
Loss of 15-Lipoxygenase in Retinodegenerative RCS Rats

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
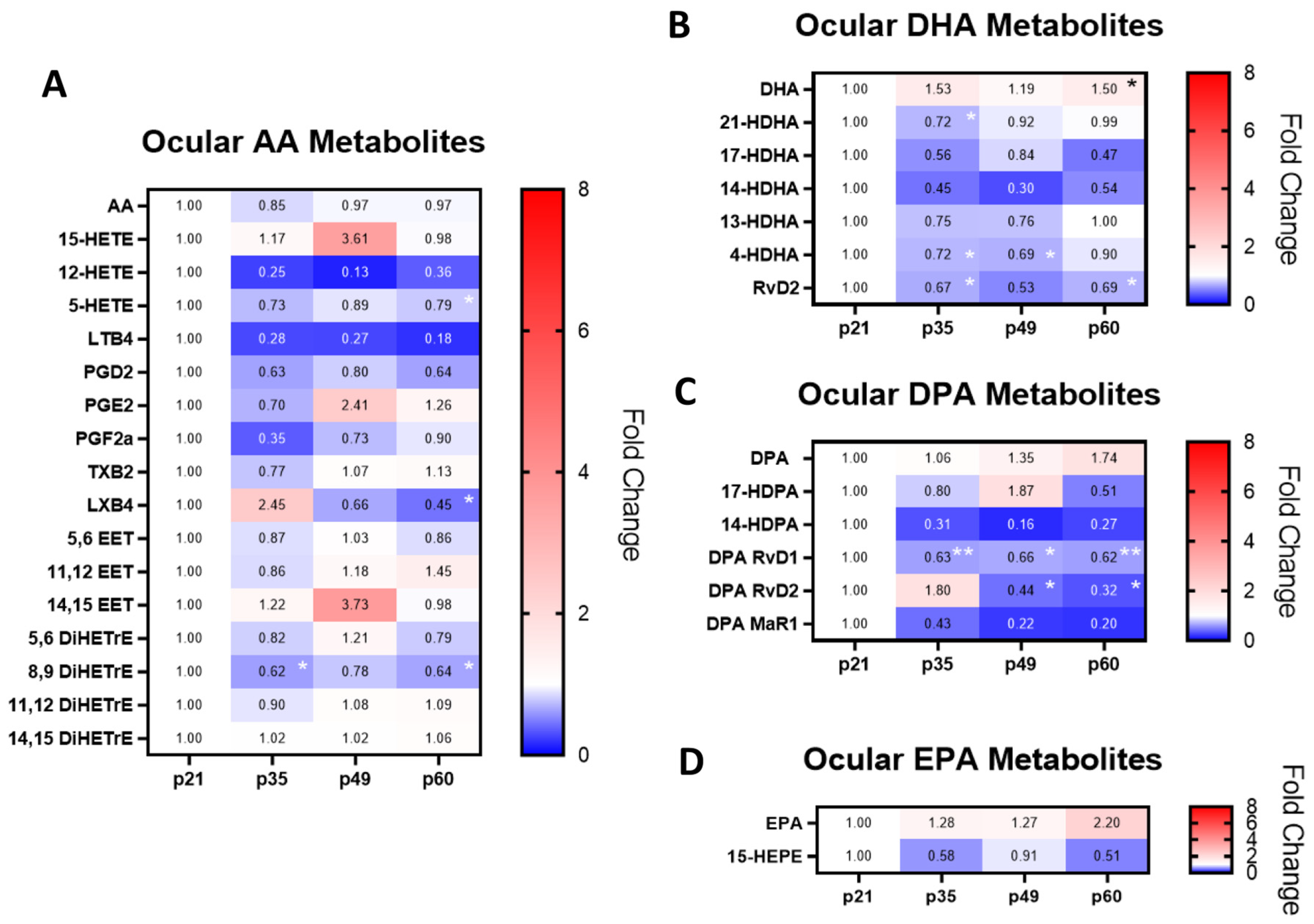
2.1. Ocular PUFAs and Their Metabolites in Relation to Retinodegeneration
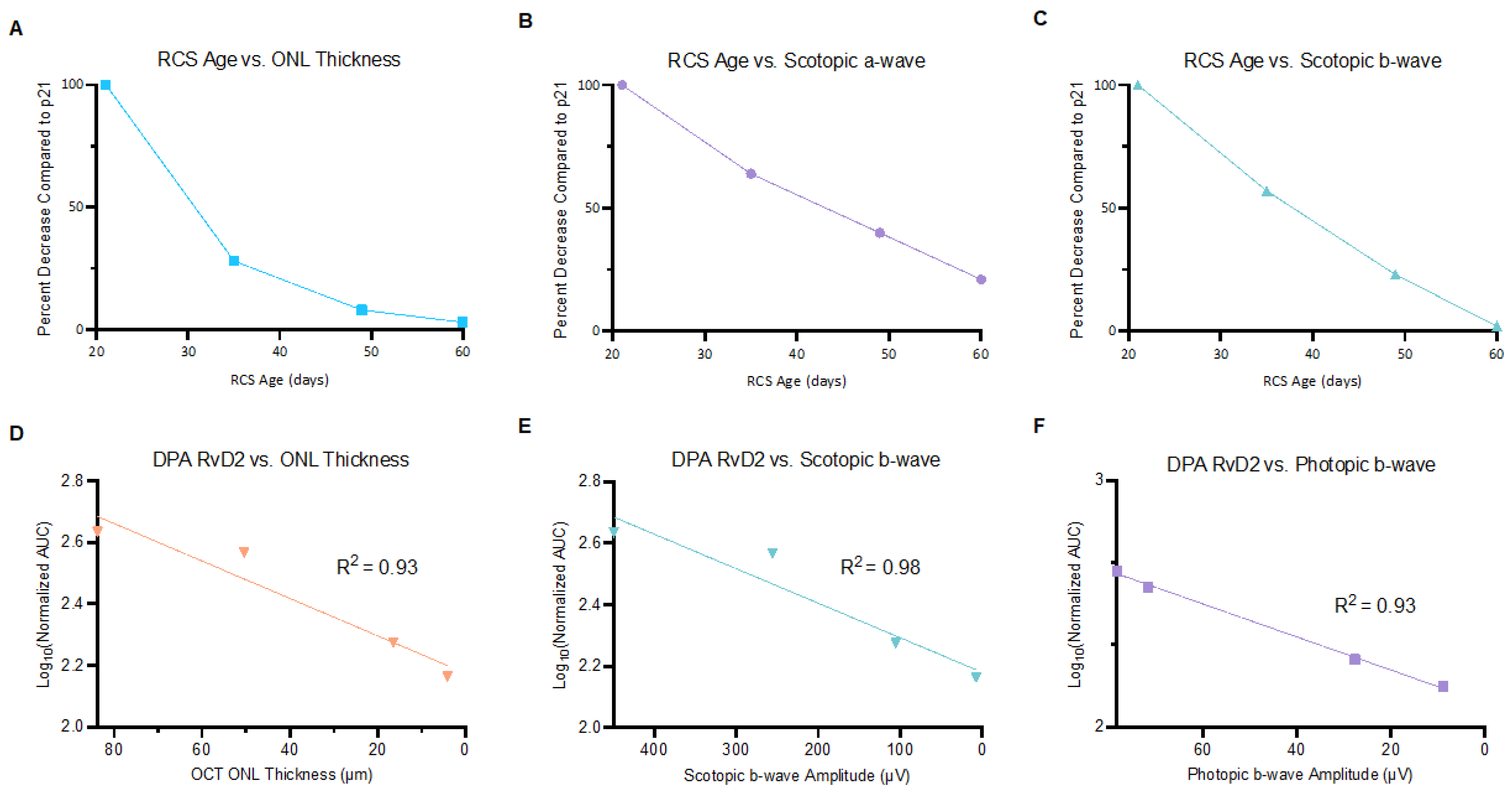
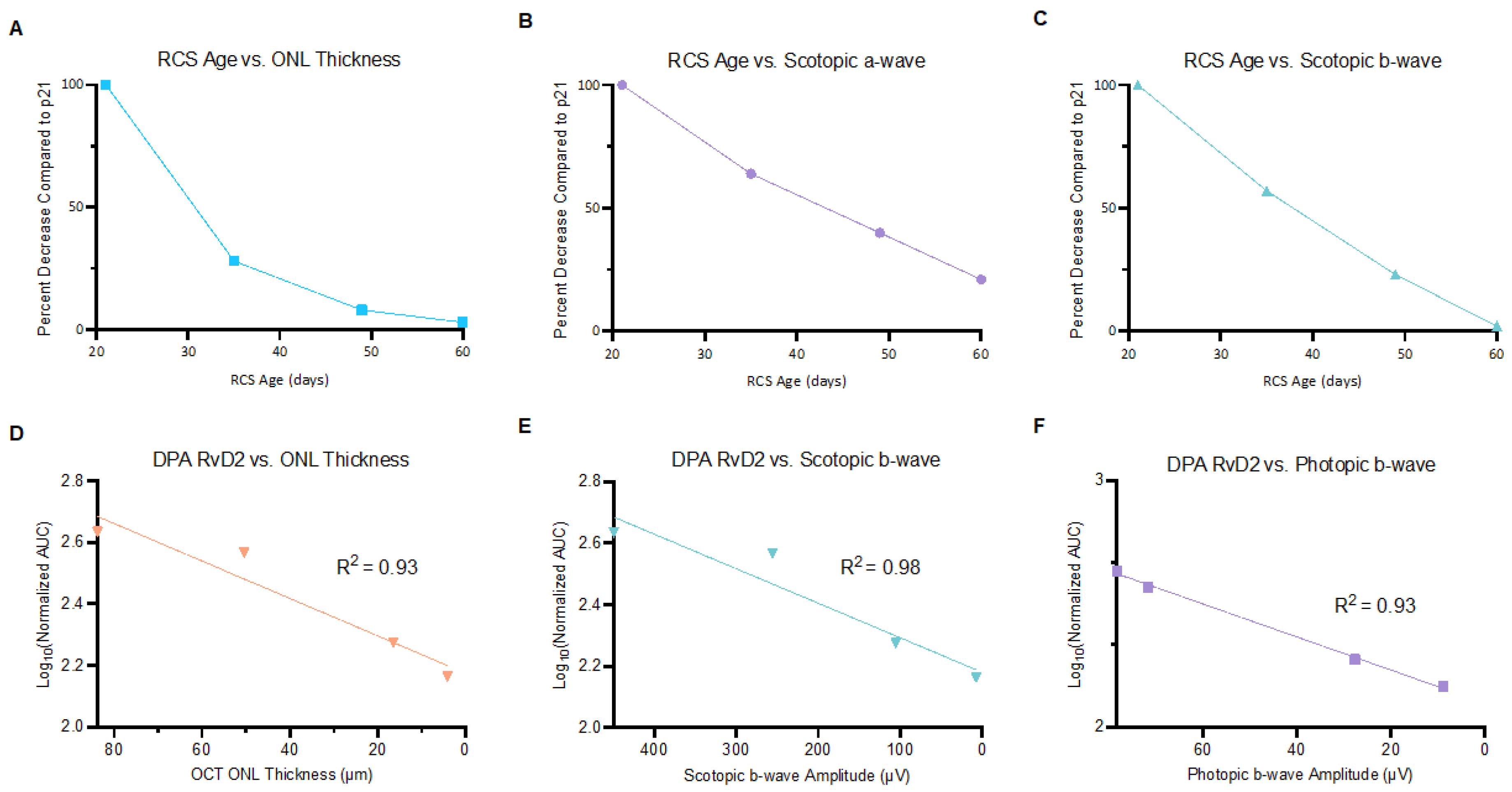
2.2. Correlations of Ocular PUFA Metabolites with Changes in Retinal Structure and Function
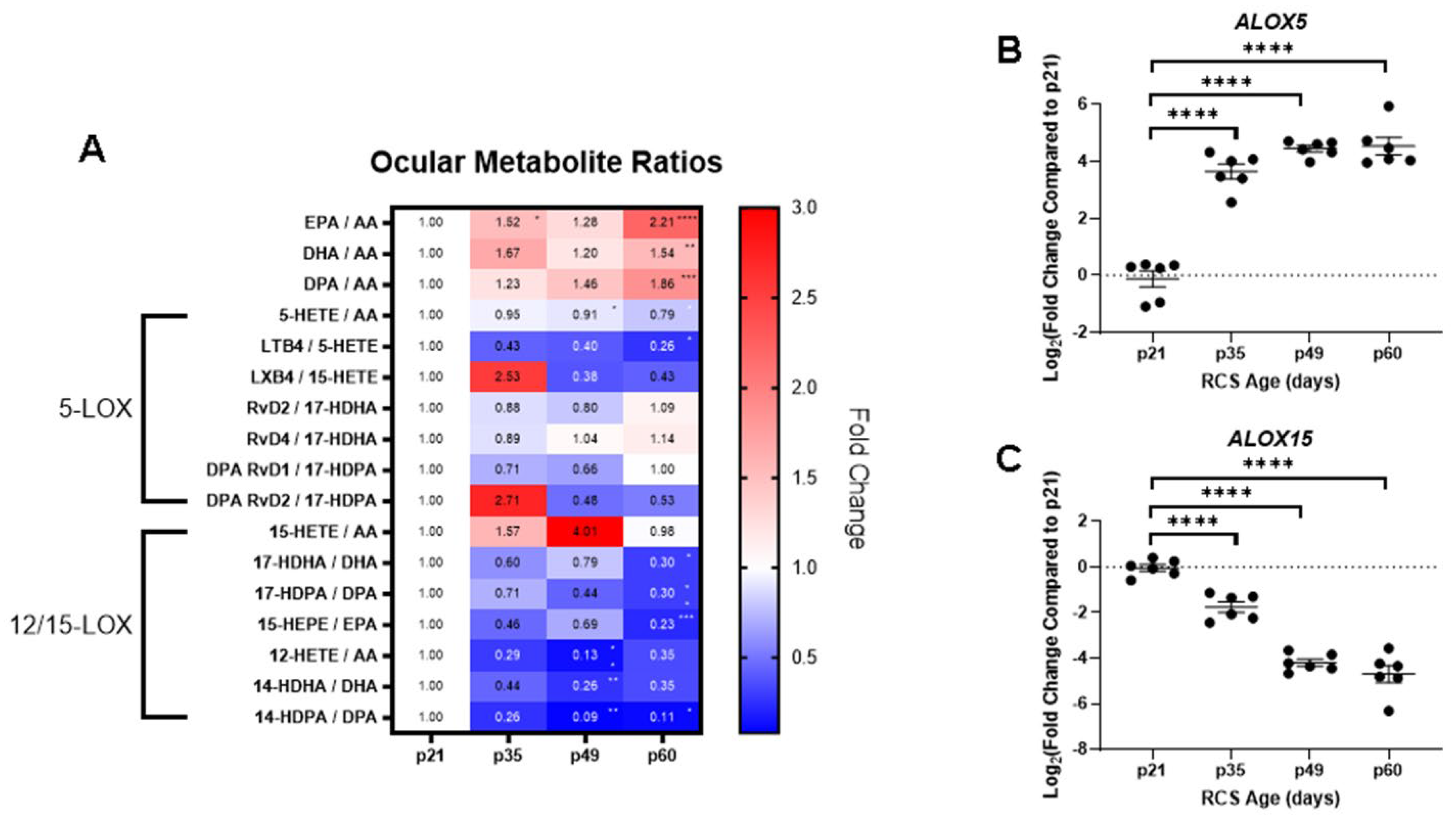
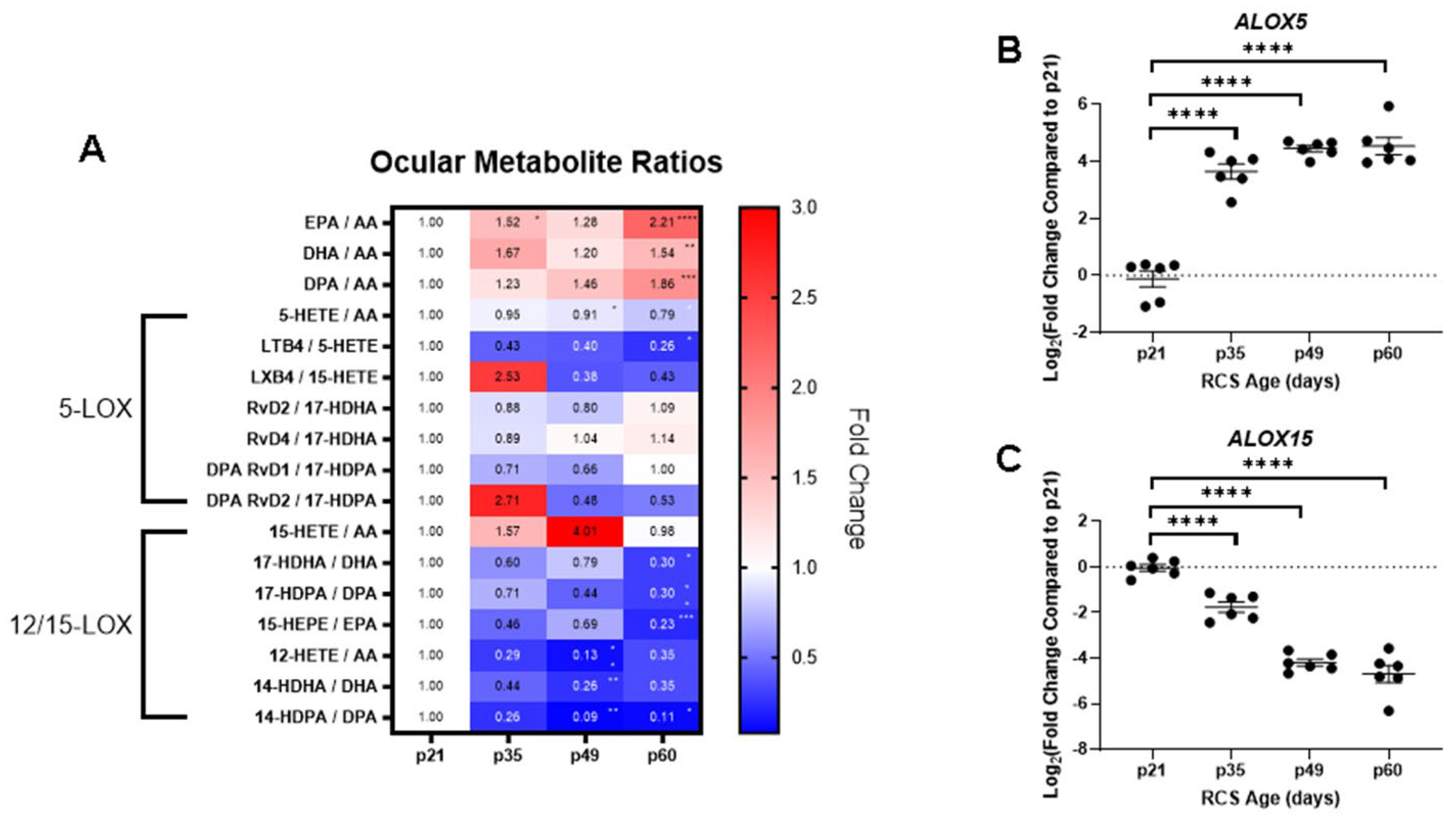
2.3. Ocular Metabolite Ratios and Retinal Lipoxygenase Gene Expression
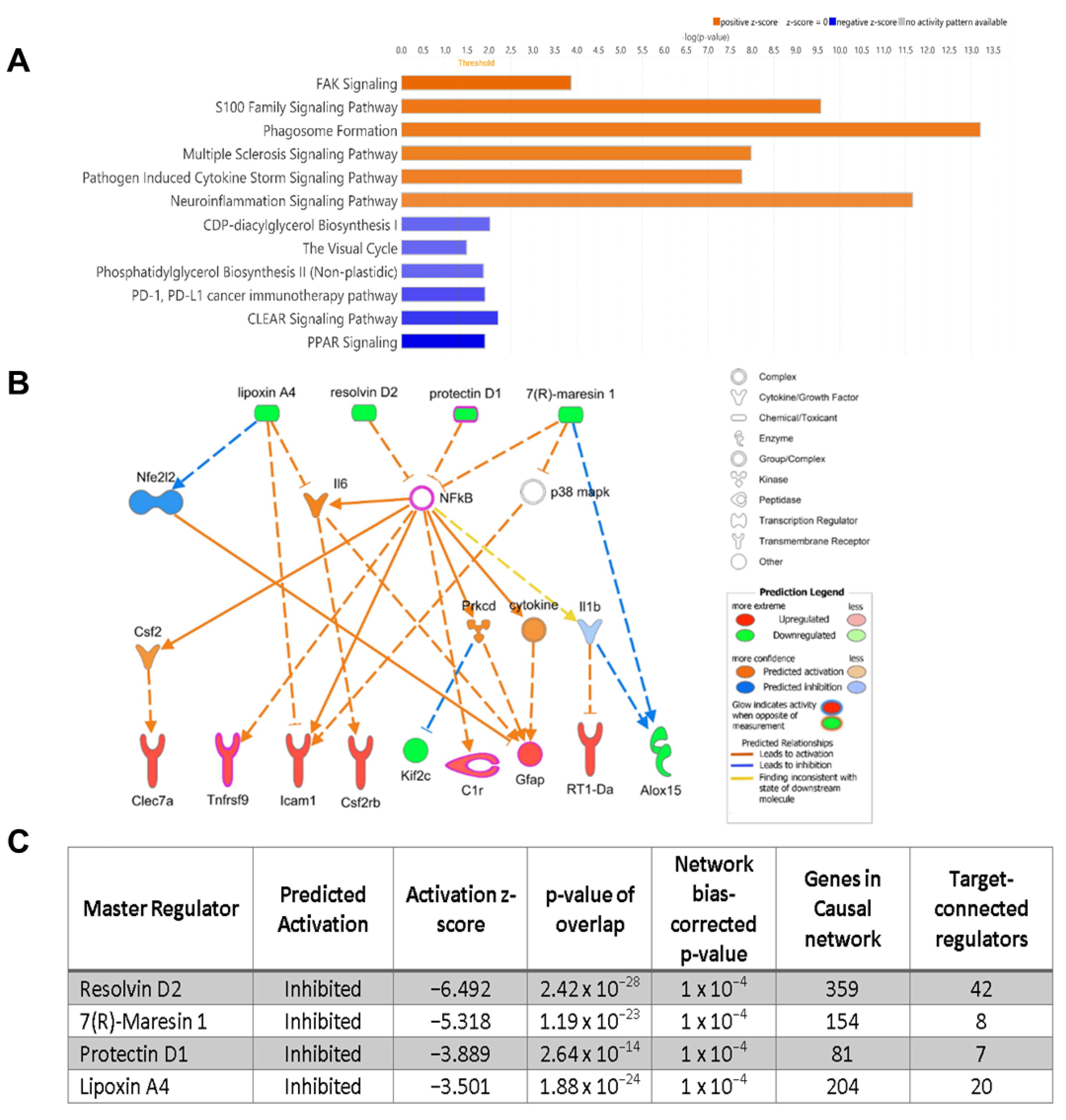
2.4. RNA Sequencing and Ingenuity Pathway Analysis Predicts Declines of SPMs with Disease Progression in RCS Rats
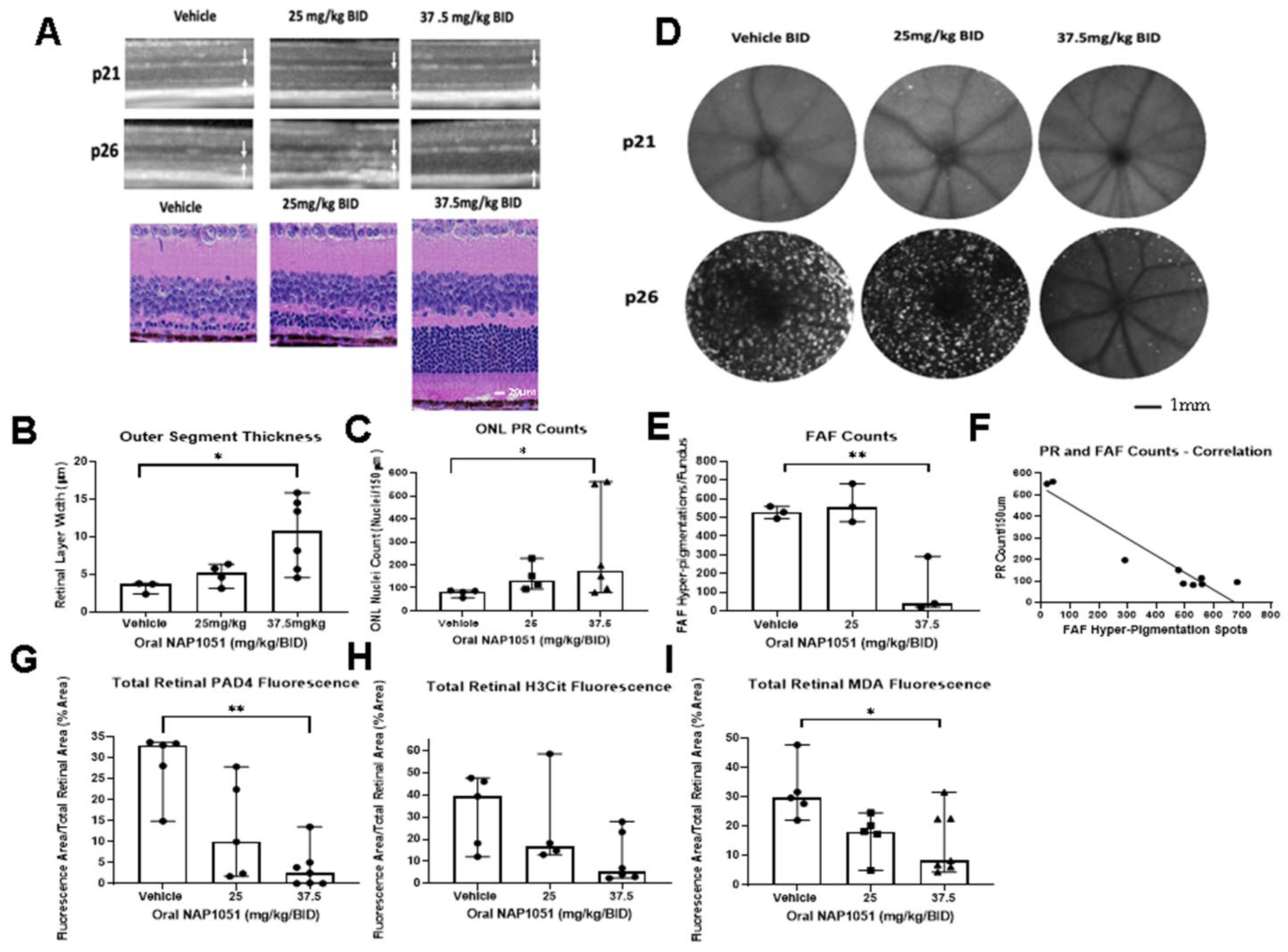
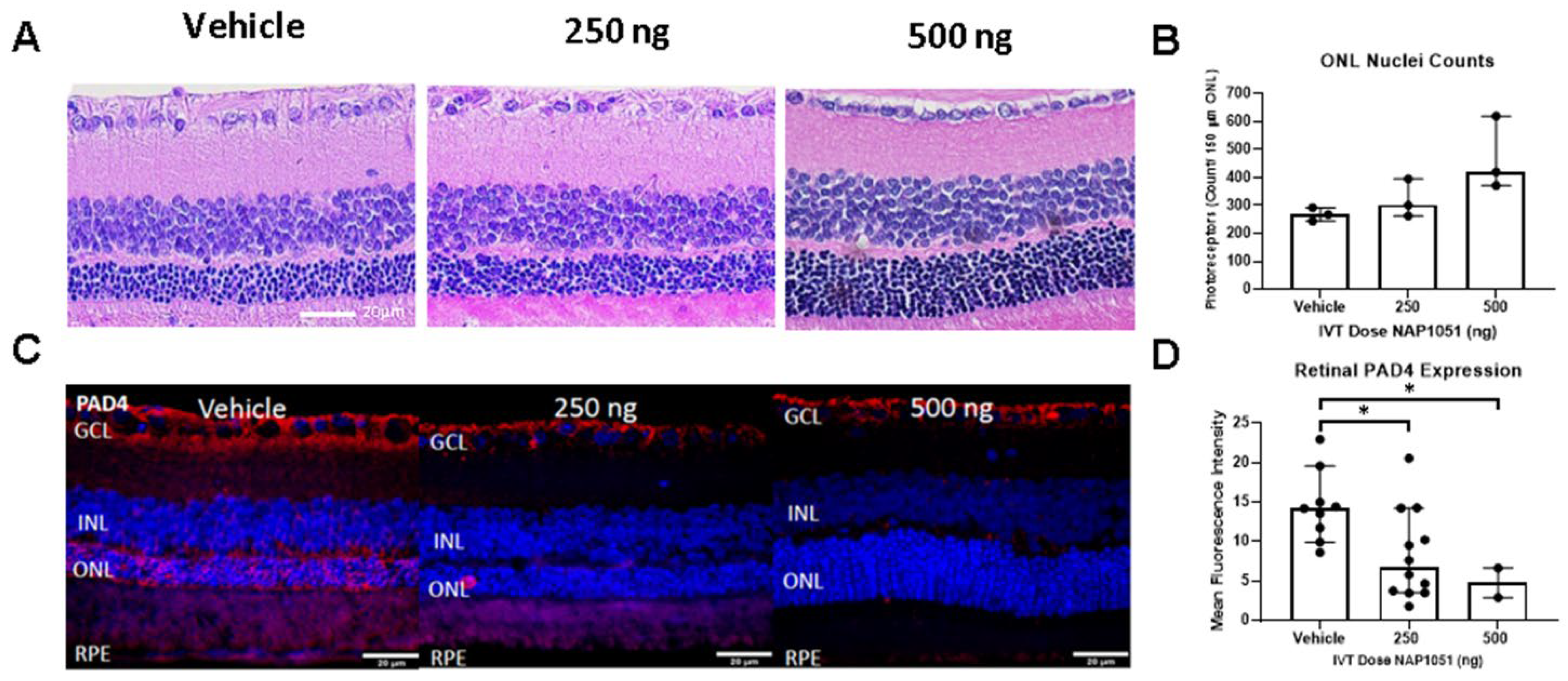
2.5. NAP1051 IVT Administration Demonstrates Preservation of ONL Nuclei and Reductions in PAD4
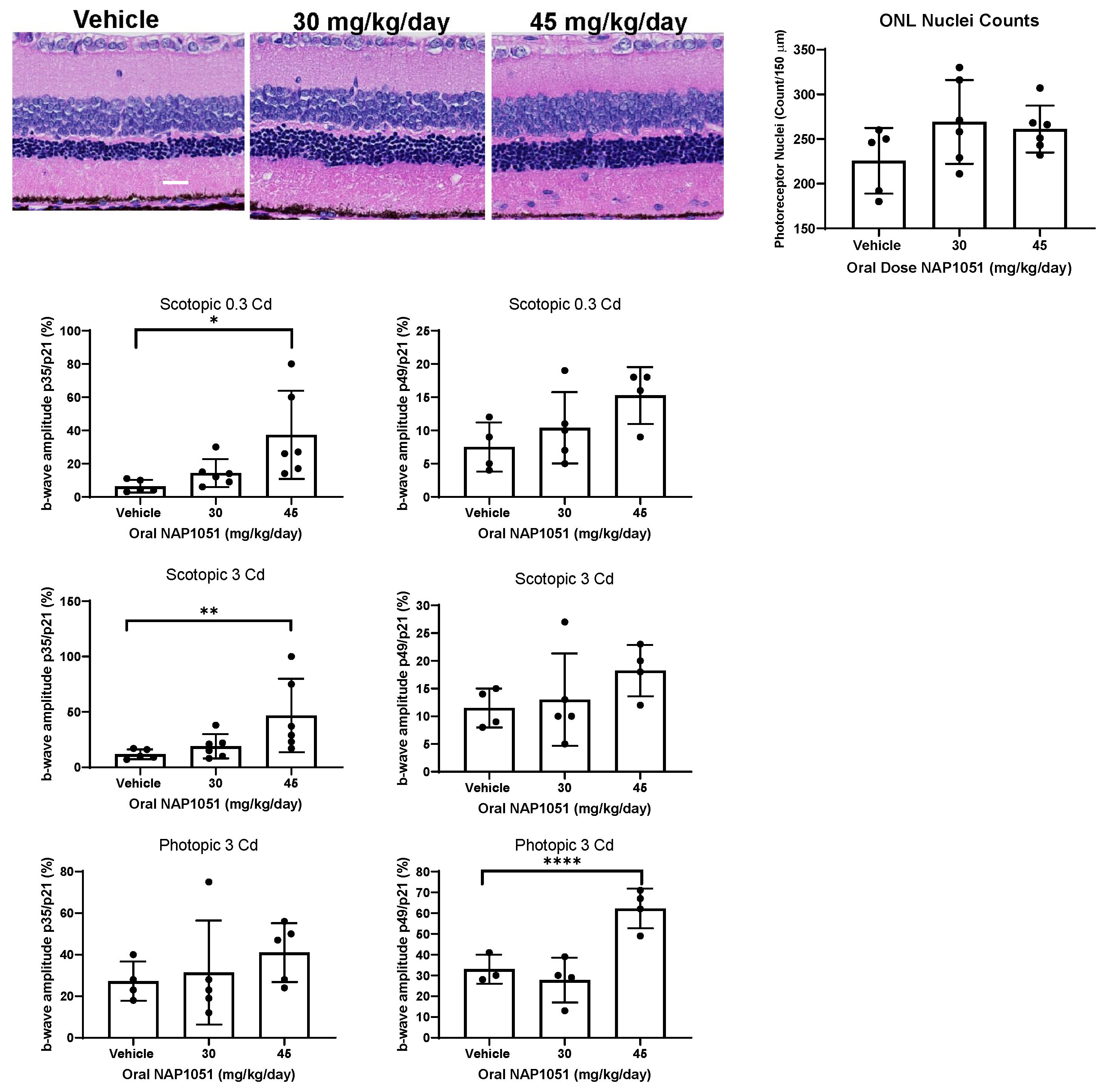
2.6. Oral NAP1051 Administration Preserved Retinal Structure and Function
