

Association between Immune Checkpoint Inhibitors and Atherosclerotic Cardiovascular Disease Risk: Another Brick in the Wall
 , , , ,
, , , ,
Abstract
1. Introduction
2. Methodology
3. Role of Inflammation in Atherosclerosis
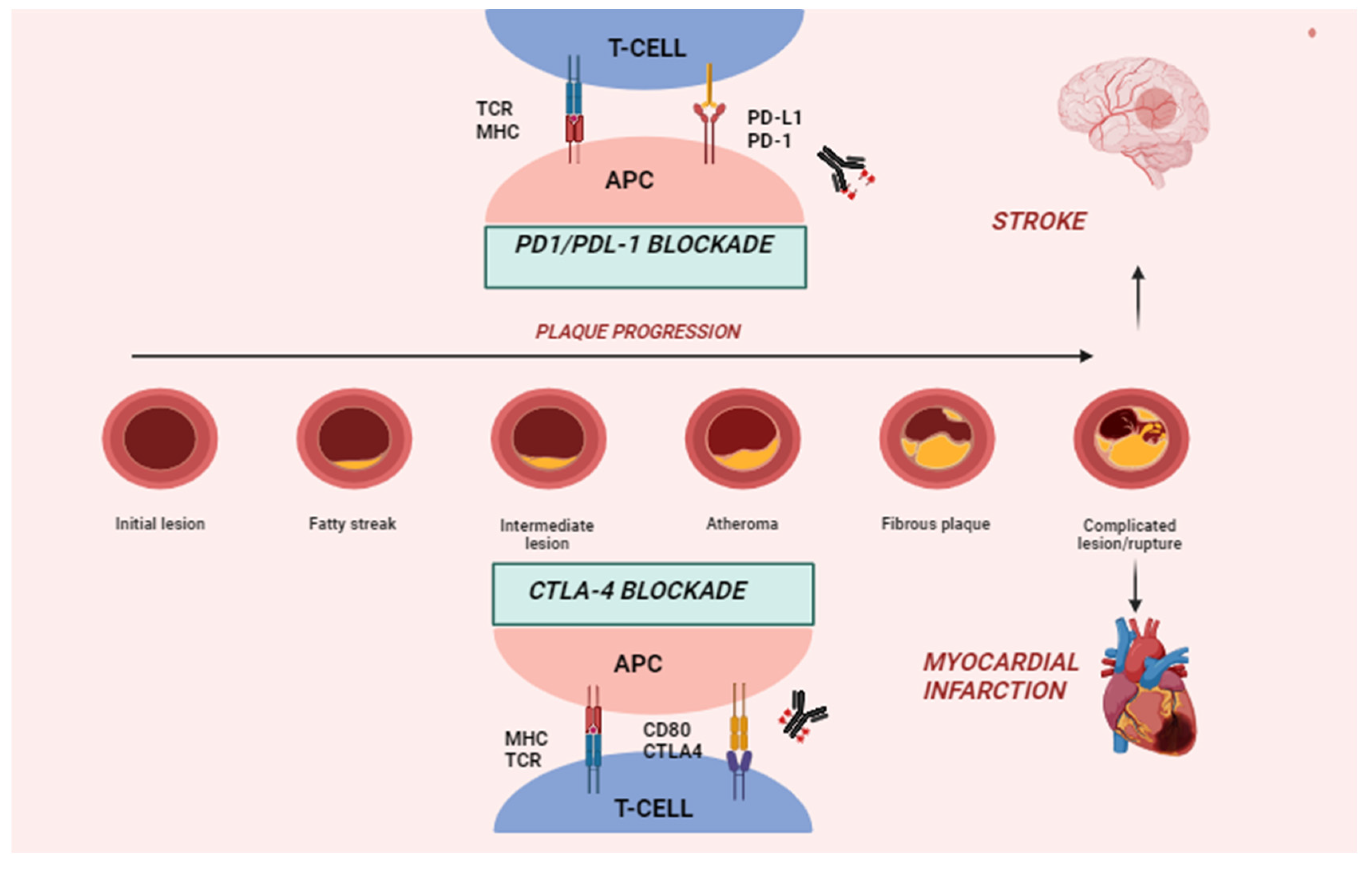
4. Immune Checkpoints and Pathophysiology of ICI-Related CVD

{kind=link}
{kind=link}
{kind=link}
| Models | Atherosclerosis | Effect on Plaque |
|---|---|---|
| Pdl1/2−/− Ldlr−/− mice [52] | ↑ | ↑ CD4, CD8, macrophages |
| Pd1−/− Ldlr−/− mice [53] | ↑ | ↑ CD4, CD8, macrophages and apoptotic cells |
| Antibody-mediated PD-1 inhibition in Ldlr−/− mice [53] | ↑ | ↑ CD4, CD8 |
| Anti-CTLA-4 antibody in ApoE3 Leiden mice [60] | ↑ | ↑ intimal thickening and intimal leukocytes |
| Anti-CTLA-4 antibody in Ldlr−/− mice [59] | ↑ | ↑ advanced lesions, necrotic core and CD3 cells |
| Combined anti-CTLA-4 and anti-PD-1 antibodies in Ldlr−/− mice [60] | ↑ | ↑ CD3, CD8, advanced lesions, necrotic core and apoptotic macrophages |
5. Cholesterol Metabolism and T-Cell Function in Cancer
6. ICIs Therapy and Atherosclerotic Cardiovascular Disease: Clinical Implications
7. Pharmacotherapy for Cardiovascular Risk Reduction in Cancer Patients Treated with ICIs
8. Limitations of Data So Far and Future Perspectives
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, L.; Reynolds, K.L.; Lyon, A.R.; Palaskas, N.; Neilan, T.G. The Evolving Immunotherapy Landscape and the Epidemiology, Diagnosis, and Management of Cardiotoxicity. JACC CardioOncology 2021, 3, 35–47. [Google Scholar] [CrossRef]
- Ribas, J.D.W.A. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Vuong, J.T.; Stein-Merlob, A.F.; Nayeri, A.; Sallam, T.; Neilan, T.G.; Yang, E.H. Immune Checkpoint Therapies and Atherosclerosis: Mechanisms and Clinical Implications. J. Am. Coll. Cardiol. 2022, 79, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune_related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Moslehi, J.J.; Salem, J.E.; Sosman, J.A.; Lebrun-Vignes, B.; Johnson, D.B. Increased reporting of fatal immune checkpoint inhibitor-associated myocar_ditis. Lancet 2018, 391, 933. [Google Scholar] [CrossRef] [PubMed]
- Laenens, D.; Yu, Y. Incidence of cardiovascular events in patients treated with immune checkpoint inhibitors. J. Clin. Oncol. 2022, 40, 3430–3438. [Google Scholar] [CrossRef]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D.; et al. Myocarditis in Patients Treated With Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zlotoff, D.A.; Awadalla, M.; Mahmood, S.S.; Nohria, A.; Hassan, M.Z.; Thuny, F.; Zubiri, L.; Chen, C.L.; Sullivan, R.J.; et al. Major Adverse Cardiovascular Events and the Timing and Dose of Corticosteroids in Immune Checkpoint Inhibitor–Associated Myocarditis. Circ. 2020, 141, 2031–2034. [Google Scholar] [CrossRef]
- Hu, Y.B.; Zhang, Q.; Li, H.J.; Michot, J.M.; Liu, H.B.; Zhan, P.; Lv, T.-F.; Song, Y. Evaluation of rare but severe immune related adverse effects in PD-1 and PD-L1 inhibitors in non–small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2017, 6 (Suppl 1), S8. [Google Scholar] [CrossRef]
