Anti-IL-6R antibody (tocilizumab) | ||||
| Statins | ||||
| SGLT2i | ||||
| Tumor necrosis factor alpha (TNF-α) |
Pro-inflammatory
| <6 pg/mL | TNF-α inhibitors
| [110,163] |
| SGLT2i | ||||
| Monocyte chemotactic protein-1 (MCP-1) (also known as CCR2) |
| <150 pg/mL | MCP-1 antagonist (propagermanium), anti-CCR2 antibody (MLN1202) | [164,165] |
| Statins PPARα activators (fenofibrate) | ||||
| Most data are from pre-clinical studies | ||||
| CD47 |
Cell-surface receptor
| not measured | CD47 monoclonal antibody (magrolimab) | [166,167] |
| Signal regulatory protein alpha (SIRP-α) | Transmembrane protein, modulates leukocyte immune responses, e.g., adhesion, migration, and phagocytosis, CD47/SIRPα axis, “don’t eat me” signal | not measured | CD47-SIRPα/Fc fusion proteins
|
7. Therapeutic Prospects for CRS
Recently Established Therapies
8. Novel Treatment Approaches in CRS
9. Emerging Perspectives in CRS
Efferocytosis
10. Sex Differences in CRS
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANP | atrial natriuretic peptide |
| BNP | brain natriuretic peptide |
| CHIP | clonal haematopoiesis of indeterminate potential |
| CKD | chronic kidney disease |
| CRS | cardiorenal syndrome |
| CVD | cardiovascular disease |
| DAMP | danger-associated molecular pattern |
| HDL | high-density lipoproteins |
| HF | heart failure |
| HFpEF | heart failure with preserved ejection fraction |
| HFrEF | heart failure with reduced ejection fraction |
| hs-CRP | high-sensitivity C-reactive protein |
| IL | interleukin |
| LDL | low-density lipoproteins |
| LVH | left ventricular hypertrophy |
| NLRP3 | nucleotide-binding oligomerisation domain (NOD), leucine rich repeat (LRR)-containing protein 3 |
| MV | microvesicles |
| PAMP | pathogen-associated molecular pattern |
| PRR | pattern recognition receptors |
| RRT | renal replacement therapy |
| SASP | senescence-associated secretory phenotype |
| SGLT2 | sodium glucose co-transporter 2 |
| TLR | Toll-like receptor |
| TNF | tumour necrosis factor |
References
- Xie, Y.; Bowe, B.; Mokdad, A.H.; Xian, H.; Yan, Y.; Li, T.; Maddukuri, G.; Tsai, C.Y.; Floyd, T.; Al-Aly, Z. Analysis of the Global Burden of Disease study highlights the global, regional, and national trends of chronic kidney disease epidemiology from 1990 to 2016. Kidney Int. 2018, 94, 567–581. [Google Scholar] [CrossRef]
- Collaboration, G.B.D.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Charytan, D.M. Introduction: Cardiovascular Disease in Chronic Kidney Disease. Semin. Nephrol. 2018, 38, 541. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Henry, R.M.; Kostense, P.J.; Bos, G.; Dekker, J.M.; Nijpels, G.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D. Mild renal insufficiency is associated with increased cardiovascular mortality: The Hoorn Study. Kidney Int. 2002, 62, 1402–1407. [Google Scholar] [CrossRef]
- Chen, L.; Booley, S.; Keates, A.K.; Stewart, S. A public/private collaboration to reduce the burden of preventable hospitalisations in patients with heart failure in Tasmania. In Mary MacKillop Institute for Health Research; Australian Catholic University: Melbourne, Australia, 2017. [Google Scholar]
- Ronco, C.; McCullough, P.A.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardiorenal syndromes: An executive summary from the consensus conference of the Acute Dialysis Quality Initiative (ADQI). In Cardiorenal Syndromes in Critical Care; Ronco, C., Bellomo, R., McCullough, P.A., Eds.; Book Series: Contributions to Nephrology; Karger Publishers: Basel, Switzerland, 2010; Volume 165, pp. 54–67. [Google Scholar] [CrossRef]
- Zannad, F.; Rossignol, P. Cardiorenal Syndrome Revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, I.; Nadkarni, G.N.; Yacoub, R.; Saha, A.; Simoes, P.; Parikh, C.R.; Coca, S.G. Representation of Patients With Kidney Disease in Trials of Cardiovascular Interventions: An Updated Systematic Review. JAMA Intern. Med. 2016, 176, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement From the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.C.; Kuo, K.L.; Wu, C.C.; Tarng, D.C. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e005022. [Google Scholar] [CrossRef] [PubMed]
- Bright, R. Cases and observations illustrative of renal disease accompanied by the secretion of albuminous urine. Guys Hosp. Rep. 1836, 1, 338–400. [Google Scholar]
- National Heart, Lung, and Blood Institute. NHLBI Working Group: Cardio-Renal Connections in Heart Failure and Cardiovascular Disease. 2004. Available online: https://www.nhlbi.nih.gov/events/2004/cardio-renal-connections-heart-failure-and-cardiovascular-disease (accessed on 3 January 2024).