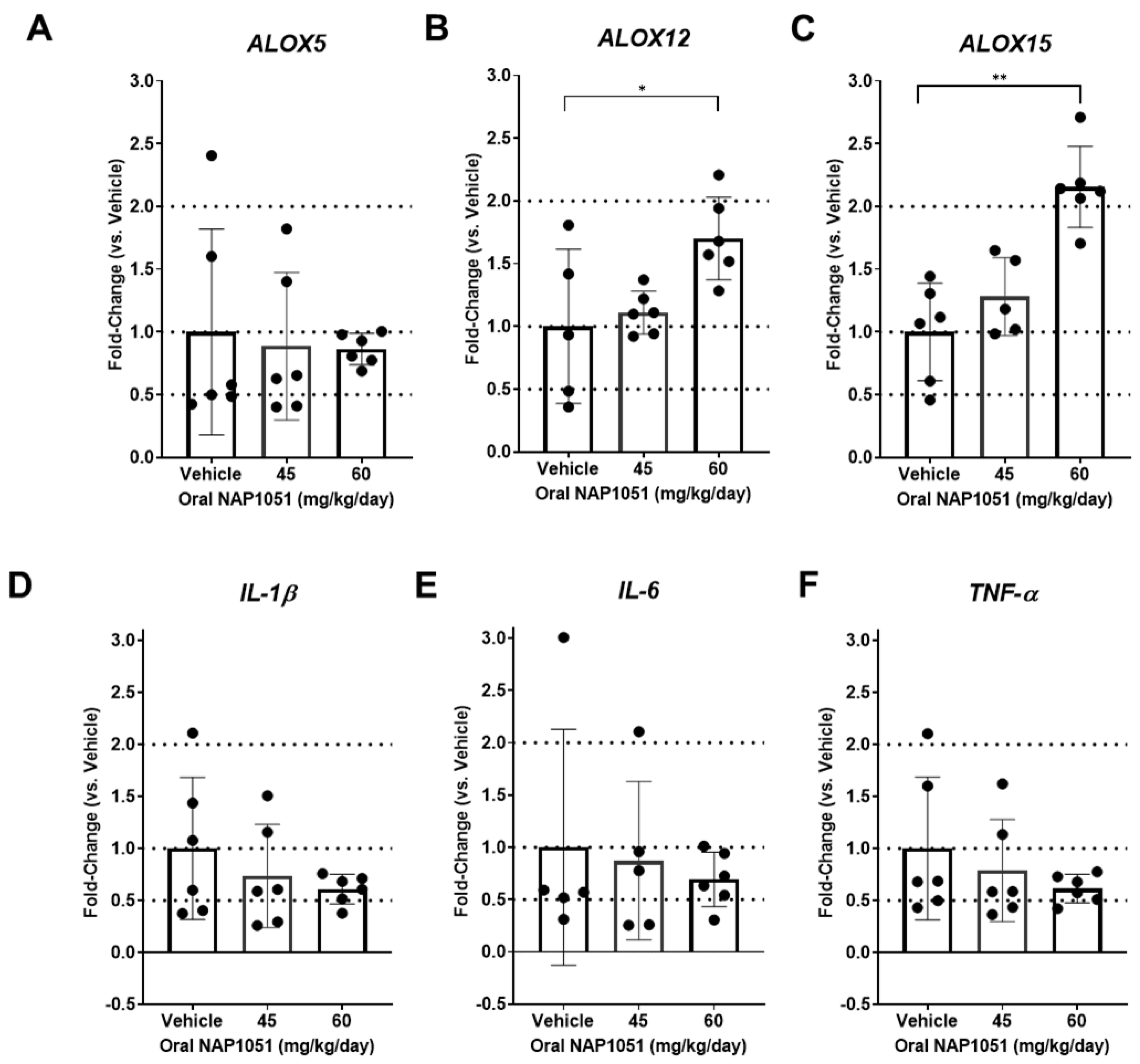
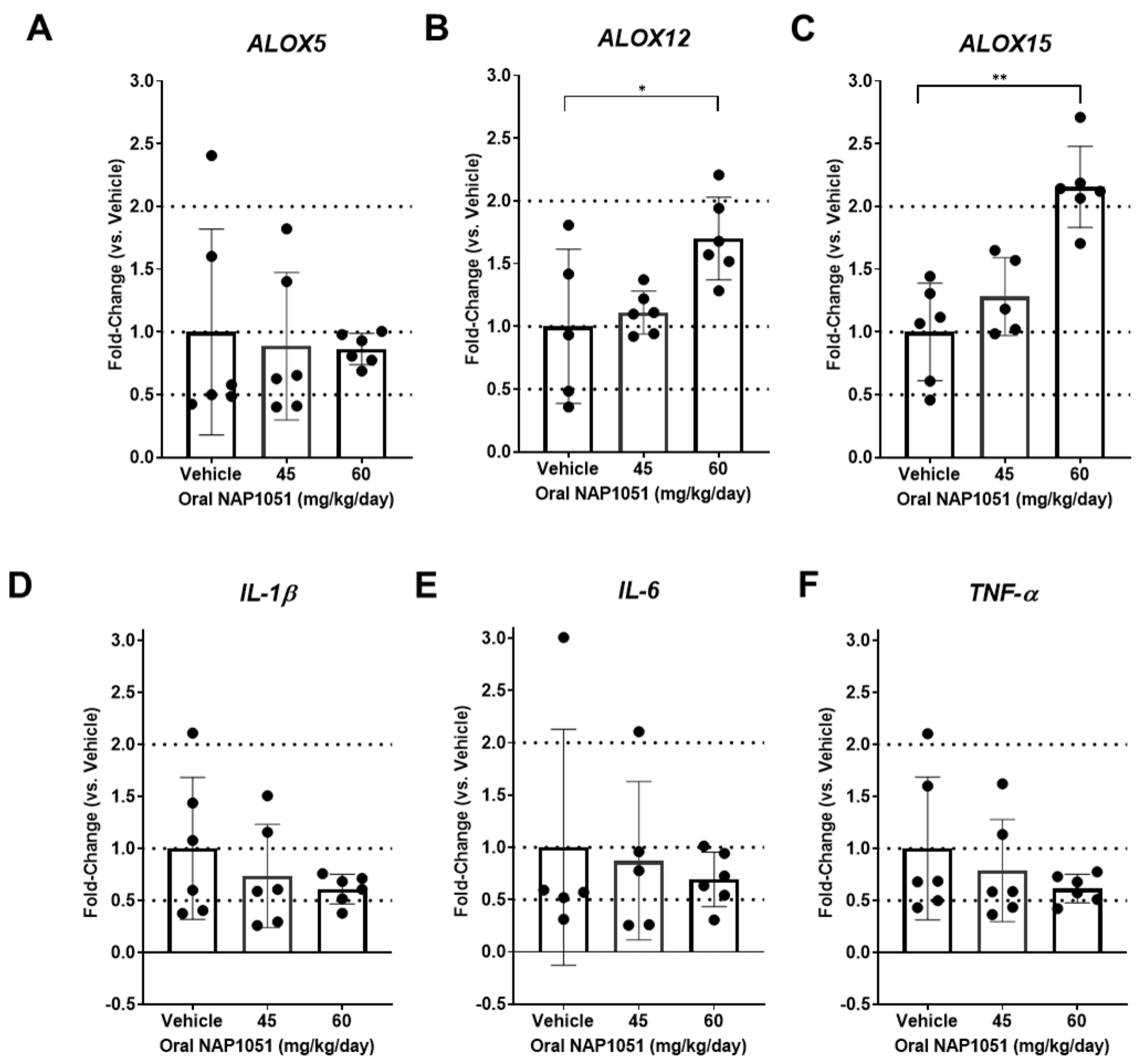
2.7. Effects of NAP1051 on Ocular Gene Expression
2.8. NAP1051 Impact on Lipidomics Analyses of RCS Rat Ocular Lipids
2.9. Oral NAP1051 Administration to rd10 Mice Preserved Retinal ONL PRs, Reduced FAF Appearance, and Reduced Markers of Inflammation and Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Natural Progression Study Design
4.3. Retinal RNA Sequencing and IPA Analyses
4.4. Intravitreal NAP1051 Study Design
4.5. Electroretinogram (ERG) Recordings
4.6. Tissue Isolation and Preparation of Histological Sections
4.7. ImageJ/FIJI Quantification of H&E ONL Nuclei, FAF Hyperpigmentation, and Retinal Layer Thickness
4.8. Immunofluorescent Staining
4.9. RNA Extraction from Ocular Tissue
4.10. cDNA Synthesis
4.11. Ocular qPCR
4.12. LC-MS/MS Lipidomics Assay
4.13. Statistical Analyses
4.14. Ethical Approval