- Heinzerling, L.; Ott, P.A.; Hodi, F.S.; Husain, A.N.; Tajmir-Riahi, A.; Tawbi, H.; Pauschinger, M.; Gajewski, T.F.; Lipson, E.J.; Luke, J.J. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J. Immunother. Cancer 2016, 4, 50. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Leitinger, N.; Schulman, I.G. Phenotypic Polarization of Macrophages in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2013, 33, 1120–1126. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis as an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef]
- Gupta, S.; Pablo, A.M.; Jiang, X.C.; Wang, N.; Tall, A.R.; Schindler, C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J. Clin. Investig. 1997, 99, 2752–2761. [Google Scholar] [CrossRef]
- Brånén, L.; Hovgaard, L.; Nitulescu, M.; Bengtsson, E.; Nilsson, J.; Jovinge, S.; L, P.; H, D.; R, W.; D, G.; et al. Inhibition of Tumor Necrosis Factor-α Reduces Atherosclerosis in Apolipoprotein E Knockout Mice. Arter. Thromb. Vasc. Biol. 2004, 24, 2137–2142. [Google Scholar] [CrossRef]
- Boesten, L.S.; Zadelaar, A.S.M.; Vannieuwkoop, A.; Gijbels, M.J.; Dewinther, M.; Havekes, L.M.; Vanvlijmen, B. Tumor necrosis factor-? promotes atherosclerotic lesion progression in APOE*3-leiden transgenic mice. Cardiovasc. Res. 2005, 66, 179–185. [Google Scholar] [CrossRef]
- Gotsman, I.; Lichtman, A.H. Targeting Interferon-γ to Treat Atherosclerosis. Circ. Res. 2007, 101, 333–334. [Google Scholar] [CrossRef][Green Version]
- Laurat, E.; Poirier, B.; Tupin, E.; Caligiuri, G.; Hansson, G.; Bariéty, J.; Nicoletti, A. In Vivo Downregulation of T Helper Cell 1 Immune Responses Reduces Atherogenesis in Apolipoprotein E-Knockout Mice. Circulation 2001, 104, 197–202. [Google Scholar] [CrossRef]
- Robertson, A.-K.L.; Rudling, M.; Zhou, X.; Gorelik, L.; Flavell, R.A.; Hansson, G.K. Disruption of TGF-β signaling in T cells accelerates atherosclerosis. J. Clin. Investig. 2003, 112, 1342–1350. [Google Scholar] [CrossRef]
- Lin, J.; Li, M.; Wang, Z.; He, S.; Ma, X.; Li, D. The role of CD4+CD25+ regulatory T cells in macrophage-derived foam-cell formation. J. Lipid Res. 2010, 51, 1208–1217. [Google Scholar] [CrossRef]
- Mallat, Z.; Besnard, S.; Duriez, M.; Deleuze, V.; Emmanuel, F.; Bureau, M.F.; Soubrier, F.; Esposito, B.; Duez, H.; Fievet, C.; et al. Protective Role of Interleukin-10 in Atherosclerosis. Circ. Res. 1999, 85, e17–e24. [Google Scholar] [CrossRef]
- Grainger, D.J. Transforming Growth Factor β and Atherosclerosis: So Far, So Good for the Protective Cytokine Hypothesis. Arter. Thromb. Vasc. Biol. 2004, 24, 399–404. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.-K.L.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef]
- Dietel, B.; Cicha, I.; Voskens, C.J.; Verhoeven, E.; Achenbach, S.; Garlichs, C.D. Decreased numbers of regulatory T cells are associated with human atherosclerotic lesion vulnerability and inversely correlate with infiltrated mature dendritic cells. Atherosclerosis 2013, 230, 92–99. [Google Scholar] [CrossRef]
- Binder, C.J.; Hartvigsen, K.; Chang, M.-K.; Miller, M.; Broide, D.; Palinski, W.; Curtiss, L.K.; Corr, M.; Witztum, J.L. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J. Clin. Investig. 2004, 114, 427–437. [Google Scholar] [CrossRef]
- Ketelhuth, D.F.; Hansson, G.K. Adaptive Response of T and B Cells in Atherosclerosis. Circ. Res. 2016, 118, 668–678. [Google Scholar] [CrossRef]
