
The Structure of the Cardiac Mitochondria Respirasome Is Adapted for the β-Oxidation of Fatty Acids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
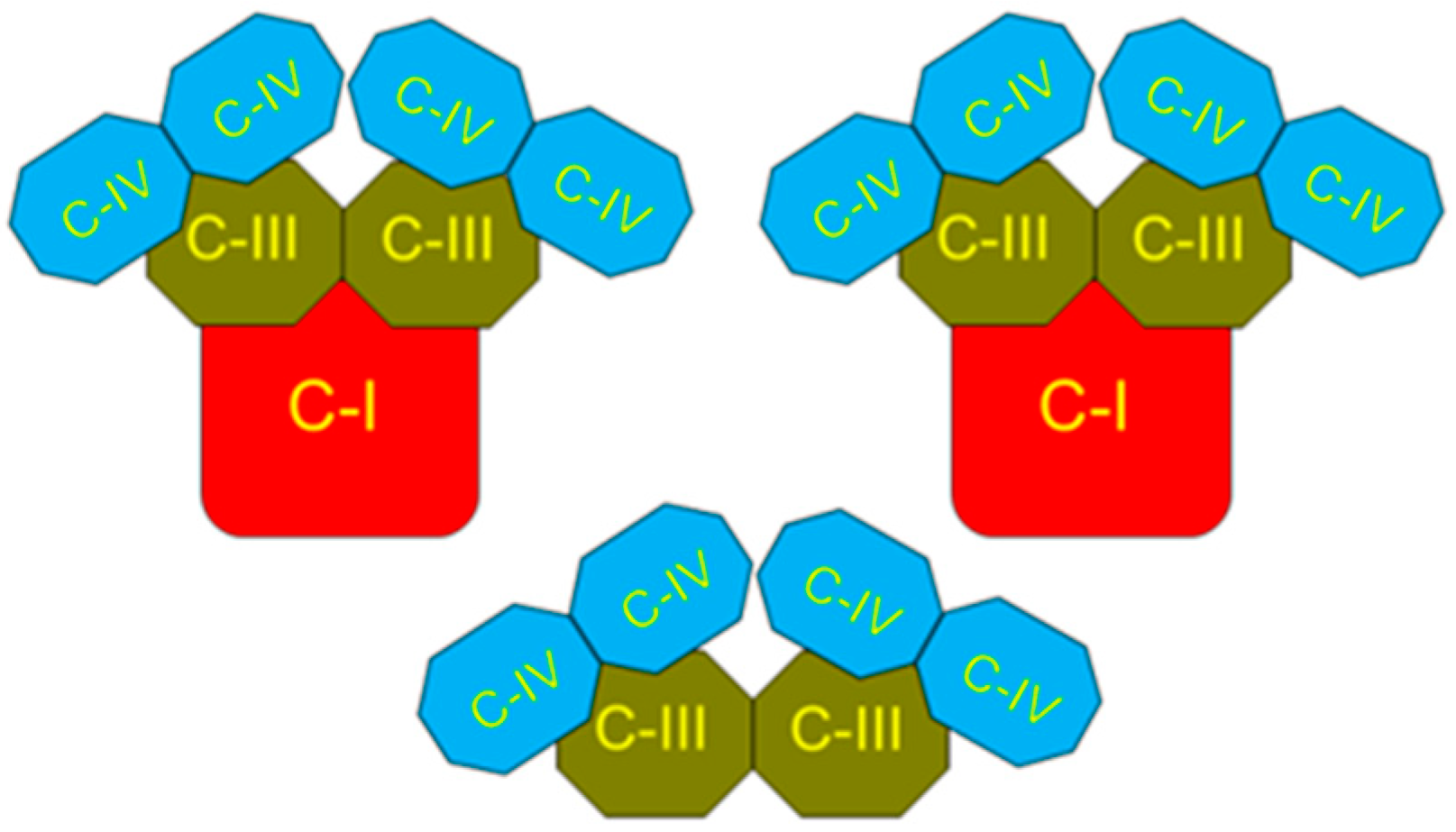
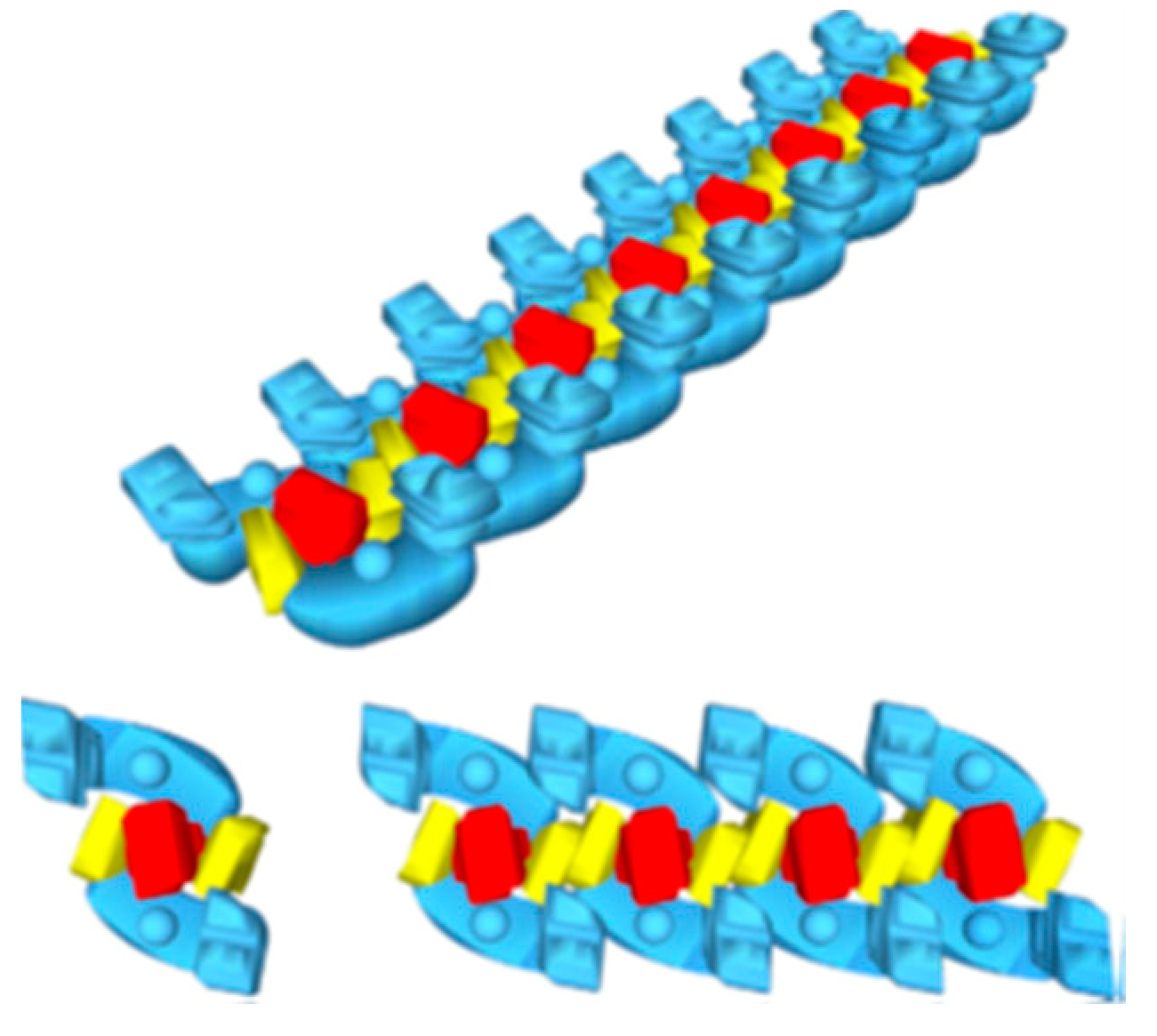
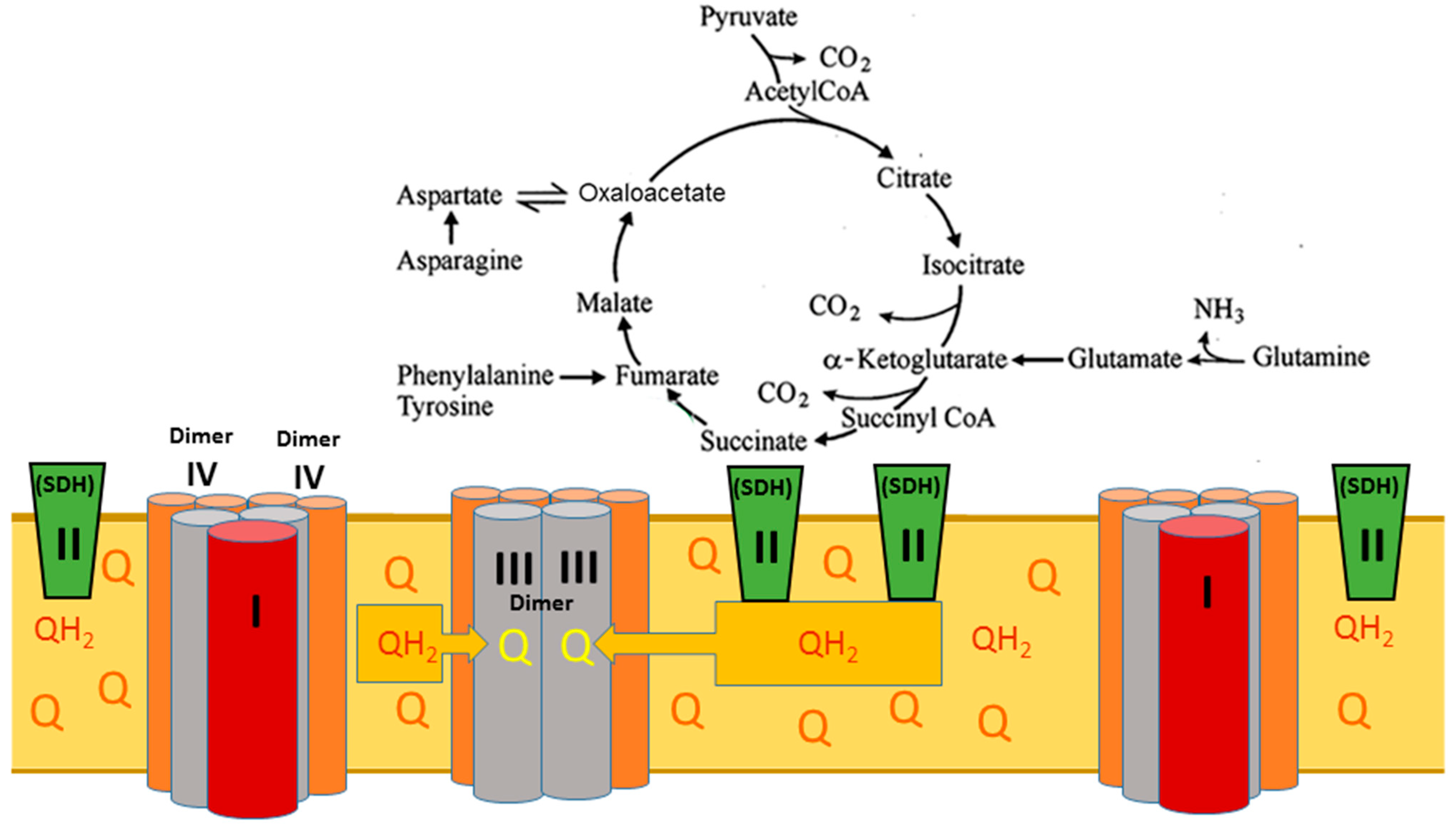
2. The Superstructural Organization of the Respiratory Chain
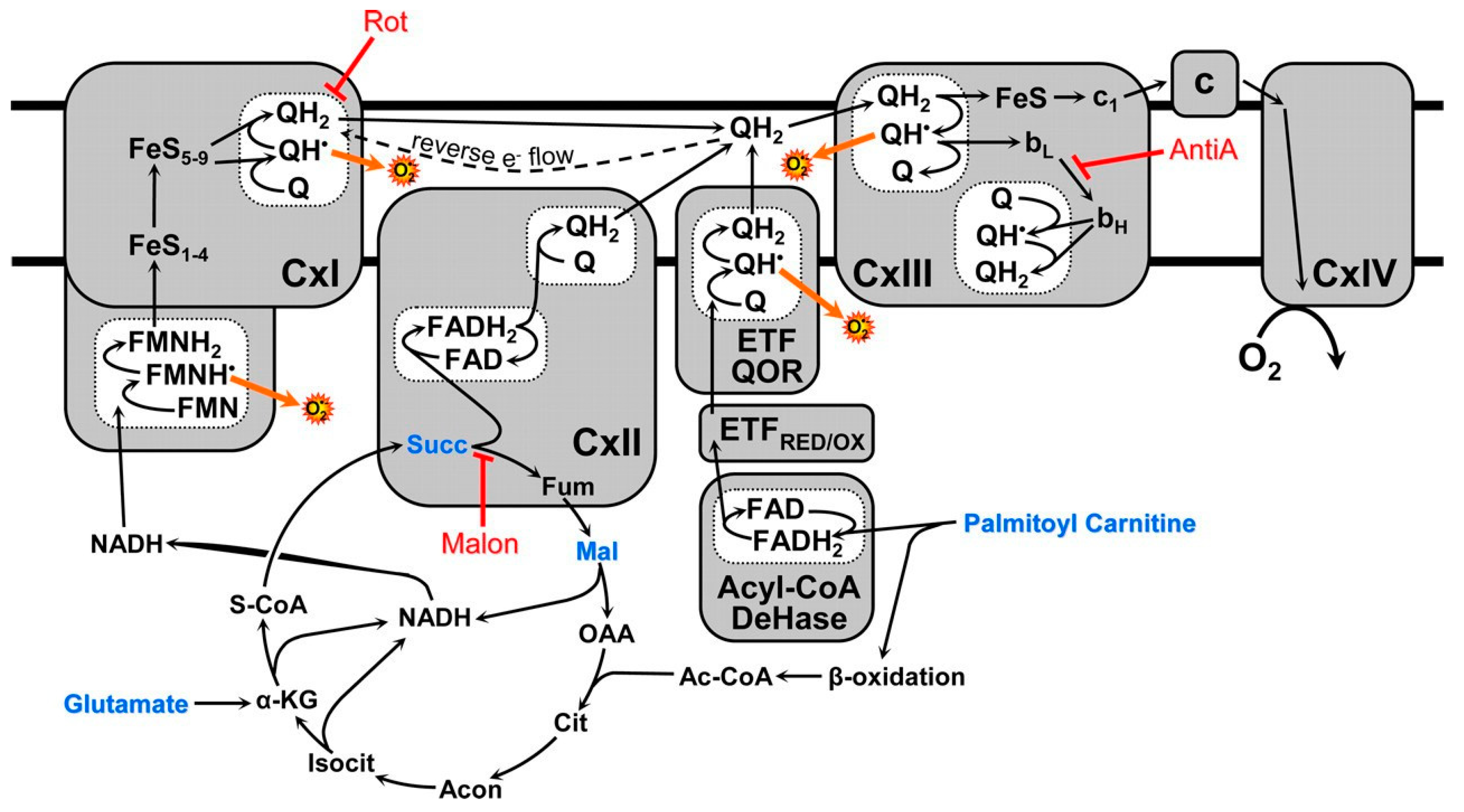
3. The Structural–Functional Properties of the Cardiac Mitochondrial Respirasome Evidence That the Respirasome Is Specifically Adapted for the β-Oxidation of Fatty Acids
3.1. In the Absence of β-Oxidation of Long-Chain and Middle-Chain Fatty Acids, the Respirasome Predominantly Supports the Catabolic Reactions
3.2. β-Oxidation of Long-Chain Fatty Acids in the Presence of Other Mitochondrial Substrates Supports a High Rate of ATP Production and Anabolic Metabolism in Cardiomyocytes
4. Stimulation of β-Oxidation of Fatty Acids by Supporting Substrates
5. The Critical Roles of the Mitochondrial Phospholipids Cardiolipin and Phosphatidylethanolamine in Mitochondria’s Structural Organization and Functioning
6. Diseases Caused by Abnormalities of Fatty Acid Metabolism and Changes during Embryonic and Postembryonic Ontogenesis
6.1. Cardiolipin Abnormalities
6.2. Enzyme Deficiencies
6.3. Postnatal Maturation of the Mitochondrial Fatty Acid β-Oxidation
7. Conclusions
Funding
Conflicts of Interest
References
- Bertermann, H.; Gronow, G.; Schirmer, A.; Weiss, C. Contribution of long chain fatty acids to the energy supply of the rat kidney cortex. Pflug. Arch. 1975, 356, 9–17. [Google Scholar] [CrossRef]
- Wirthensohn, G.; Guder, W.G. Triacylglycerol metabolism in isolated rat kidney cortex tubules. Biochem. J. 1980, 186, 317–324. [Google Scholar] [CrossRef]
- Spitzer, J.J. CNS, and fatty acid metabolism. Physiologist 1973, 16, 55–68. [Google Scholar]