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-renal syndromes: Report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J. 2010, 31, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Hatamizadeh, P.; Fonarow, G.C.; Budoff, M.J.; Darabian, S.; Kovesdy, C.P.; Kalantar-Zadeh, K. Cardiorenal syndrome: Pathophysiology and potential targets for clinical management. Nat. Rev. Nephrol. 2013, 9, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, T.; Kobayashi, S.; Moriya, H.; Negishi, K.; Okamoto, K.; Maesato, K.; Saito, S. High prevalence of occult coronary artery stenosis in patients with chronic kidney disease at the initiation of renal replacement therapy: An angiographic examination. J. Am. Soc. Nephrol. 2005, 16, 1141–1148. [Google Scholar] [CrossRef]
- Tanaka, Y.; Joki, N.; Hase, H. Ischemic Heart Disease in Patients with End-Stage Kidney Disease. Blood Purif. 2015, 40, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Gillespie, I.A.; Kronenberg, F.; Richards, S.; Stenvinkel, P.; Anker, S.D.; Wheeler, D.C.; de Francisco, A.L.; Marcelli, D.; Froissart, M.; et al. High cardiovascular event rates occur within the first weeks of starting hemodialysis. Kidney Int. 2015, 88, 1117–1125. [Google Scholar] [CrossRef]
- Herzog, C.A.; Ma, J.Z.; Collins, A.J. Poor long-term survival after acute myocardial infarction among patients on long-term dialysis. N. Engl. J. Med. 1998, 339, 799–805. [Google Scholar] [CrossRef]
- Ng, C.H.; Ong, Z.H.; Sran, H.K.; Wee, T.B. Comparison of cardiovascular mortality in hemodialysis versus peritoneal dialysis. Int. Urol. Nephrol. 2021, 53, 1363–1371. [Google Scholar] [CrossRef]
- Burton, J.O.; Jefferies, H.J.; Selby, N.M.; McIntyre, C.W. Hemodialysis-induced repetitive myocardial injury results in global and segmental reduction in systolic cardiac function. Clin. J. Am. Soc. Nephrol. 2009, 4, 1925–1931. [Google Scholar] [CrossRef]
- Burton, J.O.; Jefferies, H.J.; Selby, N.M.; McIntyre, C.W. Hemodialysis-induced cardiac injury: Determinants and associated outcomes. Clin. J. Am. Soc. Nephrol. 2009, 4, 914–920. [Google Scholar] [CrossRef]
- Iwasaki, M.; Joki, N.; Tanaka, Y.; Hayashi, T.; Kubo, S.; Asakawa, T.; Matsukane, A.; Takahashi, Y.; Hirahata, K.; Imamura, Y.; et al. Declining prevalence of coronary artery disease in incident dialysis patients over the past two decades. J. Atheroscler. Thromb. 2014, 21, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.L.; Meyer, H.P.; Ziebart, H.; Rieger, P.; Wenzel, U.; Amann, K.; Berger, I.; Adamczak, M.; Schirmacher, P.; Ritz, E. Calcification of coronary intima and media: Immunohistochemistry, backscatter imaging, and x-ray analysis in renal and nonrenal patients. Clin. J. Am. Soc. Nephrol. 2007, 2, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, U.; Buzello, M.; Ritz, E.; Stein, G.; Raabe, G.; Wiest, G.; Mall, G.; Amann, K. Morphology of coronary atherosclerotic lesions in patients with end-stage renal failure. Nephrol. Dial. Transplant. 2000, 15, 218–223. [Google Scholar] [CrossRef]
- Amann, K.; Breitbach, M.; Ritz, E.; Mall, G. Myocyte/capillary mismatch in the heart of uremic patients. J. Am. Soc. Nephrol. 1998, 9, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Bansal, N.; Chandra, M.; Lathon, P.V.; Fortmann, S.P.; Iribarren, C.; Hsu, C.Y.; Hlatky, M.A.; Investigators, A.S. Chronic kidney disease and risk for presenting with acute myocardial infarction versus stable exertional angina in adults with coronary heart disease. J. Am. Coll. Cardiol. 2011, 58, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Corrao, S.; Battaglia, Y.; Andreucci, M.; Caiazza, A.; Carlomagno, A.; Lamberti, M.; Pezone, N.; Pota, A.; Russo, L.; et al. Progression of coronary artery calcification and cardiac events in patients with chronic renal disease not receiving dialysis. Kidney Int. 2011, 80, 112–118. [Google Scholar] [CrossRef]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef]
- Hillege, H.L.; Nitsch, D.; Pfeffer, M.A.; Swedberg, K.; McMurray, J.J.; Yusuf, S.; Granger, C.B.; Michelson, E.L.; Ostergren, J.; Cornel, J.H.; et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 2006, 113, 671–678. [Google Scholar] [CrossRef]
- Lofman, I.; Szummer, K.; Dahlstrom, U.; Jernberg, T.; Lund, L.H. Associations with and prognostic impact of chronic kidney disease in heart failure with preserved, mid-range, and reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 1606–1614. [Google Scholar] [CrossRef]
- Lai, W.; Zhao, X.; Yu, S.; Mai, Z.; Zhou, Y.; Huang, Z.; Li, Q.; Huang, H.; Li, H.; Wei, H.; et al. Chronic Kidney Disease Increases Risk of Incident HFrEF Following Percutaneous Coronary Intervention. Front. Cardiovasc. Med. 2022, 9, 856602. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Cherney, D.; Postmus, D.; Stefansson, B.V.; Chertow, G.M.; Dwyer, J.P.; Greene, T.; Kosiborod, M.; Langkilde, A.M.; McMurray, J.J.V.; et al. A pre-specified analysis of the Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease (DAPA-CKD) randomized controlled trial on the incidence of abrupt declines in kidney function. Kidney Int. 2022, 101, 174–184. [Google Scholar] [CrossRef]
- Wheeler, D.C.; Stefansson, B.V.; Jongs, N.; Chertow, G.M.; Greene, T.; Hou, F.F.; McMurray, J.J.V.; Correa-Rotter, R.; Rossing, P.; Toto, R.D.; et al. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: A prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. 2021, 9, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Hoevelmann, J.; Mahfoud, F.; Lauder, L.; Scheller, B.; Bohm, M.; Ewen, S. Valvular heart disease in patients with chronic kidney disease. Herz 2021, 46, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Ternacle, J.; Cote, N.; Krapf, L.; Nguyen, A.; Clavel, M.A.; Pibarot, P. Chronic Kidney Disease and the Pathophysiology of Valvular Heart Disease. Can. J. Cardiol. 2019, 35, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Saran, R.; Robinson, B.; Abbott, K.C.; Bragg-Gresham, J.; Chen, X.; Gipson, D.; Gu, H.; Hirth, R.A.; Hutton, D.; Jin, Y.; et al. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2020, 75, A6–A7. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Russell, G.B.; Satko, S.G. Sudden and cardiac death rates in hemodialysis patients. Kidney Int. 1999, 55, 1553–1559. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Hartman, J.; Brannon, P.C.; Reeves-Daniel, A.; Satko, S.G.; Russell, G. Characteristics of sudden death in hemodialysis patients. Kidney Int. 2006, 69, 2268–2273. [Google Scholar] [CrossRef]