- King, V.L.; Szilvassy, S.J.; Daugherty, A. Interleukin-4 Deficiency Decreases Atherosclerotic Lesion Formation in a Site-Specific Manner in Female LDL Receptor−/− Mice. Arter. Thromb. Vasc. Biol. 2002, 22, 456–461. [Google Scholar] [CrossRef]
- Eid, R.E.; Rao, D.A.; Zhou, J.; Lo, S.-F.L.; Ranjbaran, H.; Gallo, A.; Sokol, S.I.; Pfau, S.; Pober, J.S.; Tellides, G.; et al. Interleukin-17 and Interferon-γ Are Produced Concomitantly by Human Coronary Artery–Infiltrating T Cells and Act Synergistically on Vascular Smooth Muscle Cells. Circ. 2009, 119, 1424–1432. [Google Scholar] [CrossRef]
- Smith, E.; Prasad, K.-M.R.; Butcher, M.; Dobrian, A.; Kolls, J.K.; Ley, K.; Galkina, E.; K, T.; H, d.l.F.; M, R.; et al. Blockade of Interleukin-17A Results in Reduced Atherosclerosis in Apolipoprotein E–Deficient Mice. Circ. 2010, 121, 1746–1755. [Google Scholar] [CrossRef]
- Gisterå, A.; Robertson, A.-K.L.; Andersson, J.; Ketelhuth, D.F.J.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming Growth Factor–β Signaling in T Cells Promotes Stabilization of Atherosclerotic Plaques Through an Interleukin-17–Dependent Pathway. Sci. Transl. Med. 2013, 5, 196ra100. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Dwyer, C.J.; Bailey, S.R.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Herrero-Sánchez, M.C.; Rodríguez-Serrano, C.; Almeida, J.; San Segundo, L.; Inogés, S.; Santos-Briz, Á.; García-Briñón, J.; Corchete, L.A.; San Miguel, J.F.; Del Cañizo, C.; et al. Targeting of PI3K/AKT/mTOR pathway to inhibit T cell activation and prevent graft-versus-host disease development. J. Hematol. Oncol. 2016, 9, 113. [Google Scholar] [CrossRef]
- Yousif, L.I.; Tanja, A.A.; de Boer, R.A.; Teske, A.J.; Meijers, W.C. The role of immune checkpoints in cardiovascular disease. Front. Pharmacol. 2022, 13, 989431. [Google Scholar] [CrossRef]
- Ghiotto, M.; Gauthier, L.; Serriari, N.; Pastor, S.; Truneh, A.; Nunès, J.A.; Olive, D. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int. Immunol. 2010, 22, 651–660. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; Ibañez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Ronen, D.; Bsoul, A.; Lotem, M.; Abedat, S.; Yarkoni, M.; Amir, O.; Asleh, R. Exploring the Mechanisms Underlying the Cardiotoxic Effects of Immune Checkpoint Inhibitor Therapies. Vaccines 2022, 10, 540. [Google Scholar] [CrossRef]
- Willsmore, Z.N.; Coumbe, B.G.T.; Crescioli, S.; Reci, S.; Gupta, A.; Harris, R.J.; Chenoweth, A.; Chauhan, J.; Bax, H.J.; McCraw, A.; et al. Combined anti-PD-1 and anti-CTLA-4 checkpoint blockade: Treatment of melanoma and immune mechanisms of action. Eur. J. Immunol. 2021, 51, 544–556. [Google Scholar] [CrossRef]
- Zhang, Q.; Chikina, M.; Szymczak-Workman, A.L.; Horne, W.; Kolls, J.K.; Vignali, K.M.; Normolle, D.; Bettini, M.; Workman, C.J.; Vignali, D.A.A. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-cell Responses. Cancer Res 2014, 74, 3418–3428. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Ball, S.; Ghosh, R.K.; Wongsaengsak, S.; Bandyopadhyay, D.; Ghosh, G.C.; Aronow, W.S.; Fonarow, G.C.; Lenihan, D.J.; Bhatt, D.L. Cardiovascular Toxicities of Immune Checkpoint Inhibitors. J. Am. Coll. Cardiol. 2019, 74, 1714–1727. [Google Scholar] [CrossRef]
