
Assessment of RAS-RAF-MAPK Pathway Mutation Status in Healthy Skin, Benign Nevi, and Cutaneous Melanomas: Pilot Study Using Droplet Digital PCR

 , ,
, ,  and
and 
Abstract
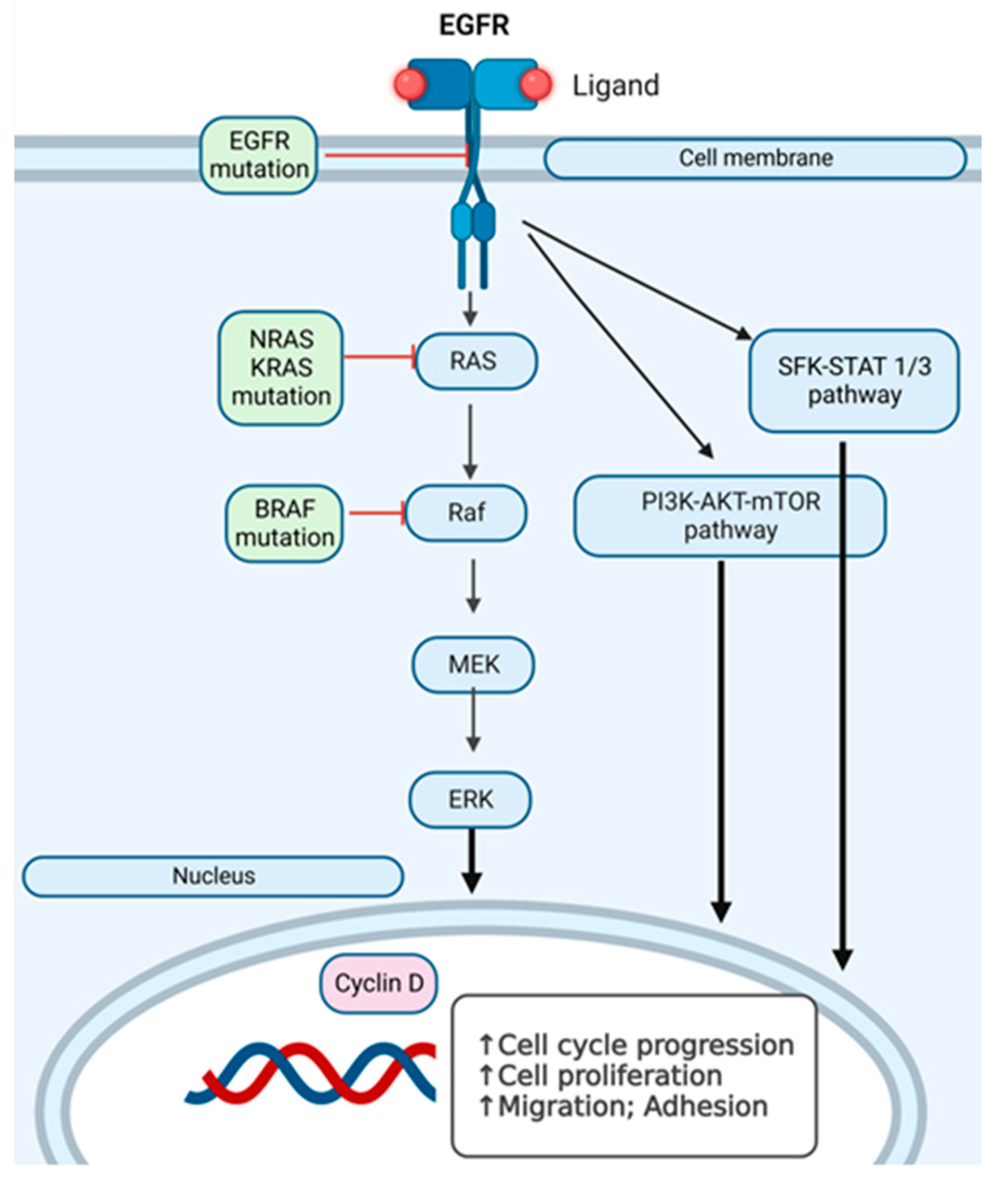
1. Introduction
2. Results
2.1. Histopathological Evaluation of Tissue Samples
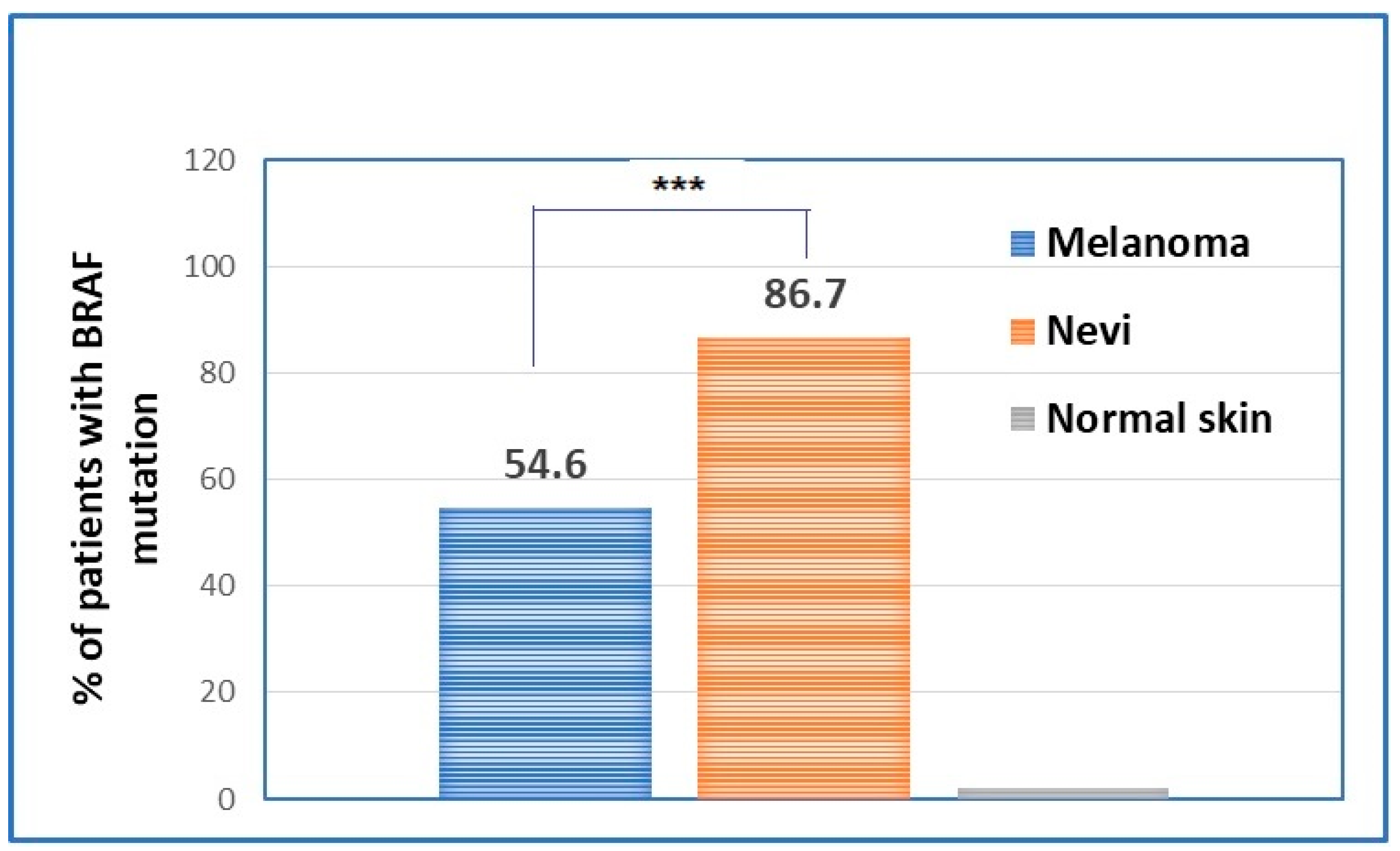
2.2. Evaluation of BRAF Mutational Status in Nevi, Perilesional Skin, and Melanomas
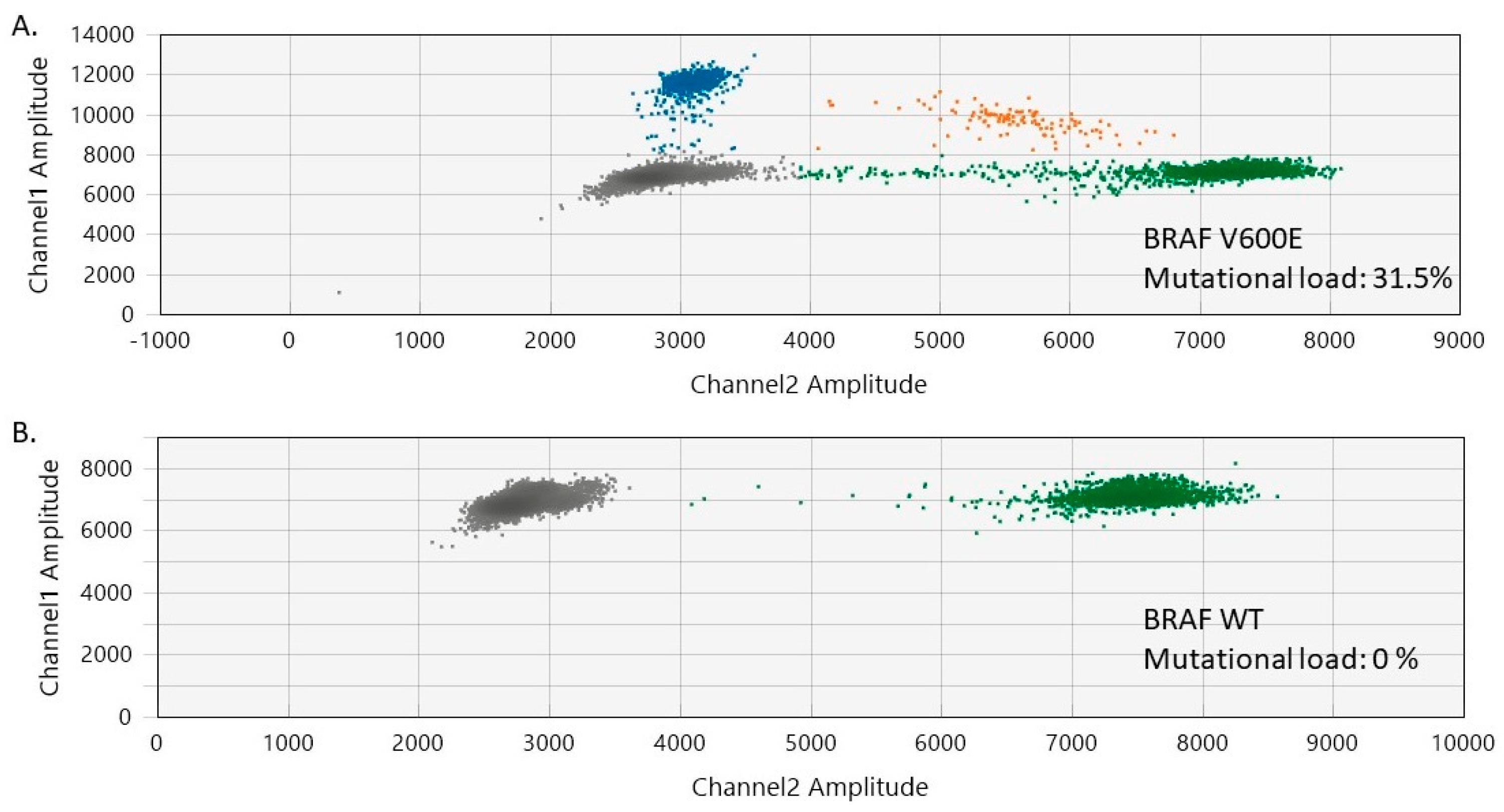
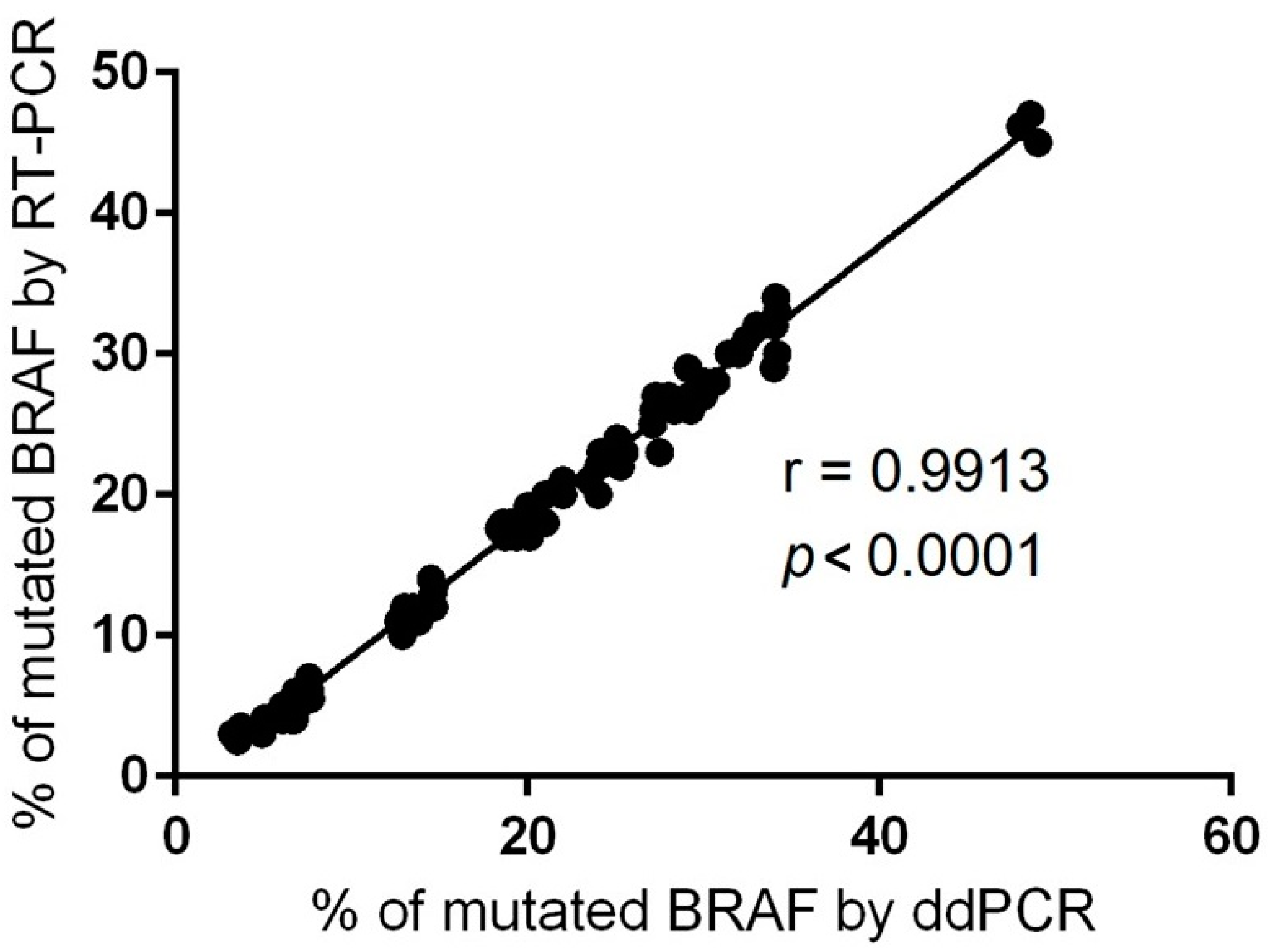
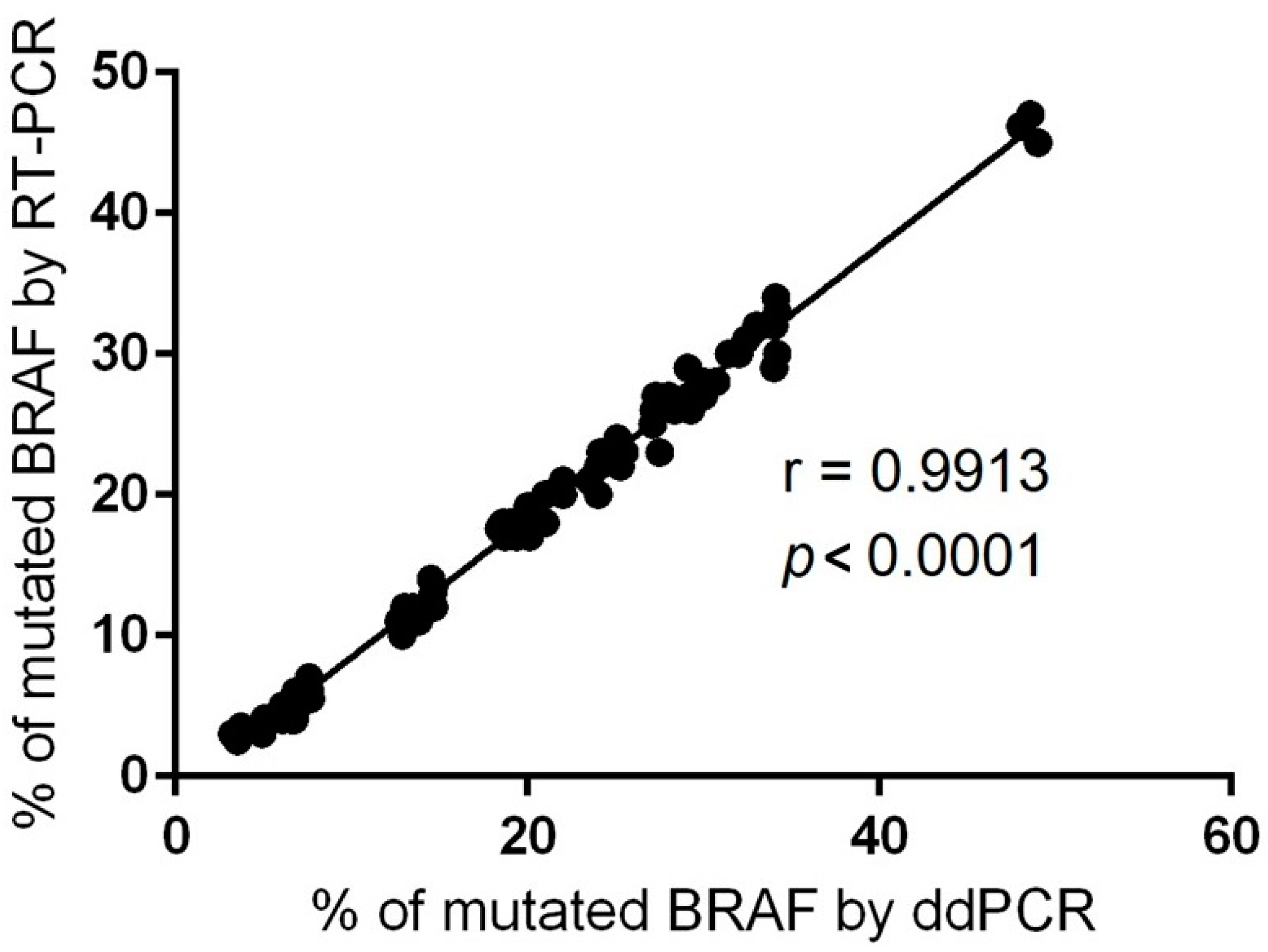
2.3. Evaluation of BRAF Mutational Status in Cutaneous Melanomas
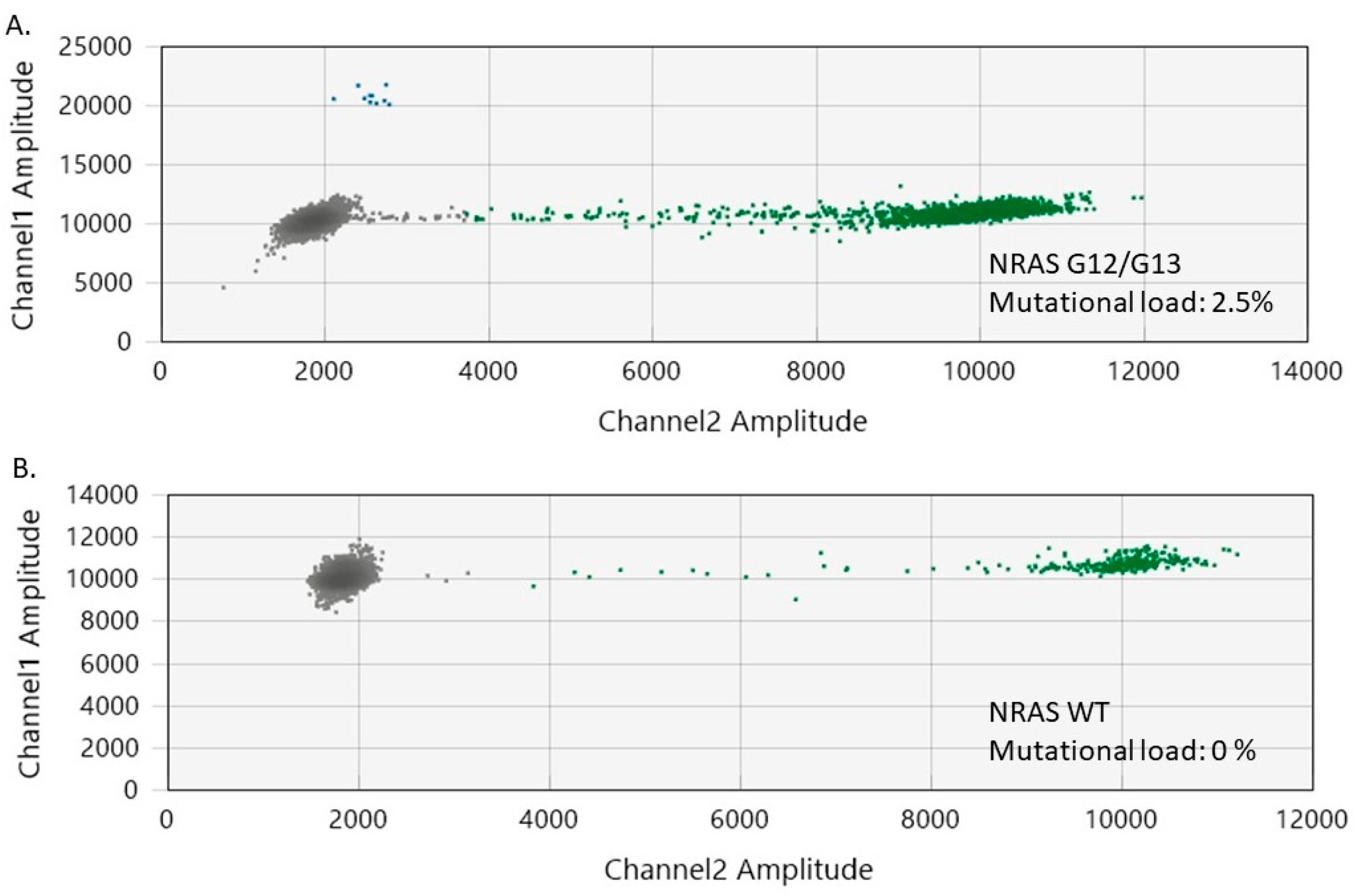
2.4. Evaluation of NRAS Mutational Status in Nevi, Perilesional Skin, and Melanomas
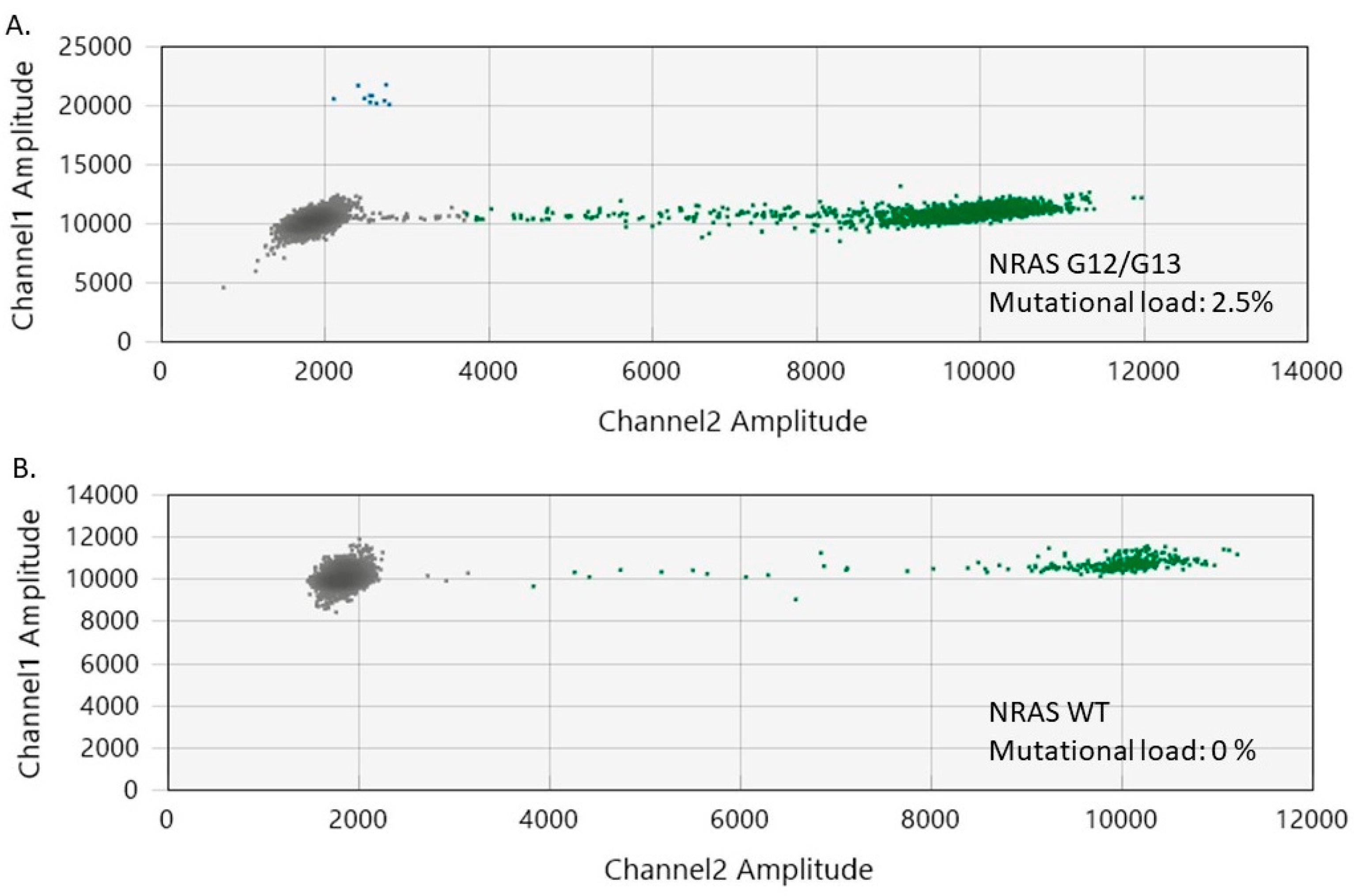
2.5. Evaluation of NRAS Mutational Status in Cutaneous Melanomas
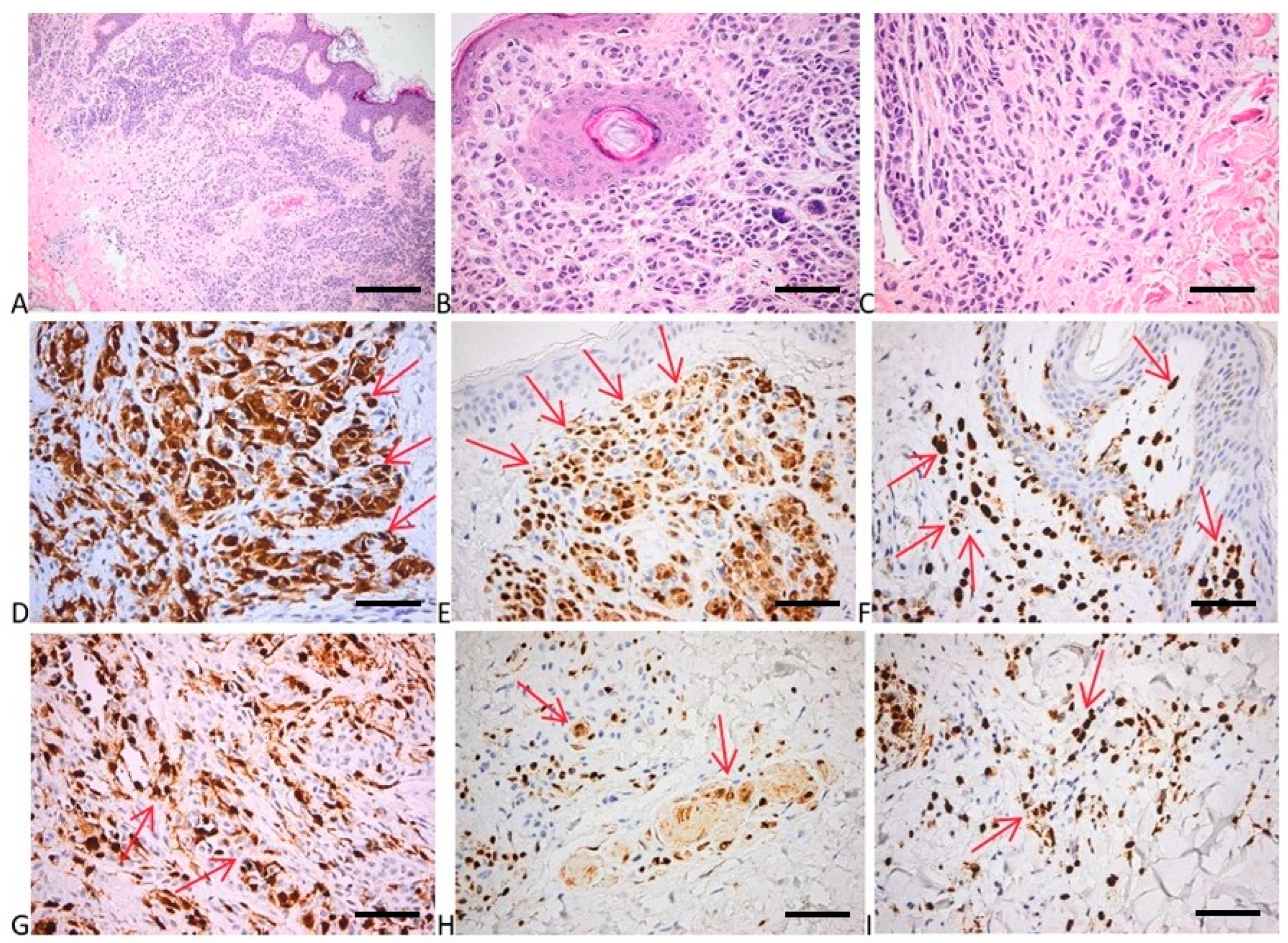
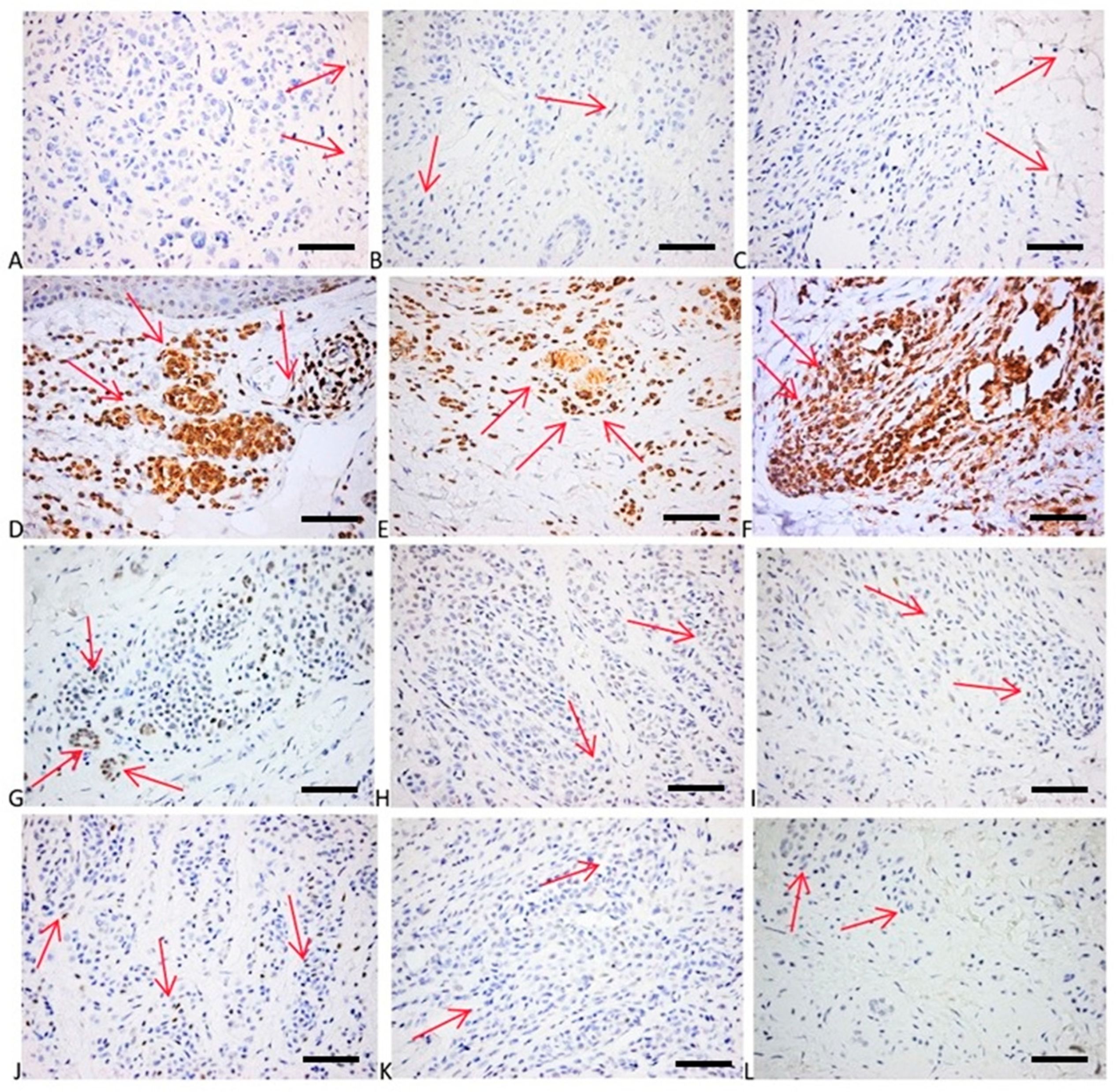
2.6. Immunohistochemical Evaluation of p16, p21, bcl2, p53, and Cyclin D1 Expression
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Histopathological Evaluation of Tumor Samples
4.3. Assessment of the Inflammatory Infiltrate of Cutaneous Tumors
4.4. Immunohistochemical Analysis of p16, p21, bcl2, p53, and Cyclin D1 Expression
4.5. Genomic DNA (gDNA) Isolation from FFPE Tissue Samples
4.6. Uracil-DNA Glycosylase (UDG) Treatment of FFPE gDNA Samples
4.7. Restriction Digestion of the gDNA Samples Prior to ddPCR
4.8. ddPCR Reaction Setup
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Urban, K.; Mehrmal, S.; Uppal, P.; Giesey, R.L.; Delost, G.R. The global burden of skin cancer: A longitudinal analysis from the Global Burden of Disease Study, 1990–2017. JAAD Int. 2021, 2, 98–108. [Google Scholar] [CrossRef]