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parmeggiani, F.S.; Sorrentino, F.; Ponzin, D.; Barbaro, V.; Ferrari, S.; Di Iorio, E. Retinitis Pigmentosa: Genes and Disease Mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Vaccarella, L.; Olatunji, S.; Cebulla, C.; Christoforidis, J. Diagnostic Challenges in Retinitis Pigmentosa: Genotypic Multiplicity and Phenotypic Variability. Curr. Genom. 2011, 12, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical Evidence of Sustained Chronic Inflammatory Reaction in Retinitis Pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Vingolo, E.M.; Casillo, L.; Contento, L.; Toja, F.; Florido, A. Retinitis Pigmentosa (RP): The Role of Oxidative Stress in the Degenerative Process Progression. Biomedicines 2022, 10, 582. [Google Scholar] [CrossRef]
- Tylek, K.; Trojan, E.; Regulska, M.; Lacivita, E.; Leopoldo, M.; Basta-Kaim, A. Formyl peptide receptor 2, as an important target for ligands triggering the inflammatory response regulation: A link to brain pathology. Pharmacol. Rep. 2021, 73, 1004–1019. [Google Scholar] [CrossRef]
- Dong, T.; Dave, P.; Yoo, E.; Ebright, B.; Ahluwalia, K.; Zhou, E.; Asante, I.; Salimova, M.; Pei, H.; Lin, T.; et al. NAP1051, a Lipoxin A4 Biomimetic Analogue, Demonstrates Antitumor Activity Against the Tumor Microenvironment. Mol. Cancer Ther. 2021, 20, 2384–2397. [Google Scholar] [CrossRef]
- Gal, A.; Li, Y.; Thompson, D.A.; Weir, J.; Orth, U.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Vollrath, D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef]
- Han, J.; Dinculescu, A.; Dai, X.; Du, W.; Smith, W.C.; Pang, J. Review: The history and role of naturally occurring mouse models with Pde6b mutations. Mol. Vis. 2013, 19, 2579–2589. [Google Scholar]
- Ahluwalia, K.; Martinez-Camarillo, J.-C.; Thomas, B.B.; Naik, A.; Gonzalez-Calle, A.; Pollalis, D.; Lebkowski, J.; Lee, S.Y.; Mitra, D.; Louie, S.G.; et al. Polarized RPE Secretome Preserves Photoreceptors in Retinal Dystrophic RCS Rats. Cells 2023, 12, 1689. [Google Scholar] [CrossRef]