- Watanabe, R.; Zhang, H.; Berry, G.; Goronzy, J.J.; Weyand, C.M. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am. J. Physiol. Circ. Physiol. 2017, 312, H1052–H1059. [Google Scholar] [CrossRef]
- Suero-Abreu, G.A.; Zanni, M.V.; Neilan, T.G. Atherosclerosis With Immune Checkpoint Inhibitor Therapy. JACC CardioOncology 2022, 4, 598–615. [Google Scholar] [CrossRef]
- Gotsman, I.; Grabie, N.; Dacosta, R.; Sukhova, G.; Sharpe, A.; Lichtman, A.H. Proatherogenic immune responses are regulated by the PD-1/PD-L pathway in mice. J. Clin. Investig. 2007, 117, 2974–2982. [Google Scholar] [CrossRef]
- Bu, D.-X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.A.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the Programmed Cell Death-1 Pathway Increases Atherosclerotic Lesion Development and Inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef]
- Lee, J.; Zhuang, Y.; Wei, X.; Shang, F.; Wang, J.; Zhang, Y.; Liu, X.; Yang, Y.; Liu, L. Contributions of PD-1/PD-L1 pathway to interactions of myeloid DCs with T cells in atherosclerosis. J. Mol. Cell Cardiol. 2009, 46, 169–176. [Google Scholar] [CrossRef]
- Michel, L.; Helfrich, I.; Hendgen-Cotta, U.B.; Mincu, R.-I.; Korste, S.; Mrotzek, S.M.; Spomer, A.; Odersky, A.; Rischpler, C.; Herrmann, K.; et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur. Hear. J. 2021, 43, 316–329. [Google Scholar] [CrossRef]
- Michel, L.; Korste, S.; Spomer, A.; Hendgen-Cotta, U.B.; Rassaf, T.; Totzeck, M. PD1 Deficiency Modifies Cardiac Immunity during Baseline Conditions and in Reperfused Acute Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 7533. [Google Scholar] [CrossRef]
- Hess, C.N.; Roe, M.T.; Clare, R.M.; Chiswell, K.; Kelly, J.; Tcheng, J.E.; Hagstrom, E.; James, S.K.; Khouri, M.G.; Hirsch, B.R.; et al. Relationship Between Cancer and Cardiovascular Outcomes Following Percutaneous Coronary Intervention. J. Am. Hear. Assoc. 2015, 4, 001779. [Google Scholar] [CrossRef]
- Matsumoto, T.; Sasaki, N.; Yamashita, T.; Emoto, T.; Kasahara, K.; Mizoguchi, T.; Hayashi, T.; Yodoi, K.; Kitano, N.; Saito, T.; et al. Overexpression of Cytotoxic T-Lymphocyte–Associated Antigen-4 Prevents Atherosclerosis in Mice. Arter. Thromb. Vasc. Biol. 2016, 36, 1141–1151. [Google Scholar] [CrossRef]
- Poels, K.; van Leent, M.M.T.; Reiche, M.E.; Kusters, P.J.H.; Huveneers, S.; de Winther, M.P.J.; Mulder, W.J.M.; Lutgens, E.; Seijkens, T.T.P. Antibody-Mediated Inhibition of CTLA4 Aggravates Atherosclerotic Plaque Inflammation and Progression in Hyperlipidemic Mice. Cells 2020, 9, 1987. [Google Scholar] [CrossRef]
- Ewing, M.; Karper, J.; Abdul, S.; de Jong, R.; Peters, H.; de Vries, M.; Redeker, A.; Kuiper, J.; Toes, R.; Arens, R.; et al. T-cell co-stimulation by CD28–CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int. J. Cardiol. 2013, 168, 1965–1974. [Google Scholar] [CrossRef]
- Ma, K.; Lv, S.; Liu, B.; Liu, Z.; Luo, Y.; Kong, W.; Xu, Q.; Feng, J.; Wang, X. CTLA4-IgG ameliorates homocysteine-accelerated atherosclerosis by inhibiting T-cell overactivation in apoE−/− mice. Cardiovasc. Res. 2012, 97, 349–359. [Google Scholar] [CrossRef]
- Golden, D.; Kolmakova, A.; Sura, S.; Vella, A.T.; Manichaikul, A.; Wang, X.-Q.; Bielinski, S.J.; Taylor, K.D.; Chen, Y.-D.I.; Rich, S.S.; et al. Lymphocyte activation gene 3 and coronary artery disease. J. Clin. Investig. 2016, 1, e88628. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef]