- Stanley, W.C.; Chandler, M.P. Energy metabolism in the normal and failing heart: Potential for therapeutic interventions. Heart Fail. Rev. 2002, 7, 115–130. [Google Scholar] [CrossRef]
- Schagger, H. Respiratory chain supercomplexes. IUBMB Life 2001, 52, 119–128. [Google Scholar] [CrossRef]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef]
- Schagger, H.; Pfeiffer, K. The Ratio of Oxidative Phosphorylation Complexes I–V in Bovine Heart Mitochondria and the Composition of Respiratory Chain Supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signaling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty Acids in Energy Metabolism of the Central Nervous System. Bio. Med. Res. Intern. 2014, 2014, 472459. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.; Dikalov, S. Brain energy Metabolism. In Encyclopedia in Biochemistry, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 1–16. [Google Scholar]
- Panov, A.V.; Mayorov, V.I.; Dikalov, S.I. Metabolic properties of murine kidney mitochondria. bioRxiv 2021. [Google Scholar] [CrossRef]
- Panov, A.; Mayorov, V.I.; Dikalov, S. Metabolic Syndrome and β-oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. Int. J. Mol. Sci. 2022, 23, 4047. [Google Scholar] [CrossRef]
- Panov, A.V.; Mayorov, V.I.; Dikalova, A.E.; Dikalov, S.I. Long-chain and medium-chain fatty acids in the energy metabolism of murine kidney mitochondria. Int. J. Mol. Sci. 2023, 24, 379. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Hemendinger, R.; Greenamyre, J.T.; Rosenfeld, J. Species and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am. J. Physiol. Cell Physiol. 2007, 292, C708–C718. [Google Scholar] [CrossRef]