- Balicka, A.; Trbolová, A.; Vrbovská, T. Electroretinography (A Review). Folia Vet. 2016, 60, 53–58. [Google Scholar] [CrossRef]
- The Data Discussed in This Publication Have Been Deposited in NCBI’s Gene Expression Omnibus (Edgaret al., 2002) and Are Accessible through GEO Series Accession Number GSE237804. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE237804 (accessed on 8 December 2022).
- Olivares-González, L.; Velasco, S.; Campillo, I.; Rodrigo, R. Retinal Inflammation, Cell Death and Inherited Retinal Dystrophies. Int. J. Mol. Sci. 2021, 22, 2096. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ali, S.A. Multifunctional Role of S100 Protein Family in the Immune System: An Update. Cells 2022, 11, 2274. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Y.; Wang, X.; Lin, Y.; Zhao, W.; Wu, D.; Pan, J.; Luo, W.; Wang, Y.; Liang, G. FAK mediates LPS-induced inflammatory lung injury through interacting TAK1 and activating TAK1-NFκB pathway. Cell Death Dis. 2022, 13, 589. [Google Scholar] [CrossRef] [PubMed]
- Remels, A.H.V.; Langen, R.C.J.; Gosker, H.R.; Russell, A.P.; Spaapen, F.; Voncken, J.W.; Schrauwen, V.P.; Schols, A.M.W.J. PPARγ inhibits NFκB-dependent transcriptional activation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E174–E183. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, L.; Li, J.; Fang, Z.; Li, S.; Peng, Y.; Zhang, M.; Wang, X. Effect of RvD1/FPR2 on inflammatory response in chorioamnionitis. J. Cell. Mol. Med. 2020, 24, 13397–13407. [Google Scholar] [CrossRef]
- Palmieri, M.; Impey, S.; Kang, H.; Di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef]
- Shi, H.; Yin, Z.; Koronyo, Y.; Fuchs, D.T.; Sheyn, J.; Davis, M.R.; Wilson, J.W.; Margeta, M.A.; Pitts, K.M.; Herron, S.; et al. Regulating microglial miR-155 transcriptional phenotype alleviates Alzheimer’s-induced retinal vasculopathy by limiting Clec7a/Galectin-3+ neurodegenerative microglia. Acta Neuropathol. Commun. 2022, 10, 136. [Google Scholar] [CrossRef]
- Yeo, Y.A.; Martínez Gómez, J.M.; Croxford, J.L.; Gasser, S.; Ling, E.A.; Schwarz, H. CD137 ligand activated microglia induces oligodendrocyte apoptosis via reactive oxygen species. J. Neuroinflamm. 2012, 9, 173. [Google Scholar] [CrossRef]
- Saita, K.; Moriuchi, Y.; Iwagawa, T.; Aihara, M.; Takai, Y.; Uchida, K.; Watanabe, S. Roles of CSF2 as a modulator of inflammation during retinal degeneration. Cytokine 2022, 158, 155996. [Google Scholar] [CrossRef]
- Lewis, G.P.; Fisher, S.K. Up-Regulation of Glial Fibrillary Acidic Protein in Response to Retinal Injury: Its Potential Role in Glial Remodeling and a Comparison to Vimentin Expression. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 263–290. Available online: https://linkinghub.elsevier.com/retrieve/pii/S0074769603300051 (accessed on 9 January 2024).