- Wan, C.; Herzog, C.A.; Zareba, W.; Szymkiewicz, S.J. Sudden cardiac arrest in hemodialysis patients with wearable cardioverter defibrillator. Ann. Noninvasive Electrocardiol. 2014, 19, 247–257. [Google Scholar] [CrossRef]
- Sacher, F.; Jesel, L.; Borni-Duval, C.; De Precigout, V.; Lavainne, F.; Bourdenx, J.P.; Haddj-Elmrabet, A.; Seigneuric, B.; Keller, A.; Ott, J.; et al. Cardiac Rhythm Disturbances in Hemodialysis Patients: Early Detection Using an Implantable Loop Recorder and Correlation With Biological and Dialysis Parameters. JACC Clin. Electrophysiol. 2018, 4, 397–408. [Google Scholar] [CrossRef]
- Jukema, J.W.; Timal, R.J.; Rotmans, J.I.; Hensen, L.C.R.; Buiten, M.S.; de Bie, M.K.; Putter, H.; Zwinderman, A.H.; van Erven, L.; Krol-van Straaten, M.J.; et al. Prophylactic Use of Implantable Cardioverter-Defibrillators in the Prevention of Sudden Cardiac Death in Dialysis Patients. Circulation 2019, 139, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Pun, P.H.; Hellkamp, A.S.; Sanders, G.D.; Middleton, J.P.; Hammill, S.C.; Al-Khalidi, H.R.; Curtis, L.H.; Fonarow, G.C.; Al-Khatib, S.M. Primary prevention implantable cardioverter defibrillators in end-stage kidney disease patients on dialysis: A matched cohort study. Nephrol. Dial. Transplant. 2015, 30, 829–835. [Google Scholar] [CrossRef]
- Zannad, F.; Rossignol, P. Cardiovascular Outcome Trials in Patients With Advanced Kidney Disease: Time for Action. Circulation 2017, 135, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Hamzaoui, M.; Djerada, Z.; Brunel, V.; Mulder, P.; Richard, V.; Bellien, J.; Guerrot, D. 5/6 nephrectomy induces different renal, cardiac and vascular consequences in 129/Sv and C57BL/6JRj mice. Sci. Rep. 2020, 10, 1524. [Google Scholar] [CrossRef]
- Tan, R.Z.; Zhong, X.; Li, J.C.; Zhang, Y.W.; Yan, Y.; Liao, Y.; Wen, D.; Diao, H.; Wang, L.; Shen, H.C. An optimized 5/6 nephrectomy mouse model based on unilateral kidney ligation and its application in renal fibrosis research. Ren. Fail. 2019, 41, 555–566. [Google Scholar] [CrossRef]
- Wang, X.; Chaudhry, M.A.; Nie, Y.; Xie, Z.; Shapiro, J.I.; Liu, J. A Mouse 5/6th Nephrectomy Model That Induces Experimental Uremic Cardiomyopathy. J. Vis. Exp. 2017, 129, e55825. [Google Scholar]
- Decleves, A.E.; Sharma, K. Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat. Rev. Nephrol. 2014, 10, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Li, W.J.; Chen, X.M.; Nie, X.Y.; Zhang, J.; Cheng, Y.J.; Lin, X.X.; Wu, S.H. Cardiac troponin and C-reactive protein for predicting all-cause and cardiovascular mortality in patients with chronic kidney disease: A meta-analysis. Clinics 2015, 70, 301–311. [Google Scholar] [CrossRef]
- Jager, A.; van Hinsbergh, V.W.; Kostense, P.J.; Emeis, J.J.; Nijpels, G.; Dekker, J.M.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D. C-reactive protein and soluble vascular cell adhesion molecule-1 are associated with elevated urinary albumin excretion but do not explain its link with cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 593–598. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef]
- Himmelfarb, J. Linking oxidative stress and inflammation in kidney disease: Which is the chicken and which is the egg? Semin. Dial. 2004, 17, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, L.S.; Go, A.S. Epidemiology of acute infections among patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1487–1493. [Google Scholar] [CrossRef]
- Jazani, N.H.; Savoj, J.; Lustgarten, M.; Lau, W.L.; Vaziri, N.D. Impact of Gut Dysbiosis on Neurohormonal Pathways in Chronic Kidney Disease. Diseases 2019, 7, 21. [Google Scholar] [CrossRef]
- Eustace, J.A.; Astor, B.; Muntner, P.M.; Ikizler, T.A.; Coresh, J. Prevalence of acidosis and inflammation and their association with low serum albumin in chronic kidney disease. Kidney Int. 2004, 65, 1031–1040. [Google Scholar] [CrossRef]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstadt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. Proinflammatory cytokines lowering erythropoietin production. J. Interferon Cytokine Res. 1998, 18, 555–559. [Google Scholar] [CrossRef]
- Avesani, C.M.; Draibe, S.A.; Kamimura, M.A.; Colugnati, F.A.; Cuppari, L. Resting energy expenditure of chronic kidney disease patients: Influence of renal function and subclinical inflammation. Am. J. Kidney Dis. 2004, 44, 1008–1016. [Google Scholar] [CrossRef]
- Slee, A.D. Exploring metabolic dysfunction in chronic kidney disease. Nutr. Metab. 2012, 9, 36. [Google Scholar] [CrossRef]
- Fouque, D.; Kalantar-Zadeh, K.; Kopple, J.; Cano, N.; Chauveau, P.; Cuppari, L.; Franch, H.; Guarnieri, G.; Ikizler, T.A.; Kaysen, G.; et al. A proposed nomenclature and diagnostic criteria for protein-energy wasting in acute and chronic kidney disease. Kidney Int. 2008, 73, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.H.; Ronco, C.; Okusa, M.D. The role of inflammation in the cardio-renal syndrome: A focus on cytokines and inflammatory mediators. Semin. Nephrol. 2012, 32, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Linhart, C.; Ulrich, C.; Greinert, D.; Dambeck, S.; Wienke, A.; Girndt, M.; Pliquett, R.U. Systemic inflammation in acute cardiorenal syndrome: An observational pilot study. ESC Heart Fail. 2018, 5, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1beta by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Tuttle, K.R.; Perkovic, V.; Libby, P.; MacFadyen, J.G. Inflammation drives residual risk in chronic kidney disease: A CANTOS substudy. Eur. Heart J. 2022, 43, 4832–4844. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Devalaraja, M.; Fishbane, S.; Chonchol, M.; Mathur, V.S.; Smith, M.T.; Lo, L.; Herzog, K.; Kakkar, R.; Davidson, M.H. Ziltivekimab for Treatment of Anemia of Inflammation in Patients on Hemodialysis: Results from a Phase 1/2 Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial. J. Am. Soc. Nephrol. 2021, 32, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef]