- A Fadok, V.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef]
- Rayner, K.J. Cell Death in the Vessel Wall. Arter. Thromb. Vasc. Biol. 2017, 37, e75–e81. [Google Scholar] [CrossRef]
- Jarr, K.-U.; Nakamoto, R.; Doan, B.H.; Kojima, Y.; Weissman, I.L.; Advani, R.H.; Iagaru, A.; Leeper, N.J. Effect of CD47 Blockade on Vascular Inflammation. New Engl. J. Med. 2021, 384, 382–383. [Google Scholar] [CrossRef]
- Flores, A.M.; Hosseini-Nassab, N.; Jarr, K.-U.; Ye, J.; Zhu, X.; Wirka, R.; Koh, A.L.; Tsantilas, P.; Wang, Y.; Nanda, V.; et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat. Nanotechnol. 2020, 15, 154–161. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Proto, J.D.; Doran, A.C.; Gusarova, G.; Yurdagul, A.; Sozen, E.; Subramanian, M.; Islam, M.N.; Rymond, C.C.; Du, J.; Hook, J.; et al. Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 2018, 49, 560–565. [Google Scholar] [CrossRef]
- Porta, C.; Marino, A.; Consonni, F.M.; Bleve, A.; Mola, S.; Storto, M.; Riboldi, E.; Sica, A. Metabolic influence on the differentiation of suppressive myeloid cells in cancer. Carcinog. 2018, 39, 1095–1104. [Google Scholar] [CrossRef]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e5. [Google Scholar] [CrossRef]
- Perrone, F.; Minari, R.; Bersanelli, M.; Bordi, P.; Tiseo, M.; Favari, E.; Sabato, R.; Buti, S. The Prognostic Role of High Blood Cholesterol in Advanced Cancer Patients Treated With Immune Checkpoint Inhibitors. J. Immunother. 2020, 43, 196–203. [Google Scholar] [CrossRef]
- Young, A.C.; Quach, H.T.; Song, H.; Davis, E.J.; Moslehi, J.J.; Ye, F.; Williams, G.R.; Johnson, D.B. Impact of body composition on outcomes from anti-PD1 +/− anti-CTLA-4 treatment in melanoma. J. Immunother. Cancer 2020, 8, e000821. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K.; et al. Association Between Immune Checkpoint Inhibitors With Cardiovascular Events and Atherosclerotic Plaque. Circ. 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Bar, J.; Markel, G.; Gottfried, T.; Percik, R.; Leibowitz-Amit, R.; Berger, R.; Golan, T.; Daher, S.; Taliansky, A.; Dudnik, E.; et al. Acute vascular events as a possibly related adverse event of immunotherapy: A single-institute retrospective study. Eur. J. Cancer 2019, 120, 122–131. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, S.; Yang, F.; Qi, X.; Wang, X.; Guan, X.; Shen, C.; Duma, N.; Vera Aguilera, J.; Chintakuntlawar, A.; et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-analysis. JAMA Oncol. 2019, 5, 1008–1019. [Google Scholar] [CrossRef]
- Oren, O.; Yang, E.H.; Molina, J.R.; Bailey, K.R.; Blumenthal, R.S.; Kopecky, S.L. Cardiovascular Health and Outcomes in Cancer Patients Receiving Immune Checkpoint Inhibitors. Am. J. Cardiol. 2020, 125, 1920–1926. [Google Scholar] [CrossRef]
- Nichetti, F.; Ligorio, F.; Zattarin, E.; Signorelli, D.; Prelaj, A.; Proto, C.; Galli, G.; Marra, A.; Apollonio, G.; Porcu, L.; et al. Is There an Interplay between Immune Checkpoint Inhibitors, Thromboprophylactic Treatments and Thromboembolic Events? Mechanisms and Impact in Non-Small Cell Lung Cancer Patients. Cancers 2019, 12, 67. [Google Scholar] [CrossRef]