- Panov, A.V.; Darenskaya, M.A.; Dikalov, S.I.; Kolesnikov, S.I. Metabolic syndrome as the first stage of eldership; the beginning of real aging. In Update in Geriatrics; Somarnyotin, S., Ed.; Intech Open: London, UK, 2021; pp. 37–67. ISBN 978-1-83962-309-7. [Google Scholar]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef]
- Hoffman, D.L.; Brookes, P.S. Oxygen sensitivity of mitochondrial reactive oxygen species generation depends on metabolic conditions. J. Biol. Chem. 2009, 284, 16236–16245. [Google Scholar] [CrossRef]
- Brzezinski, P.; Moe, A.; Adelroth, P. Structure and Mechanism of Respiratory III–IV Supercomplexes in Bioenergetic Membranes. Chem. Rev. 2021, 121, 9644–9673. [Google Scholar] [CrossRef] [PubMed]
- Dudkina, N.V.; Kouril, R.; Peters, K.; Braun, H.P.; Boekema, E.J. Structure and function of mitochondrial supercomplexes. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, S.; Chesnokov, Y.; Kamyshinsky, R.; Panteleeva, A.; Lyamzaev, K.; Vasilov, R.; Yaguzhinsky, L. Ordered Clusters of the Complete Oxidative Phosphorylation System in Cardiac Mitochondria. Int. J. Mol. Sci. 2021, 22, 1462. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, S.V.; Yaguzhinsky, L.S.; Vasilov, R.G.; Kadantsev, V.N.; Goltsov, A.N. Contribution of the Collective Excitations to the Coupled Proton and Energy Transport along Mitochondrial Cristae Membrane in Oxidative Phosphorylation System. Entropy 2022, 24, 1813. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; de Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. Subcell. Biochem. 2018, 87, 167–227. [Google Scholar]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell. 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Chapa-Dubocq, X.R.; Khuchua, Z.; Camara, A.K. Mitochondrial respiratory supercomplexes in mammalian cells: Structural versus functional role. J. Mol. Med. 2021, 99, 57–73. [Google Scholar] [CrossRef]