- Guy, G.P., Jr.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982–2030. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 591–596. [Google Scholar]
- Dimitriou, F.; Krattinger, R.; Ramelyte, E.; Barysch, M.J.; Micaletto, S.; Dummer, R.; Goldinger, S.M. The World of Melanoma: Epidemiologic, Genetic, and Anatomic Differences of Melanoma Across the Globe. Curr. Oncol. Rep. 2018, 20, 87–97. [Google Scholar] [CrossRef]
- Neagu, M.; Constantin, C.; Zurac, S. Immune parameters in the prognosis and therapy monitoring of cutaneous melanoma patients: Experience, role, and limitations. Biomed Res. Int. 2013, 2013, 107940. [Google Scholar] [CrossRef]
- Lopes, J.; Rodrigues, C.M.P.; Gaspar, M.M.; Reis, C.P. Melanoma Management: From Epidemiology to Treatment and Latest Advances. Cancers 2022, 14, 4652. [Google Scholar] [CrossRef]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology 2009, 23, 488–496. [Google Scholar]
- Rebecca, V.W.; Somasundaram, R.; Herlyn, M. Pre-clinical modeling of cutaneous melanoma. Nat. Commun. 2020, 11, 2858–2868. [Google Scholar] [CrossRef]
- Larribère, L.; Utikal, J. Stem Cell-Derived Models of Neural Crest Are Essential to Understand Melanoma Progression and Therapy Resistance. Front. Mol. Neurosci. 2019, 12, 111–120. [Google Scholar] [CrossRef]
- Zhao, X.; Little, P.; Hoyle, A.P.; Pegna, G.J.; Hayward, M.C.; Ivanova, A.; Parker, J.S.; Marron, D.L.; Soloway, M.G.; Jo, H.; et al. The Prognostic Significance of Low-Frequency Somatic Mutations in Metastatic Cutaneous Melanoma. Front. Oncol. 2019, 8, 584–598. [Google Scholar] [CrossRef]