- Munoz Mendoza, J.; Isakova, T.; Cai, X.; Bayes, L.Y.; Faul, C.; Scialla, J.J.; Lash, J.P.; Chen, J.; He, J.; Navaneethan, S.; et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017, 91, 711–719. [Google Scholar] [CrossRef]
- Batra, G.; Ghukasyan Lakic, T.; Lindback, J.; Held, C.; White, H.D.; Stewart, R.A.H.; Koenig, W.; Cannon, C.P.; Budaj, A.; Hagstrom, E.; et al. Interleukin 6 and Cardiovascular Outcomes in Patients With Chronic Kidney Disease and Chronic Coronary Syndrome. JAMA Cardiol. 2021, 6, 1440–1445. [Google Scholar] [CrossRef]
- Schunk, S.J.; Triem, S.; Schmit, D.; Zewinger, S.; Sarakpi, T.; Becker, E.; Hutter, G.; Wrublewsky, S.; Kuting, F.; Hohl, M.; et al. Interleukin-1alpha Is a Central Regulator of Leukocyte-Endothelial Adhesion in Myocardial Infarction and in Chronic Kidney Disease. Circulation 2021, 144, 893–908. [Google Scholar] [CrossRef]
- Emerging Risk Factors Collaboration; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar] [CrossRef]
- Elliott, P.; Chambers, J.C.; Zhang, W.; Clarke, R.; Hopewell, J.C.; Peden, J.F.; Erdmann, J.; Braund, P.; Engert, J.C.; Bennett, D.; et al. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA 2009, 302, 37–48. [Google Scholar] [CrossRef]
- Said, S.; Pazoki, R.; Karhunen, V.; Vosa, U.; Ligthart, S.; Bodinier, B.; Koskeridis, F.; Welsh, P.; Alizadeh, B.Z.; Chasman, D.I.; et al. Genetic analysis of over half a million people characterises C-reactive protein loci. Nat. Commun. 2022, 13, 2198. [Google Scholar] [CrossRef]
- IL6R Genetics Consortium Emerging Risk Factors Collaboration; Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; et al. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar] [CrossRef]
- Schunk, S.J.; Kleber, M.E.; Marz, W.; Pang, S.; Zewinger, S.; Triem, S.; Ege, P.; Reichert, M.C.; Krawczyk, M.; Weber, S.N.; et al. Genetically determined NLRP3 inflammasome activation associates with systemic inflammation and cardiovascular mortality. Eur. Heart J. 2021, 42, 1742–1756. [Google Scholar] [CrossRef]
- Spoto, B.; Mattace-Raso, F.; Sijbrands, E.; Leonardis, D.; Testa, A.; Pisano, A.; Pizzini, P.; Cutrupi, S.; Parlongo, R.M.; D’Arrigo, G.; et al. Association of IL-6 and a functional polymorphism in the IL-6 gene with cardiovascular events in patients with CKD. Clin. J. Am. Soc. Nephrol. 2015, 10, 232–240. [Google Scholar] [CrossRef]
- Susilo, H.; Thaha, M.; Pikir, B.S.; Alsagaff, M.Y.; Suryantoro, S.D.; Wungu, C.D.K.; Pratama, N.R.; Pakpahan, C.; Oceandy, D. The Role of Plasma Interleukin-6 Levels on Atherosclerotic Cardiovascular Disease and Cardiovascular Mortality Risk Scores in Javanese Patients with Chronic Kidney Disease. J. Pers. Med. 2022, 12, 1122. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zekavat, S.M.; Honigberg, M.C.; Natarajan, P. Genetic IL-6 Signaling Modifies Incident Coronary Artery Disease Risk in Chronic Kidney Disease. J. Am. Coll. Cardiol. 2022, 79, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.; Valente, M.J.; Coimbra, S.; Catarino, C.; Rocha-Pereira, P.; Oliveira, J.G.; Madureira, J.; Fernandes, J.C.; do Sameiro-Faria, M.; Miranda, V.; et al. Interleukin 6 (rs1800795) and pentraxin 3 (rs2305619) polymorphisms-association with inflammation and all-cause mortality in end-stage-renal disease patients on dialysis. Sci. Rep. 2021, 11, 14768. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, J.B.; Hung, A.M.; Lovett, D.H.; Sen, S.; Quershy, O.; Johansen, K.L. Interleukin-1 gene cluster polymorphisms predict risk of ESRD. Kidney Int. 2005, 68, 278–284. [Google Scholar] [CrossRef][Green Version]
- Horibe, H.; Fujimaki, T.; Oguri, M.; Kato, K.; Matsuoka, R.; Abe, S.; Tokoro, F.; Arai, M.; Noda, T.; Watanabe, S.; et al. Association of a polymorphism of the interleukin 6 receptor gene with chronic kidney disease in Japanese individuals. Nephrology (Carlton) 2015, 20, 273–278. [Google Scholar] [CrossRef]
- Ridker, P.M.; Bhatt, D.L.; Pradhan, A.D.; Glynn, R.J.; MacFadyen, J.G.; Nissen, S.E.; Prominent, R.-I.; Investigators, S. Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: A collaborative analysis of three randomised trials. Lancet 2023, 401, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Schumann, T.; Fliser, D.; Speer, T. Innate immunity in CKD-associated vascular diseases. Nephrol. Dial. Transplant. 2016, 31, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, M.M.L.; Trentin-Sonoda, M.; Panico, K.; Schleier, Y.; Duque, T.; Moreno-Loaiza, O.; de Yurre, A.R.; Ferreira, F.; Caio-Silva, W.; Coury, P.R.; et al. Cardiac arrhythmias after renal I/R depend on IL-1beta. J. Mol. Cell Cardiol. 2019, 131, 101–111. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef]
- Monnerat, G.; Alarcon, M.L.; Vasconcellos, L.R.; Hochman-Mendez, C.; Brasil, G.; Bassani, R.A.; Casis, O.; Malan, D.; Travassos, L.H.; Sepulveda, M.; et al. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016, 7, 13344. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Vogel, P.; Johnson, G.R.; Kelliher, M.A.; Iwakura, Y.; Lamkanfi, M.; Kanneganti, T.D. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature 2013, 498, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Lee, Y.; Kim, E.; Kwak, A.; Ryoo, S.; Bae, S.H.; Azam, T.; Kim, S.; Dinarello, C.A. The Interleukin-1alpha Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front. Immunol. 2013, 4, 391. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Shayakhmetov, D.M. Interleukin 1alpha and the inflammatory process. Nat. Immunol. 2016, 17, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Luthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1alpha. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef]