- Nath, S. Supercomplex supercomplexes: Raison d’etre and functional significance of supramolecular organization in oxidative phosphorylation. Biomol. Concepts 2022, 13, 272–288. [Google Scholar] [CrossRef]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilization. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, S.A.; Morley, G.E. Gap junctions and propagation of the cardiac action potential. Adv. Cardiol. 2006, 42, 71–85. [Google Scholar] [PubMed]
- Wang, Y.; Mohsen, A.W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef]
- MacDonald, M.J. High content of mitochondrial glycerol-3-phosphate dehydrogenase in pancreatic islets and its inhibition by diazoxide. J. Biol. Chem. 1981, 256, 8287–8290. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free. Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Perevoshchikova, I.V.; Quinlan, C.L.; Orr, A.O.; Gerencser, A.A.; Brand, M.D. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2013, 61C, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V. Synergistic Oxidation of Fatty Acids, Glucose, and Amino Acids Metabolites by Isolated Rat Heart Mitochondria. EC Cardiol. 2018, 5, 98–208. [Google Scholar]
- Panov, A.; Orynbayeva, Z. Determination of mitochondrial metabolic phenotype through investigation of the intrinsic inhibition of succinate dehydrogenase. Analyt. Biochem. 2018, 552, 30–37. [Google Scholar] [CrossRef]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta Mol. Basis Dis. 1995, 1271, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 721–726. [Google Scholar] [CrossRef]
- Francisco, A.; Ronchi, J.A.; Navarro, C.D.; Figueira, T.R.; Castilho, R.F. Nicotinamide nucleotide transhydrogenase is required for brain mitochondrial redox balance under hampered energy substrate metabolism and a high-fat diet. J. Neurochem. 2018, 147, 663–677. [Google Scholar] [CrossRef]