- Wei, L.; Christensen, S.R.; Fitzgerald, M.E.; Graham, J.; Hutson, N.D.; Zhang, C.; Huang, Z.; Hu, Q.; Zhan, F.; Xie, J.; et al. Ultradeep sequencing differentiates patterns of skin clonal mutations associated with sun-exposure status and skin cancer burden. Sci. Adv. 2021, 7, eabd7703. [Google Scholar] [CrossRef]
- Sanna, A.; Harbst, K.; Johansson, I.; Christensen, G.; Lauss, M.; Mitra, S.; Rosengren, F.; Häkkinen, J.; Vallon-Christersson, J.; Olsson, H.; et al. Tumor genetic heterogeneity analysis of chronic sun-damaged melanoma. Pigment Cell Melanoma Res. 2020, 33, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Fewings, E.; Chang, D.; Zeng, H.; Liu, S.; Jorapur, A.; Belote, R.L.; McNeal, A.S.; Tan, T.M.; Yeh, I.; et al. The genomic landscapes of individual melanocytes from human skin. Nature 2020, 586, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; Bastian, B.C. Melanoma pathology: New approaches and classification. Br. J. Dermatol. 2021, 185, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Dobre, E.G.; Constantin, C.; Costache, M.; Neagu, M. Interrogating Epigenome toward Personalized Approach in Cutaneous Melanoma. J. Pers. Med. 2021, 11, 901. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, M.; Tartar, D.M.; Qi, L.; Chen, D.; Yu, L.; Konia, T.; McPherson, J.D.; Murphy, W.J.; Fung, M.A. Improving classification of melanocytic nevi: Association of BRAF V600E expression with distinct histomorphologic features. J. Am. Acad. Dermatol. 2018, 79, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Loras, A.; Gil-Barrachina, M.; Marqués-Torrejón, M.Á.; Perez-Pastor, G.; Martinez-Cadenas, C. UV-Induced Somatic Mutations Driving Clonal Evolution in Healthy Skin, Nevus, and Cutaneous Melanoma. Life 2022, 12, 1339. [Google Scholar] [CrossRef]