- Queck, A.; Fink, A.F.; Sirait-Fischer, E.; Rüschenbaum, S.; Thomas, D.; Snodgrass, R.G.; Geisslinger, G.; Baba, H.A.; Trebicka, J.; Zeuzem, S.; et al. Alox12/15 Deficiency Exacerbates, While Lipoxin A4 Ameliorates Hepatic Inflammation in Murine Alcoholic Hepatitis. Front. Immunol. 2020, 11, 1447. [Google Scholar] [CrossRef]
- Ripps, H. Cell Death in Retinitis Pigmentosa: Gap Junctions and the ‘Bystander’ Effect. Exp. Eye Res. 2002, 74, 327–336. [Google Scholar] [CrossRef]
- Gargini, C.; Terzibasi, E.; Mazzoni, F.; Strettoi, E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: A morphological and ERG study. J. Comp. Neurol. 2007, 500, 222–238. [Google Scholar] [CrossRef]
- Nair, A.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic. Clin. Pharm. 2016, 7, 27. [Google Scholar] [CrossRef]
- Taubitz, T.; Fang, Y.; Biesemeier, A.; Julien-Schraermeyer, S.; Schraermeyer, U. Age, lipofuscin and melanin oxidation affect fundus near-infrared autofluorescence. EBioMedicine 2019, 48, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. Deciphering Complex Interactions in Bioactive Lipid Signaling. Molecules 2023, 28, 2622. [Google Scholar] [CrossRef]
- Rossi, S.; Di Filippo, C.; Gesualdo, C.; Potenza, N.; Russo, A.; Trotta, M.C.; Zippo, M.V.; Maisto, R.; Ferraraccio, F.; Simonelli, F.; et al. Protection from Endotoxic Uveitis by Intravitreal Resolvin D1: Involvement of Lymphocytes, miRNAs, Ubiquitin-Proteasome, and M1/M2 Macrophages. Mediat. Inflamm. 2015, 2015, 149381. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Ding, D.H.; Wang, X.Y.; Sun, Y.Y.; Wu, J. Lipoxin A4 regulates microglial M1/M2 polarization after cerebral ischemia-reperfusion injury via the Notch signaling pathway. Exp. Neurol. 2021, 339, 113645. [Google Scholar] [CrossRef] [PubMed]
- Gronert, K.; Maheshwari, N.; Khan, N.; Hassan, I.R.; Dunn, M.; Laniado Schwartzman, M. A Role for the Mouse 12/15-Lipoxygenase Pathway in Promoting Epithelial Wound Healing and Host Defense. J. Biol. Chem. 2005, 280, 15267–15278. [Google Scholar] [CrossRef]
- Calandria, J.M.; Marcheselli, V.L.; Mukherjee, P.K.; Uddin, J.; Winkler, J.W.; Petasis, N.A.; Bazan, N.G. Selective Survival Rescue in 15-Lipoxygenase-1-deficient Retinal Pigment Epithelial Cells by the Novel Docosahexaenoic Acid-derived Mediator, Neuroprotectin D1. J. Biol. Chem. 2009, 284, 17877–17882. [Google Scholar] [CrossRef]
- Fu, T.; Mohan, M.; Brennan, E.P.; Woodman, O.L.; Godson, C.; Kantharidis, P.; Ritchie, R.G.; Qin, C.X. Therapeutic Potential ofLipoxin A 4 in Chronic Inflammation: Focus on Cardiometabolic Disease. ACS Pharmacol. Transl. Sci. 2020, 3, 43–55. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Serhan, C.N. Novel n-3 Immunoresolvents: Structures and Actions. Sci. Rep. 2013, 3, 1940. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fan, D.; Lei, Q.; Lu, A.; He, X. Roles of Resolvins in Chronic Inflammatory Response. Int. J. Mol. Sci. 2022, 23, 14883. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Zaqout, S.; Becker, L.L.; Kaindl, A.M. Immunofluorescence Staining of Paraffin Sections Step by Step. Front. Neuroanat. 2020, 14, 582218. [Google Scholar] [CrossRef] [PubMed]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA Using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef] [PubMed]
- Ebright, B.; Assante, I.; Poblete, R.A.; Wang, S.; Duro, M.V.; Bennett, D.A.; Arvanitakis, Z.; Louie, S.G.; Yassine, H.N. Eicosanoid lipidome activation in post-mortem brain tissues of individuals with APOE4 and Alzheimer’s dementia. Alzheimer’s Res. Ther. 2022, 14, 152. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mead, A.J.; Ahluwalia, K.; Ebright, B.; Zhang, Z.; Dave, P.; Li, Z.; Zhou, E.; Naik, A.A.; Ngu, R.; Chester, C.; et al. Loss of 15-Lipoxygenase in Retinodegenerative RCS Rats. Int. J. Mol. Sci. 2024, 25, 2309. https://doi.org/10.3390/ijms25042309
Mead AJ, Ahluwalia K, Ebright B, Zhang Z, Dave P, Li Z, Zhou E, Naik AA, Ngu R, Chester C, et al. Loss of 15-Lipoxygenase in Retinodegenerative RCS Rats. International Journal of Molecular Sciences. 2024; 25(4):2309. https://doi.org/10.3390/ijms25042309
Chicago/Turabian StyleMead, Andrew James, Kabir Ahluwalia, Brandon Ebright, Zeyu Zhang, Priyal Dave, Zeyang Li, Eugene Zhou, Aditya Anil Naik, Rachael Ngu, Catherine Chester, and et al. 2024. "Loss of 15-Lipoxygenase in Retinodegenerative RCS Rats" International Journal of Molecular Sciences 25, no. 4: 2309. https://doi.org/10.3390/ijms25042309
APA StyleMead, A. J., Ahluwalia, K., Ebright, B., Zhang, Z., Dave, P., Li, Z., Zhou, E., Naik, A. A., Ngu, R., Chester, C., Lu, A., Asante, I., Pollalis, D., Martinez, J. C., Humayun, M., & Louie, S. (2024). Loss of 15-Lipoxygenase in Retinodegenerative RCS Rats. International Journal of Molecular Sciences, 25(4), 2309. https://doi.org/10.3390/ijms25042309