- Calabretta, R.; Hoeller, C.; Pichler, V.; Mitterhauser, M.; Karanikas, G.; Haug, A.; Li, X.; Hacker, M. Immune Checkpoint Inhibitor Therapy Induces Inflammatory Activity in Large Arteries. Circ. 2020, 142, 2396–2398. [Google Scholar] [CrossRef]
- Drobni, Z.D.; Gongora, C.; Taron, J.; A Suero-Abreu, G.; Karady, J.; Gilman, H.K.; Supraja, S.; Nikolaidou, S.; Leeper, N.; Merkely, B.; et al. Impact of immune checkpoint inhibitors on atherosclerosis progression in patients with lung cancer. J. Immunother. Cancer 2023, 11, e007307. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Y.; Zhang, Y.; Wang, W.; Wang, Y.; Lu, Z.; Zhang, Y.; Lei, H.; Li, D.; Long, B.; et al. Association of immune checkpoint inhibitors therapy with arterial thromboembolic events in cancer patients: A retrospective cohort study. Cancer Med. 2023, 12, 18531–18541. [Google Scholar] [CrossRef]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts). Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef] [PubMed]
- Inno, A.; Chiampan, A.; Lanzoni, L.; Verzè, M.; Molon, G.; Gori, S. Immune Checkpoint Inhibitors and Atherosclerotic Vascular Events in Cancer Patients. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Liu, X.; Bao, X.; Hu, M.; Chang, H.; Jiao, M.; Cheng, J.; Xie, L.; Huang, Q.; Li, F.; Li, C.-Y. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 2020, 588, 693–698. [Google Scholar] [CrossRef]
- Quagliariello, V.; Bisceglia, I.; Berretta, M.; Iovine, M.; Canale, M.L.; Maurea, C.; Giordano, V.; Paccone, A.; Inno, A.; Maurea, N. PCSK9 Inhibitors in Cancer Patients Treated with Immune-Checkpoint Inhibitors to Reduce Cardiovascular Events: New Frontiers in Cardioncology. Cancers 2023, 15, 1397. [Google Scholar] [CrossRef]
- Fu, T.; Guan, Y.; Xu, J.; Wang, Y. APP, APLP2 and LRP1 interact with PCSK9 but are not required for PCSK9-mediated degradation of the LDLR in vivo. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2017, 1862, 883–889. [Google Scholar] [CrossRef]
- Carey, S.T.; Bridgeman, C.; Jewell, C.M. Biomaterial Strategies for Selective Immune Tolerance: Advances and Gaps. Adv. Sci. 2023, 10, e2205105. [Google Scholar] [CrossRef]
- Douna, H.; Smit, V.; van Puijvelde, G.H.; Kiss, M.G.; Binder, C.J.; Bot, L.; Kuchroo, V.K.; Lichtman, A.H.; Kuiper, J.; Foks, A.C. Tim-1 mucin domain-mutant mice display exacerbated atherosclerosis. Atherosclerosis 2022, 352, 1–9. [Google Scholar] [CrossRef]
- Abdelwahed, K.S.; Siddique, A.B.; Qusa, M.H.; King, J.A.; Souid, S.; Elmageed, Z.Y.A.; El Sayed, K.A. PCSK9 Axis-Targeting Pseurotin A as a Novel Prostate Cancer Recurrence Suppressor Lead. ACS Pharmacol. Transl. Sci. 2021, 4, 1771–1781. [Google Scholar] [CrossRef]
- Saha, S.; Singh, A.; Kumar, P.; Sonkar, A.B.; Gautam, A.K.; Verma, A.; Maity, B.; Tiwari, H.; Sahoo, N.G.; Keshari, A.K.; et al. A Comprehensive Review on PCSK9 as Mechanistic Target Approach in Cancer Therapy. Mini-Reviews Med. Chem. 2023, 23, 24–32. [Google Scholar] [CrossRef]
- Xia, X.-D.; Peng, Z.-S.; Gu, H.-M.; Wang, M.; Wang, G.-Q.; Zhang, D.-W. Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Bonaventura, A.; Grossi, F.; Carbone, F.; Vecchié, A.; Minetti, S.; Bardi, N.; Elia, E.; Ansaldo, A.M.; Ferrara, D.; Rijavec, E.; et al. Serum PCSK9 levels at the second nivolumab cycle predict overall survival in elderly patients with NSCLC: A pilot study. Cancer Immunol. Immunother. 2019, 68, 1351–1358. [Google Scholar] [CrossRef]