- Scolyer, R.A.; Prieto, V.G.; Elder, D.E.; Cochran, A.J.; Mihm, M.C. Classification and Histopathology of Melanoma. In Cutaneous Melanoma; Balch, C.M., Atkins, M.B., Garbe, C., Gershenwald, J.E., Halpern, A.C., Kirkwood, J.M., McArthur, G.A., Thompson, J.F., Sober, A.J., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 317–379. [Google Scholar]
- Gutiérrez-Castañeda, L.D.; Gamboa, M.; Nova, J.A.; Pulido, L.; Tovar-Parra, J.D. Mutations in the BRAF, NRAS, and C-KIT Genes of Patients Diagnosed with Melanoma in Colombia Population. Biomed. Res. Int. 2020, 2020, 2046947. [Google Scholar] [CrossRef]
- Tschandl, P.; Berghoff, A.S.; Preusser, M.; Burgstaller-Muehlbacher, S.; Pehamberger, H.; Okamoto, I.; Kittler, H. NRAS and BRAF mutations in melanoma-associated nevi and uninvolved nevi. PLoS ONE 2013, 8, e69639. [Google Scholar] [CrossRef]
- Schulz, A.; Raetz, J.; Karitzky, P.C.; Dinter, L.; Tietze, J.K.; Kolbe, I.; Käubler, T.; Renner, B.; Beissert, S.; Meier, F.; et al. Head-to-Head Comparison of BRAF/MEK Inhibitor Combinations Proposes Superiority of Encorafenib Plus Trametinib in Melanoma. Cancers 2022, 14, 4930. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Ottaviano, M.; Giunta, E.F.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef]