- Burzynski, L.C.; Humphry, M.; Pyrillou, K.; Wiggins, K.A.; Chan, J.N.E.; Figg, N.; Kitt, L.L.; Summers, C.; Tatham, K.C.; Martin, P.B.; et al. The Coagulation and Immune Systems Are Directly Linked through the Activation of Interleukin-1alpha by Thrombin. Immunity 2019, 50, 1033–1042.e1036. [Google Scholar] [CrossRef] [PubMed]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130. [Google Scholar] [CrossRef]
- Dutta, P.; Sager, H.B.; Stengel, K.R.; Naxerova, K.; Courties, G.; Saez, B.; Silberstein, L.; Heidt, T.; Sebas, M.; Sun, Y.; et al. Myocardial Infarction Activates CCR2+ Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2015, 16, 477–487. [Google Scholar] [CrossRef]
- Nabi, H.; Kivimaki, M.; Batty, G.D.; Shipley, M.J.; Britton, A.; Brunner, E.J.; Vahtera, J.; Lemogne, C.; Elbaz, A.; Singh-Manoux, A. Increased risk of coronary heart disease among individuals reporting adverse impact of stress on their health: The Whitehall II prospective cohort study. Eur. Heart J. 2013, 34, 2697–2705. [Google Scholar] [CrossRef]
- Barrett, T.J.; Corr, E.M.; van Solingen, C.; Schlamp, F.; Brown, E.J.; Koelwyn, G.J.; Lee, A.H.; Shanley, L.C.; Spruill, T.M.; Bozal, F.; et al. Chronic stress primes innate immune responses in mice and humans. Cell Rep. 2021, 36, 109595. [Google Scholar] [CrossRef]
- Christ, A.; Gunther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Bassler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell 2018, 172, 162–175.e114. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Rev. Nephrol. 2014, 10, 625–643. [Google Scholar] [CrossRef]
- Muller, D.N.; Shagdarsuren, E.; Park, J.K.; Dechend, R.; Mervaala, E.; Hampich, F.; Fiebeler, A.; Ju, X.; Finckenberg, P.; Theuer, J.; et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am. J. Pathol. 2002, 161, 1679–1693. [Google Scholar] [CrossRef]
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and cytokine receptors in advanced heart failure: An analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T.; ATTACH Investigators. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [PubMed]
- Anker, S.D.; Coats, A.J. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int. J. Cardiol. 2002, 86, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Smolgovsky, S.; Bayer, A.L.; Kaur, K.; Sanders, E.; Aronovitz, M.; Filipp, M.E.; Thorp, E.B.; Schiattarella, G.G.; Hill, J.A.; Blanton, R.M.; et al. Impaired T cell IRE1alpha/XBP1 signaling directs inflammation in experimental heart failure with preserved ejection fraction. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Jobin, K.; Muller, D.N.; Jantsch, J.; Kurts, C. Sodium and its manifold impact on our immune system. Trends Immunol. 2021, 42, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef]
- Madhur, M.S.; Lob, H.E.; McCann, L.A.; Iwakura, Y.; Blinder, Y.; Guzik, T.J.; Harrison, D.G. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 2010, 55, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chen, T.W.; Lu, W.L.; Liang, S.S.; Huang, H.D.; Tseng, C.P.; Tarng, D.C. Synbiotics Alleviate the Gut Indole Load and Dysbiosis in Chronic Kidney Disease. Cells 2021, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Speer, T.; Ridker, P.M.; von Eckardstein, A.; Schunk, S.J.; Fliser, D. Lipoproteins in chronic kidney disease: From bench to bedside. Eur. Heart J. 2021, 42, 2170–2185. [Google Scholar] [CrossRef]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2020, 21, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Schunk, S.J.; Hermann, J.; Sarakpi, T.; Triem, S.; Lellig, M.; Hahm, E.; Zewinger, S.; Schmit, D.; Becker, E.; Mollmann, J.; et al. Guanidinylated Apolipoprotein C3 (ApoC3) Associates with Kidney and Vascular Injury. J. Am. Soc. Nephrol. 2021, 32, 3146–3160. [Google Scholar] [CrossRef] [PubMed]
- Zewinger, S.; Speer, T.; Kleber, M.E.; Scharnagl, H.; Woitas, R.; Lepper, P.M.; Pfahler, K.; Seiler, S.; Heine, G.H.; Marz, W.; et al. HDL cholesterol is not associated with lower mortality in patients with kidney dysfunction. J. Am. Soc. Nephrol. 2014, 25, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Speer, T.; Colin, S.; Charakida, M.; Zewinger, S.; Staels, B.; Chinetti-Gbaguidi, G.; Hettrich, I.; Rohrer, L.; O’Neill, F.; et al. HDL in children with CKD promotes endothelial dysfunction and an abnormal vascular phenotype. J. Am. Soc. Nephrol. 2014, 25, 2658–2668. [Google Scholar] [CrossRef]
- Jovanovich, A.; Isakova, T.; Stubbs, J. Microbiome and Cardiovascular Disease in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 1598–1604. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Julovi, S.M.; Trinh, K.; Robertson, H.; Xu, C.; Minhas, N.; Viswanathan, S.; Patrick, E.; Horowitz, J.D.; Meijles, D.N.; Rogers, N.M. Thrombospondin-1 drives cardiac remodelling in chronic kidney disease. JACC Basic. Transl. Sci. 2024. preprint. [Google Scholar] [CrossRef]
- Docherty, M.H.; O’Sullivan, E.D.; Bonventre, J.V.; Ferenbach, D.A. Cellular Senescence in the Kidney. J. Am. Soc. Nephrol. 2019, 30, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.L.; Yiu, W.H.; Chan, K.W.; Li, Y.; Li, B.; Lok, S.W.Y.; Taketo, M.M.; Igarashi, P.; Chan, L.Y.Y.; Leung, J.C.K.; et al. Activated renal tubular Wnt/beta-catenin signaling triggers renal inflammation during overload proteinuria. Kidney Int. 2018, 93, 1367–1383. [Google Scholar] [CrossRef]
- Luo, C.; Zhou, S.; Zhou, Z.; Liu, Y.; Yang, L.; Liu, J.; Zhang, Y.; Li, H.; Liu, Y.; Hou, F.F.; et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2018, 29, 1238–1256. [Google Scholar] [CrossRef]