- Ronchi, J.A.; Francisco, A.; Passos, L.A.; Figueira, T.R.; Castilho, R.F. The Contribution of Nicotinamide Nucleotide Transhydrogenase to Peroxide Detoxification Is Dependent on the Respiratory State and Counterbalanced by Other Sources of NADPH in Liver Mitochondria. J. Biol. Chem. 2016, 291, 20173–20187. [Google Scholar] [CrossRef]
- Mühleip, A.; Flygaard, R.K.; Baradaran, R.; Haapanen, O.; Gruhl, T.; Tobiasson, V.; Maréchal, A.; Sharma, V.; Amunts, A. Structural basis of mitochondrial membrane bending by the I-II-III2-IV2 supercomplex. Nature 2023, 615, 934–938. [Google Scholar] [CrossRef]
- Muhleip, A.; McComas, S.E.; Amunts, A. Structure of a mitochondrial ATP synthase with bound native cardiolipin. Elife 2019, 8, e51179. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2084–2091. [Google Scholar] [CrossRef]
- Harris, S.I.; Balaban, R.S.; Barrett, L.; Mandel, L.J. Mitochondrial respiratory capacity and Na+- and K+-dependent adenosine triphosphatase-mediated ion transport in the intact renal cell. J. BioI. Chem. 1981, 256, 10319–10328. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. [Google Scholar] [CrossRef]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Harner, M.; Körner, C.; Walther, D.; Mokranjac, D.; Kaesmacher, J.; Welsch, U.; Griffith, J.; Mann, M.; Reggiori, F.; Neupert, W. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011, 30, 4356–4370. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, M.; Horvath, S.E.; Pfanner, N. Mitochondrial contact site and cristae organizing system. Curr. Opin. Cell Biol. 2016, 41, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.N.A.H.; McElhaney, R.N. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Panov, A. Perhydroxyl radical (HO2•) as an inducer of the isoprostane lipid peroxidation in mitochondria. Mol. Biol. 2018, 52, 295–305. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov, S.I. Cardiolipin, Perhydroxyl Radicals and Lipid Peroxidation in Mitochondrial Dysfunctions and Aging. Oxidative Med. Cell. Longev. 2020, 2020, 1323028. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M. Cardiolipin remodeling and the function of tafazzin. Biochim. Biophys. Acta 2013, 1831, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Sparagna, G.C.; Lesnefsky, E.J. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 2009, 53, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Cheng, I.F.; Chowdhury, A.; Wozny, K.; Balleininger, M.; Reinhold, R.; Grunau, S.; Callegari, S.; Toischer, K.; Wanders, R.J.; et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol. Med. 2016, 8, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Shen, Z.; Greenberg, M.L. Cardiolipin remodeling: A regulatory hub for modulating cardiolipin metabolism and function. J. Bioenerg. Biomembr. 2016, 48, 113–123. [Google Scholar] [CrossRef]
- Lee, H.J.; Mayette, J.; Rapoport, S.I.; Bazinet, R.P. Selective remodeling of cardiolipin fatty acids in the aged rat heart. Lipids Health Dis. 2006, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, J.; Yang, K.; Zhao, Z.; Abendschein, D.R.; Gross, R.W. Alterations in myocardial cardiolipin content and composition occur at the very earliest stages of diabetes: A shotgun lipidomics study. Biochemistry 2007, 46, 6417–6428. [Google Scholar] [CrossRef] [PubMed]
- Guertl, B.; Noehammer, C.; Hoefler, G. Metabolic cardiomyopathies. Int. J. Exp. Pathol. 2000, 81, 349–372. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium. 2009, 45, 643–650. [Google Scholar] [CrossRef]
- Fried, L.P.; Ferrucci, L.; Darer, J.; Williamson, J.D.; Anderson, G. Untangling the concepts of disability, frailty, and Geriatrics comorbidity: Implications for improved targeting and care. J. Gerontol. A. Biol. Sci. Med. Sci. 2004, 59, 255–263. [Google Scholar] [CrossRef]