- Zablocka, T.; Kreismane, M.; Pjanova, D.; Isajevs, S. Effects of BRAF V600E and NRAS mutational status on the progression-free survival and clinicopathological characteristics of patients with melanoma. Oncol. Lett. 2023, 25, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Rozzo, C.; Paliogiannis, P.; Casula, M.; Manca, A.; Doneddu, V.; Fedeli, M.A.; Sini, M.C.; Palomba, G.; Pisano, M.; et al. Comparison of BRAF Mutation Screening Strategies in a Large Real-Life Series of Advanced Melanoma Patients. J. Clin. Med. 2020, 9, 2430. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Erturk, K. BRAF V600E mutation as a prognostic factor in cutaneous melanoma patients. Dermatol. Ther. 2020, 33, e13270. [Google Scholar] [CrossRef]
- Bauer, J.; Büttner, P.; Murali, R.; Okamoto, I.; Kolaitis, N.A.; Landi, M.T.; Scolyer, R.A.; Bastian, B.C. BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res. 2011, 24, 345–351. [Google Scholar] [CrossRef]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Berrino, E.; Balsamo, A.; Pisacane, A.; Gallo, S.; Becco, P.; Miglio, U.; Caravelli, D.; Poletto, S.; Paruzzo, L.; Debernardi, C.; et al. High BRAF variant allele frequencies are associated with distinct pathological features and responsiveness to target therapy in melanoma patients. ESMO Open 2021, 6, 100133. [Google Scholar] [CrossRef]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco. Targets. Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef]
- Posch, C.; Sanlorenzo, M.; Vujic, I.; Oses-Prieto, J.A.; Cholewa, B.D.; Kim, S.T.; Ma, J.; Lai, K.; Zekhtser, M.; Esteve-Puig, R.; et al. Phosphoproteomic Analyses of NRAS(G12) and NRAS(Q61) Mutant Melanocytes Reveal Increased CK2α Kinase Levels in NRAS(Q61) Mutant Cells. J. Investig. Dermatol. 2016, 136, 2041–2048. [Google Scholar] [CrossRef]