- Li, M.; Yang, Y.; Wei, J.; Cun, X.; Lu, Z.; Qiu, Y.; Zhang, Z.; He, Q. Enhanced chemo-immunotherapy against melanoma by inhibition of cholesterol esterification in CD8+ T cells. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2541–2550. [Google Scholar] [CrossRef]
- Charbe, N.B.; Lagos, C.F.; Ortiz, C.A.V.; Tambuwala, M.; Palakurthi, S.S.; Zacconi, F.C. PCSK9 conjugated liposomes for targeted delivery of paclitaxel to the cancer cell: A proof-of-concept study. Biomed. Pharmacother. 2022, 153, 113428. [Google Scholar] [CrossRef]
- Xu, X.; Cui, Y.; Cao, L.; Zhang, Y.; Yin, Y.; Hu, X. PCSK9 regulates apoptosis in human lung adenocarcinoma A549 cells via endoplasmic reticulum stress and mitochondrial signaling pathways. Exp. Ther. Med. 2017, 13, 1993–1999. [Google Scholar] [CrossRef]
- Wang, L.; Li, S.; Luo, H.; Lu, Q.; Yu, S. PCSK9 promotes the progression and metastasis of colon cancer cells through regulation of EMT and PI3K/AKT signaling in tumor cells and phenotypic polarization of macrophages. J. Exp. Clin. Cancer Res. 2022, 41, 1–21. [Google Scholar] [CrossRef]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Lopez-Mattei, J.C.; Yang, E.H.; Ferencik, M.; Baldassarre, L.A.; Dent, S.; Budoff, M.J. Cardiac Computed Tomography in Cardio-Oncology. JACC: CardioOncology 2021, 3, 635–649. [Google Scholar] [CrossRef]
- Mani, V.; Woodward, M.; Samber, D.; Bucerius, J.; Tawakol, A.; Kallend, D.; Rudd, J.H.F.; Abt, M.; Fayad, Z.A. Predictors of change in carotid atherosclerotic plaque inflammation and burden as measured by 18-FDG-PET and MRI, respectively, in the dal-PLAQUE study. Int. J. Cardiovasc. Imaging 2014, 30, 571–582. [Google Scholar] [CrossRef]
- Pérez-Medina, C.; Fayad, Z.A.; Mulder, W.J. Atherosclerosis Immunoimaging by Positron Emission Tomography. Arter. Thromb. Vasc. Biol. 2020, 40, 865–873. [Google Scholar] [CrossRef]
- Kusters, P.J.H.; Lutgens, E.; Seijkens, T.T.P. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc. Res. 2018, 114, 368–377. [Google Scholar] [CrossRef]
- Meletta, R.; Herde, A.M.; Dennler, P.; Fischer, E.; Schibli, R.; Krämer, S.D. Preclinical imaging of the co-stimulatory molecules CD80 and CD86 with indium-111-labeled belatacept in atherosclerosis. EJNMMI Res. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Mach, F.; Schönbeck, U.; Sukhova, G.K.; Atkinson, E.; Libby, P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature 1998, 394, 200–203. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Stan, M.N.; Frommer, L.; Gergely, P.; Colin, L.; Amer, A.; Schuhmann, I.; Espie, P.; Rush, J.S.; Basson, C.; et al. A Novel Anti-CD40 Monoclonal Antibody, Iscalimab, for Control of Graves Hyperthyroidism—A Proof-of-Concept Trial. J. Clin. Endocrinol. Metab. 2019, 105, 696–704. [Google Scholar] [CrossRef]
- Meletta, R.; Steier, L.; Borel, N.; Mu, L.; Keller, C.; Chiotellis, A.; Russo, E.; Halin, C.; Ametamey, S.M.; Schibli, R.; et al. CD80 Is Upregulated in a Mouse Model with Shear Stress-Induced Atherosclerosis and Allows for Evaluating CD80-Targeting PET Tracers. Mol. Imaging Biol. 2016, 19, 90–99. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, H.; Yu, J.; Tian, W.; Song, Y. Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 1–18. [Google Scholar] [CrossRef]