- Gvozdjak, J.; Gvozdjakova, A.; Kucharska, J.; Bada, V.; Kovalikova, V.; Zachar, A. Metabolic disorders of cardiac muscle in alcoholic and smoke cardiomyopathy. Cor. Vasa. 1989, 31, 312–320. [Google Scholar]
- van der Vusse, G.J.; van Bilsen, M.; Glatz, J.F. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc. Res. 2000, 45, 279–293. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Mayatepek, E. Update on mitochondrial fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2000, 33, 467–468. [Google Scholar] [CrossRef]
- Winter, S.C.; Buist, N.R. Cardiomyopathy in childhood, mitochondrial dysfunction, and the role of L-carnitine. Am. Heart J. 2000, 139, S63–S69. [Google Scholar] [CrossRef] [PubMed]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.H.; Taegtmeyer, H. Metabolic gene expression in fetal and failing human heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 2010, 56, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.M.A.; Abouleisa, R.; Hill, B.G. Metabolic Determinants of Cardiomyocyte Proliferation. Stem Cells 2022, 40, 458–467. [Google Scholar] [CrossRef]
- Bartelds, B.; Knoester, H.; Beaufort-Krol, G.C.; Smid, G.B.; Takens, J.; Zijlstra, W.G.; Heymans, H.S.; Kuipers, J.R. Myocardial lactate metabolism in fetal and newborn lambs. Circulation 1999, 99, 1892–1897. [Google Scholar] [CrossRef]
- Cotter, D.G.; d’Avignon, D.A.; Wentz, A.E.; Weber, M.L.; Crawford, P.A. Obligate role for ketone body oxidation in neonatal metabolic homeostasis. J. Biol. Chem. 2011, 286, 6902–6910. [Google Scholar] [CrossRef] [PubMed]