- Ueda, S.; Tominaga, T.; Ochi, A.; Sakurai, A.; Nishimura, K.; Shibata, E.; Wakino, S.; Tamaki, M.; Nagai, K. TGF-beta1 is involved in senescence-related pathways in glomerular endothelial cells via p16 translocation and p21 induction. Sci. Rep. 2021, 11, 21643. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.H.; Du, J. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef]
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G.; et al. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef]
- Yamada, S.; Tokumoto, M.; Tatsumoto, N.; Taniguchi, M.; Noguchi, H.; Nakano, T.; Masutani, K.; Ooboshi, H.; Tsuruya, K.; Kitazono, T. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am. J. Physiol. Renal Physiol. 2014, 306, F1418–F1428. [Google Scholar] [CrossRef]
- Yamada, S.; Tsuruya, K.; Kitazono, T.; Nakano, T. Emerging cross-talks between chronic kidney disease-mineral and bone disorder (CKD-MBD) and malnutrition-inflammation complex syndrome (MICS) in patients receiving dialysis. Clin. Exp. Nephrol. 2022, 26, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, J.; Wolf, M. Regulation and Effects of FGF23 in Chronic Kidney Disease. Annu. Rev. Physiol. 2020, 82, 365–390. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Yang, B.; Zhou, C.; Dai, B.; Liu, Y.; Mao, Z.; Yu, S.; Mei, C. Fibroblast Growth Factor 23 Predicts All-Cause Mortality in a Dose-Response Fashion in Pre-Dialysis Patients with Chronic Kidney Disease. Am. J. Nephrol. 2017, 45, 149–159. [Google Scholar] [CrossRef]
- Drechsler, C.; Pilz, S.; Obermayer-Pietsch, B.; Verduijn, M.; Tomaschitz, A.; Krane, V.; Espe, K.; Dekker, F.; Brandenburg, V.; Marz, W.; et al. Vitamin D deficiency is associated with sudden cardiac death, combined cardiovascular events, and mortality in haemodialysis patients. Eur. Heart J. 2010, 31, 2253–2261. [Google Scholar] [CrossRef]
- Baylis, C. Nitric oxide deficiency in chronic kidney disease. Am. J. Physiol. Renal Physiol. 2008, 294, F1–F9. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.C.; Banchs, J.E.; Celaj, S.; Talreja, A.; Lachmann, J.; Malla, S.; DuBois, N.B.; Ashton, A.W.; Latif, F.; Jorde, U.P.; et al. Endothelial cell activation in patients with decompensated heart failure. Circulation 2005, 111, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Suda, O.; Tsutsui, M.; Morishita, T.; Tasaki, H.; Ueno, S.; Nakata, S.; Tsujimoto, T.; Toyohira, Y.; Hayashida, Y.; Sasaguri, Y.; et al. Asymmetric dimethylarginine produces vascular lesions in endothelial nitric oxide synthase-deficient mice: Involvement of renin-angiotensin system and oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar]
- Zoccali, C.; Benedetto, F.A.; Maas, R.; Mallamaci, F.; Tripepi, G.; Salvatore Malatino, L.; Boger, R. Asymmetric dimethylarginine, C-reactive protein, and carotid intima-media thickness in end-stage renal disease. J. Am. Soc. Nephrol. 2002, 13, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Mallamaci, F.; Maas, R.; Benedetto, F.A.; Tripepi, G.; Malatino, L.S.; Cataliotti, A.; Bellanuova, I.; Boger, R.; Investigators, C. Left ventricular hypertrophy, cardiac remodeling and asymmetric dimethylarginine (ADMA) in hemodialysis patients. Kidney Int. 2002, 62, 339–345. [Google Scholar] [CrossRef]
- Zoccali, C.; Bode-Boger, S.; Mallamaci, F.; Benedetto, F.; Tripepi, G.; Malatino, L.; Cataliotti, A.; Bellanuova, I.; Fermo, I.; Frolich, J.; et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: A prospective study. Lancet 2001, 358, 2113–2117. [Google Scholar] [CrossRef]
- Trocha, M.; Szuba, A.; Merwid-Lad, A.; Sozanski, T. Effect of selected drugs on plasma asymmetric dimethylarginine (ADMA) levels. Pharmazie 2010, 65, 562–571. [Google Scholar]
- Maas, R. Pharmacotherapies and their influence on asymmetric dimethylargine (ADMA). Vasc. Med. 2005, 10 (Suppl. 1), S49–S57. [Google Scholar] [CrossRef]
- Yeoh, S.E.; Docherty, K.F.; Campbell, R.T.; Jhund, P.S.; Hammarstedt, A.; Heerspink, H.J.L.; Jarolim, P.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; et al. Endothelin-1, Outcomes in Patients With Heart Failure and Reduced Ejection Fraction, and Effects of Dapagliflozin: Findings From DAPA-HF. Circulation 2023, 147, 1670–1683. [Google Scholar] [CrossRef]
- Packer, M.; McMurray, J.J.V.; Krum, H.; Kiowski, W.; Massie, B.M.; Caspi, A.; Pratt, C.M.; Petrie, M.C.; DeMets, D.; Kobrin, I.; et al. Long-Term Effect of Endothelin Receptor Antagonism With Bosentan on the Morbidity and Mortality of Patients With Severe Chronic Heart Failure: Primary Results of the ENABLE Trials. JACC Heart Fail. 2017, 5, 317–326. [Google Scholar] [CrossRef]
- Bohm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.; Bollmann, M. Soluble syndecans: Biomarkers for diseases and therapeutic options. Br. J. Pharmacol. 2019, 176, 67–81. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Starling, R.C.; Hernandez, A.F.; Armstrong, P.W.; Dickstein, K.; Hasselblad, V.; Heizer, G.M.; Komajda, M.; Massie, B.M.; McMurray, J.J.; et al. Effect of nesiritide in patients with acute decompensated heart failure. N. Engl. J. Med. 2011, 365, 32–43. [Google Scholar] [CrossRef]
- Loppnow, H.; Zhang, L.; Buerke, M.; Lautenschlager, M.; Chen, L.; Frister, A.; Schlitt, A.; Luther, T.; Song, N.; Hofmann, B.; et al. Statins potently reduce the cytokine-mediated IL-6 release in SMC/MNC cocultures. J. Cell Mol. Med. 2011, 15, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Perco, P.; Mulder, S.; Leierer, J.; Hansen, M.K.; Heinzel, A.; Mayer, G. Canagliflozin reduces inflammation and fibrosis biomarkers: A potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia 2019, 62, 1154–1166. [Google Scholar] [CrossRef]
- Gilbert, J.; Lekstrom-Himes, J.; Donaldson, D.; Lee, Y.; Hu, M.; Xu, J.; Wyant, T.; Davidson, M.; Group, M.L.N.S. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 2011, 107, 906–911. [Google Scholar] [CrossRef]