- Vanni, I.; Tanda, E.T.; Dalmasso, B.; Pastorino, L.; Andreotti, V.; Bruno, W.; Boutros, A.; Spagnolo, F.; Ghiorzo, P. Non-BRAF Mutant Melanoma: Molecular Features and Therapeutical Implications. Front. Mol. Biosci. 2020, 7, 172–202. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.Y.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef]
- Ellerhorst, J.A.; Greene, V.R.; Ekmekcioglu, S.; Warneke, C.L.; Johnson, M.M.; Cooke, C.P.; Wang, L.E.; Prieto, V.G.; Gershenwald, J.E.; Wei, Q.; et al. Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin. Cancer Res. 2011, 17, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Chiappetta, C.; Proietti, I.; Soccodato, V.; Puggioni, C.; Zaralli, R.; Pacini, L.; Porta, N.; Skroza, N.; Petrozza, V.; Potenza, C.; et al. BRAF and NRAS mutations are heterogeneous and not mutually exclusive in nodular melanoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 172–177. [Google Scholar] [CrossRef]
- Raaijmakers, M.I.G.; Widmer, D.S.; Narechania, A.; Eichhoff, O.; Freiberger, S.N.; Wenzina, J.; Cheng, P.F.; Mihic-Probst, D.; Desalle, R.; Dummer, R.; et al. Co-existence of BRAF and NRAS driver mutations in the same melanoma cells results in heterogeneity of targeted therapy resistance. Oncotarget 2016, 7, 77163–77174. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Rabbie, R.; Ferguson, P.; Wong, K.; Couturier, D.L.; Moran, U.; Turner, C.; Emanuel, P.; Haas, K.; Saunus, J.M.; Davidson, M.R.; et al. The mutational landscape of melanoma brain metastases presenting as the first visceral site of recurrence. Br. J. Cancer 2021, 124, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Pastwińska, J.; Karaś, K.; Karwaciak, I.; Ratajewski, M. Targeting EGFR in melanoma—The sea of possibilities to overcome drug resistance. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188754. [Google Scholar] [CrossRef]