| Author | Study | Major Findings | ICI Effect |
|---|---|---|---|
| Hu et al. [9] (2017) | Meta-analysis (22 studies) | 1.0% incidence of MI (95% CI:0–13%) | ASCVD RATE ↑ |
| Bar et al. [75] (2019) | Single center retrospective | Single cohort: acute vascular events within 6 mo post-ICIs: 2.6% (95% CI: 1.8%–3.6%); event rate was 5.2% (95% CI: 2.8%–9.2%); acute vascular events incidence higher within 6 mo (OR: 3.49; 95% CI 1.45–8.41; p = 0.002). 1% of cases were MI or ischemic stroke. OS worse in post-Id patients with acute vascular events 3 mo vs. 14 mo; HR: 3.01; 95% CI:2–4.39; p < 0.0001 | ASCVD RATE ↑ |
| Wang et al. [76] (2019) | Meta-analysis (125 studies) | 9.8% incidence of ASCVD rate treatment-related deaths due to CV irAEs, including MI, HF and CM | ASCVD RATE ↑ |
| Nichetti et al. [78] (2020) | Prospective observational | 6.5% incidence of acute vascular events (2 ACS, 9 strokes, 3 visceral arterial thromboses) within 16 mo | ASCVD RATE ↑ |
| Oren et al. [77] (2020) | Single center retrospective | Rate of MI was 213 (7%) and stroke was 227 (7%) patients | ASCVD RATE↑ |
| Drobni et al. [74] (2020) | Single center retrospective | Matched cohort, case crossover, and imaging study: higher risk of acute vascular events in ICIs (HR: 3.3; 95% Cl: 2.0– 5.5; p < 0.001) Case crossover: higher incidence of acute vascular events at 2 years after ICIs vs 2 years before ICIs (adjusted HR: 4.8; 95% CI: 3.6–6.5; p < 0.001) Imaging: Higher aortic plaque progression rate (2.1%/y before ICIs to 6.7%/y after ICIs) | ASCVD RATE andplaque ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piras, L.; Zuccanti, M.; Russo, P.; Riccio, F.; Agresti, A.; Lustri, C.; Dardani, D.; Ferrera, A.; Fiorentini, V.; Tocci, G.; et al. Association between Immune Checkpoint Inhibitors and Atherosclerotic Cardiovascular Disease Risk: Another Brick in the Wall. Int. J. Mol. Sci. 2024, 25, 2502. https://doi.org/10.3390/ijms25052502
Piras L, Zuccanti M, Russo P, Riccio F, Agresti A, Lustri C, Dardani D, Ferrera A, Fiorentini V, Tocci G, et al. Association between Immune Checkpoint Inhibitors and Atherosclerotic Cardiovascular Disease Risk: Another Brick in the Wall. International Journal of Molecular Sciences. 2024; 25(5):2502. https://doi.org/10.3390/ijms25052502
Chicago/Turabian StylePiras, Linda, Michela Zuccanti, Paola Russo, Francesca Riccio, Antonio Agresti, Camilla Lustri, Domenico Dardani, Armando Ferrera, Vincenzo Fiorentini, Giuliano Tocci, and et al. 2024. "Association between Immune Checkpoint Inhibitors and Atherosclerotic Cardiovascular Disease Risk: Another Brick in the Wall" International Journal of Molecular Sciences 25, no. 5: 2502. https://doi.org/10.3390/ijms25052502
APA StylePiras, L., Zuccanti, M., Russo, P., Riccio, F., Agresti, A., Lustri, C., Dardani, D., Ferrera, A., Fiorentini, V., Tocci, G., Tini Melato, G., Volpe, M., Barbato, E., & Battistoni, A. (2024). Association between Immune Checkpoint Inhibitors and Atherosclerotic Cardiovascular Disease Risk: Another Brick in the Wall. International Journal of Molecular Sciences, 25(5), 2502. https://doi.org/10.3390/ijms25052502