- Anmann, T.; Varikmaa, M.; Timohhina, N.; Tepp, T.; Shevchuk, I.; Chekulayev, V.; Saks, V.; Kaambre, T. Formation of highly organized intracellular structure and energy metabolism in cardiac muscle cells during postnatal development of rat heart. Biochim. Biophys. Acta 2014, 1837, 1350–1361. [Google Scholar] [CrossRef]
- Murphy, M.P.; Chouchani, E.T. Why succinate? Physiological regulation by a mitochondrial coenzyme Q sentinel. Nat. Chem. Biol. 2022, 18, 461–469. [Google Scholar] [CrossRef]
- Deen, P.M.; Robben, J.H. Succinate receptors in the kidney. J. Am. Soc. Nephrol. 2011, 22, 1416–1422. [Google Scholar] [CrossRef]
- Carter, S.L.; Rennie, C.; Tarnopolsky, M.A. Substrate utilization during endurance exercise in men and women after endurance training. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E898–E907. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Giordano, G.; Corradi, D.; Melissari, M.; Lagrasta, C.; Gambert, S.R.; Anversa, P. Gender differences and aging: Effects on the human heart. J. Am. Coll. Cardiol. 1995, 26, 1068–1079. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panov, A.V. The Structure of the Cardiac Mitochondria Respirasome Is Adapted for the β-Oxidation of Fatty Acids. Int. J. Mol. Sci. 2024, 25, 2410. https://doi.org/10.3390/ijms25042410
Panov AV. The Structure of the Cardiac Mitochondria Respirasome Is Adapted for the β-Oxidation of Fatty Acids. International Journal of Molecular Sciences. 2024; 25(4):2410. https://doi.org/10.3390/ijms25042410
Chicago/Turabian StylePanov, Alexander V. 2024. "The Structure of the Cardiac Mitochondria Respirasome Is Adapted for the β-Oxidation of Fatty Acids" International Journal of Molecular Sciences 25, no. 4: 2410. https://doi.org/10.3390/ijms25042410
APA StylePanov, A. V. (2024). The Structure of the Cardiac Mitochondria Respirasome Is Adapted for the β-Oxidation of Fatty Acids. International Journal of Molecular Sciences, 25(4), 2410. https://doi.org/10.3390/ijms25042410