- Pasceri, V.; Cheng, J.S.; Willerson, J.T.; Yeh, E.T. Modulation of C-reactive protein-mediated monocyte chemoattractant protein-1 induction in human endothelial cells by anti-atherosclerosis drugs. Circulation 2001, 103, 2531–2534. [Google Scholar] [CrossRef]
- Kojima, Y.; Volkmer, J.P.; McKenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J.; et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016, 536, 86–90. [Google Scholar] [CrossRef]
- Jarr, K.U.; Nakamoto, R.; Doan, B.H.; Kojima, Y.; Weissman, I.L.; Advani, R.H.; Iagaru, A.; Leeper, N.J. Effect of CD47 Blockade on Vascular Inflammation. N. Engl. J. Med. 2021, 384, 382–383. [Google Scholar] [CrossRef]
- Dane, M.J.; Khairoun, M.; Lee, D.H.; van den Berg, B.M.; Eskens, B.J.; Boels, M.G.; van Teeffelen, J.W.; Rops, A.L.; van der Vlag, J.; van Zonneveld, A.J.; et al. Association of kidney function with changes in the endothelial surface layer. Clin. J. Am. Soc. Nephrol. 2014, 9, 698–704. [Google Scholar] [CrossRef]
- Harper, S.J.; Tomson, C.R.; Bates, D.O. Human uremic plasma increases microvascular permeability to water and proteins in vivo. Kidney Int. 2002, 61, 1416–1422. [Google Scholar] [CrossRef][Green Version]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J. Am. Soc. Nephrol. 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Vink, H.; Constantinescu, A.A.; Spaan, J.A. Oxidized lipoproteins degrade the endothelial surface layer: Implications for platelet-endothelial cell adhesion. Circulation 2000, 101, 1500–1502. [Google Scholar] [CrossRef]
- Cooper, S.; McDonald, K.; Burkat, D.; Leask, R.L. Stenosis Hemodynamics Disrupt the Endothelial Cell Glycocalyx by MMP Activity Creating a Proinflammatory Environment. Ann. Biomed. Eng. 2017, 45, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Nijst, P.; Kiefer, K.; Tang, W.H. Endothelial Glycocalyx as Biomarker for Cardiovascular Diseases: Mechanistic and Clinical Implications. Curr. Heart Fail. Rep. 2017, 14, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Carmona, A.; Aguera, M.L.; Luna-Ruiz, C.; Buendia, P.; Calleros, L.; Garcia-Jerez, A.; Rodriguez-Puyol, M.; Arias, M.; Arias-Guillen, M.; de Arriba, G.; et al. Markers of endothelial damage in patients with chronic kidney disease on hemodialysis. Am. J. Physiol. Renal Physiol. 2017, 312, F673–F681. [Google Scholar] [CrossRef]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef]
- Finn, N.A.; Eapen, D.; Manocha, P.; Al Kassem, H.; Lassegue, B.; Ghasemzadeh, N.; Quyyumi, A.; Searles, C.D. Coronary heart disease alters intercellular communication by modifying microparticle-mediated microRNA transport. FEBS Lett. 2013, 587, 3456–3463. [Google Scholar] [CrossRef]
- Nozaki, T.; Sugiyama, S.; Koga, H.; Sugamura, K.; Ohba, K.; Matsuzawa, Y.; Sumida, H.; Matsui, K.; Jinnouchi, H.; Ogawa, H. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J. Am. Coll. Cardiol. 2009, 54, 601–608. [Google Scholar] [CrossRef]
- Carracedo, J.; Alique, M.; Vida, C.; Bodega, G.; Ceprian, N.; Morales, E.; Praga, M.; de Sequera, P.; Ramirez, R. Mechanisms of Cardiovascular Disorders in Patients With Chronic Kidney Disease: A Process Related to Accelerated Senescence. Front. Cell Dev. Biol. 2020, 8, 185. [Google Scholar] [CrossRef]
- Goicoechea, M.; de Vinuesa, S.G.; Lahera, V.; Cachofeiro, V.; Gomez-Campdera, F.; Vega, A.; Abad, S.; Luno, J. Effects of atorvastatin on inflammatory and fibrinolytic parameters in patients with chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, S231–S235. [Google Scholar] [CrossRef]
- Davignon, J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004, 109, III39–III43. [Google Scholar] [CrossRef]
- van der Aart-van der Beek, A.B.; de Boer, R.A.; Heerspink, H.J.L. Kidney and heart failure outcomes associated with SGLT2 inhibitor use. Nat. Rev. Nephrol. 2022, 18, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Alicic, R.Z.; Cox, E.J.; Neumiller, J.J.; Tuttle, K.R. Incretin drugs in diabetic kidney disease: Biological mechanisms and clinical evidence. Nat. Rev. Nephrol. 2021, 17, 227–244. [Google Scholar] [CrossRef]
- Sattar, N.; Lee, M.M.Y.; Kristensen, S.L.; Branch, K.R.H.; Del Prato, S.; Khurmi, N.S.; Lam, C.S.P.; Lopes, R.D.; McMurray, J.J.V.; Pratley, R.E.; et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021, 9, 653–662. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Laufs, U.; Ray, K.K.; Leiter, L.A.; Bays, H.E.; Goldberg, A.C.; Stroes, E.S.; MacDougall, D.; Zhao, X.; Catapano, A.L. Bempedoic acid plus ezetimibe fixed-dose combination in patients with hypercholesterolemia and high CVD risk treated with maximally tolerated statin therapy. Eur. J. Prev. Cardiol. 2020, 27, 593–603. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Mullard, A. NLRP3 inhibitors stoke anti-inflammatory ambitions. Nat. Rev. Drug Discov. 2019, 18, 405–407. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 17, 78. [Google Scholar] [CrossRef] [PubMed]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef]
- Ge, Y.; Huang, M.; Yao, Y.M. Efferocytosis and Its Role in Inflammatory Disorders. Front. Cell Dev. Biol. 2022, 10, 839248. [Google Scholar] [CrossRef]
- Korns, D.; Frasch, S.C.; Fernandez-Boyanapalli, R.; Henson, P.M.; Bratton, D.L. Modulation of macrophage efferocytosis in inflammation. Front. Immunol. 2011, 2, 57. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef] [PubMed]
- Wan, E.; Yeap, X.Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Chen, S.; Bai, P.; Luo, C.; Liu, J.; Sun, A.; Ge, J. Cardiac Resident Macrophage-Derived Legumain Improves Cardiac Repair by Promoting Clearance and Degradation of Apoptotic Cardiomyocytes After Myocardial Infarction. Circulation 2022, 145, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Howangyin, K.Y.; Zlatanova, I.; Pinto, C.; Ngkelo, A.; Cochain, C.; Rouanet, M.; Vilar, J.; Lemitre, M.; Stockmann, C.; Fleischmann, B.K.; et al. Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase and Milk Fat Globule Epidermal Growth Factor 8 Coordinately Improve Remodeling After Myocardial Infarction via Local Delivery of Vascular Endothelial Growth Factor. Circulation 2016, 133, 826–839. [Google Scholar] [CrossRef]
- Salvador, A.M.; Nevers, T.; Velazquez, F.; Aronovitz, M.; Wang, B.; Abadia Molina, A.; Jaffe, I.Z.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Intercellular Adhesion Molecule 1 Regulates Left Ventricular Leukocyte Infiltration, Cardiac Remodeling, and Function in Pressure Overload-Induced Heart Failure. J. Am. Heart Assoc. 2016, 5, e003126. [Google Scholar] [CrossRef]
- Gerlach, B.D.; Marinello, M.; Heinz, J.; Rymut, N.; Sansbury, B.E.; Riley, C.O.; Sadhu, S.; Hosseini, Z.; Kojima, Y.; Tang, D.D.; et al. Resolvin D1 promotes the targeting and clearance of necroptotic cells. Cell Death Differ. 2020, 27, 525–539. [Google Scholar] [CrossRef]
- Zhang, S.; Yeap, X.Y.; DeBerge, M.; Naresh, N.K.; Wang, K.; Jiang, Z.; Wilcox, J.E.; White, S.M.; Morrow, J.P.; Burridge, P.W.; et al. Acute CD47 Blockade During Ischemic Myocardial Reperfusion Enhances Phagocytosis-Associated Cardiac Repair. JACC Basic. Transl. Sci. 2017, 2, 386–397. [Google Scholar] [CrossRef]
- Moodley, Y.; Rigby, P.; Bundell, C.; Bunt, S.; Hayashi, H.; Misso, N.; McAnulty, R.; Laurent, G.; Scaffidi, A.; Thompson, P.; et al. Macrophage recognition and phagocytosis of apoptotic fibroblasts is critically dependent on fibroblast-derived thrombospondin 1 and CD36. Am. J. Pathol. 2003, 162, 771–779. [Google Scholar] [CrossRef]
- Julovi, S.M.; Sanganeria, B.; Minhas, N.; Ghimire, K.; Nankivell, B.; Rogers, N.M. Blocking thrombospondin-1 signaling via CD47 mitigates renal interstitial fibrosis. Lab. Investig. 2020, 100, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Ismail, O.Z.; Zhang, X.; Bonventre, J.V.; Gunaratnam, L. G protein alpha(12) (Galpha(12)) is a negative regulator of kidney injury molecule-1-mediated efferocytosis. Am. J. Physiol. Renal Physiol. 2016, 310, F607–F620. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wang, B.O.; Hou, Y.F.; Fu, Y.; Cui, S.J.; Zhu, J.H.; Zhan, X.Y.; Li, R.K.; Tang, W.; Wu, J.C.; et al. JAML promotes acute kidney injury mainly through a macrophage-dependent mechanism. JCI Insight 2022, 7. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Kompa, A.R.; Zhang, Y.; Wang, B.H.; Kelly, D.J.; Krum, H. Myocardial infarction impairs renal function, induces renal interstitial fibrosis, and increases renal KIM-1 expression: Implications for cardiorenal syndrome. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1884–H1893. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Xu, F.; Sabbisetti, V.; Grgic, I.; Movahedi Naini, S.; Wang, N.; Chen, G.; Xiao, S.; Patel, D.; Henderson, J.M.; et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J. Clin. Investig. 2013, 123, 4023–4035. [Google Scholar] [CrossRef]
- Wyld, M.L.R.; Mata, N.L.; Viecelli, A.; Swaminathan, R.; O’Sullivan, K.M.; O’Lone, E.; Rowlandson, M.; Francis, A.; Wyburn, K.; Webster, A.C. Sex-Based Differences in Risk Factors and Complications of Chronic Kidney Disease. Semin. Nephrol. 2022, 42, 153–169. [Google Scholar] [CrossRef]
- Dunn, S.E.; Perry, W.A.; Klein, S.L. Mechanisms and consequences of sex differences in immune responses. Nat. Rev. Nephrol. 2024, 20, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Veiras, L.C.; Girardi, A.C.C.; Curry, J.; Pei, L.; Ralph, D.L.; Tran, A.; Castelo-Branco, R.C.; Pastor-Soler, N.; Arranz, C.T.; Yu, A.S.L.; et al. Sexual Dimorphic Pattern of Renal Transporters and Electrolyte Homeostasis. J. Am. Soc. Nephrol. 2017, 28, 3504–3517. [Google Scholar] [CrossRef]
- Sugimoto, C.R.; Ahn, Y.Y.; Smith, E.; Macaluso, B.; Lariviere, V. Factors affecting sex-related reporting in medical research: A cross-disciplinary bibliometric analysis. Lancet 2019, 393, 550–559. [Google Scholar] [CrossRef]
- Pinho-Gomes, A.C.; Carcel, C.; Woodward, M.; Hockham, C. Women’s representation in clinical trials of patients with chronic kidney disease. Clin. Kidney J. 2023, 16, 1457–1464. [Google Scholar] [CrossRef]
- Wu, Y.; Prasanna, A.; Miller, H.N.; Ogungbe, O.; Peeler, A.; Juraschek, S.P.; Turkson-Ocran, R.A.; Plante, T.B. Female Recruitment Into Cardiovascular Disease Trials. Am. J. Cardiol. 2023, 198, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.N.; Elgendy, I.Y.; Saad, M.; Elgendy, A.Y.; Barakat, A.F.; Mentias, A.; Abuzaid, A.; Bavry, A.A. Does Gender Influence the Cardiovascular Benefits Observed with Sodium Glucose Co-Transporter-2 (SGLT-2) Inhibitors? A Meta-Regression Analysis. Cardiol. Ther. 2017, 6, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Dave, C.V.; Schneeweiss, S.; Patorno, E. Comparative risk of genital infections associated with sodium-glucose co-transporter-2 inhibitors. Diabetes Obes. Metab. 2019, 21, 434–438. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Tsihlis, G.; Chau, K.; Trinh, K.; Rogers, N.M.; Julovi, S.M. Novel Perspectives in Chronic Kidney Disease-Specific Cardiovascular Disease. Int. J. Mol. Sci. 2024, 25, 2658. https://doi.org/10.3390/ijms25052658
Xu C, Tsihlis G, Chau K, Trinh K, Rogers NM, Julovi SM. Novel Perspectives in Chronic Kidney Disease-Specific Cardiovascular Disease. International Journal of Molecular Sciences. 2024; 25(5):2658. https://doi.org/10.3390/ijms25052658
Chicago/Turabian StyleXu, Cuicui, George Tsihlis, Katrina Chau, Katie Trinh, Natasha M. Rogers, and Sohel M. Julovi. 2024. "Novel Perspectives in Chronic Kidney Disease-Specific Cardiovascular Disease" International Journal of Molecular Sciences 25, no. 5: 2658. https://doi.org/10.3390/ijms25052658
APA StyleXu, C., Tsihlis, G., Chau, K., Trinh, K., Rogers, N. M., & Julovi, S. M. (2024). Novel Perspectives in Chronic Kidney Disease-Specific Cardiovascular Disease. International Journal of Molecular Sciences, 25(5), 2658. https://doi.org/10.3390/ijms25052658