- Grahn, J.C.; Isseroff, R.R. Human melanocytes do not express EGF receptors. J. Investig. Dermatol. 2004, 123, 244–246. [Google Scholar] [CrossRef]
- Akslen, L.A.; Puntervoll, H.; Bachmann, I.M.; Straume, O.; Vuhahula, E.; Kumar, R.; Molven, A. Mutation analysis of the EGFR–NRAS–BRAF pathway in melanomas from black Africans and other subgroups of cutaneous melanoma. Melanoma Res. 2008, 18, 29–35. [Google Scholar] [CrossRef]
- De Wit, P.E.; Moretti, S.; Koenders, P.G.; Weterman, M.A.; van Muijen, G.N.; Gianotti, B.; Ruiter, D.J. Increasing epidermal growth factor receptor expression in human melanocytic tumor progression. J. Investig. Dermatol. 1992, 99, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Pietraszek-Gremplewicz, K.; Simiczyjew, A.; Dratkiewicz, E.; Podgórska, M.; Styczeń, I.; Matkowski, R.; Ziętek, M.; Nowak, D. Expression level of EGFR and MET receptors regulates invasiveness of melanoma cells. J. Cell. Mol. Med. 2019, 23, 8453–8463. [Google Scholar] [CrossRef] [PubMed]
- Simiczyjew, A.; Pietraszek-Gremplewicz, K.; Dratkiewicz, E.; Podgórska, M.; Matkowski, R.; Ziętek, M.; Nowak, D. Combination of Selected MET and EGFR Inhibitors Decreases Melanoma Cells’ Invasive Abilities. Front. Pharmacol. 2019, 10, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Dratkiewicz, E.; Simiczyjew, A.; Pietraszek-Gremplewicz, K.; Mazurkiewicz, J.; Nowak, D. Characterization of melanoma cell lines resistant to Vemurafenib and evaluation of Their responsiveness to EGFR- and MET-inhibitor treatment. Int. J. Mol. Sci. 2019, 21, 113. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Bauer, J.; Curtin, J.A.; Pinkel, D.; Bastian, B.C. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J. Investig. Dermatol. 2007, 127, 179–182. [Google Scholar] [CrossRef]
- Pedersen, M.; Viros, A.; Cook, M.; Marais, R. G12DNRAS and kinase-dead BRAF cooperate to drive naevogenesis and melanomagenesis. Pigment Cell Melanoma Res. 2014, 27, 1162–1166. [Google Scholar] [CrossRef]
- Burd, C.E.; Liu, W.; Huynh, M.V.; Waqas, M.A.; Gillahan, J.E.; Clark, K.S.; Fu, K.; Martin, B.L.; Jeck, W.R.; Souroullas, G.P.; et al. Mutation-Specific RAS Oncogenicity Explains NRAS Codon 61 Selection in Melanoma. Cancer Discov. 2014, 4, 1418–1429. [Google Scholar] [CrossRef]
- Murphy, B.M.; Terrell, E.M.; Chirasani, V.R.; Weiss, T.J.; Lew, R.E.; Holderbaum, A.M.; Dhakal, A.; Posada, V.; Fort, M.; Bodnar, M.S.; et al. Enhanced BRAF engagement by NRAS mutants capable of promoting melanoma initiation. Nat. Commun. 2022, 13, 3153–3168. [Google Scholar] [CrossRef] [PubMed]
- Hélias-Rodzewicz, Z.; Funck-Brentano, E.; Baudoux, L.; Jung, C.K.; Zimmermann, U.; Marin, C.; Clerici, T.; Le Gall, C.; Peschaud, F.; Taly, V.; et al. Variations of BRAF mutant allele percentage in melanomas. BMC Cancer 2015, 15, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Soria, X.; Vilardell, F.; Maiques, Ó.; Barceló, C.; Sisó, P.; de la Rosa, I.; Velasco, A.; Cuevas, D.; Santacana, M.; Gatius, S.; et al. BRAF(V600E) Mutant Allele Frequency (MAF) Influences Melanoma Clinicopathologic Characteristics. Cancers 2021, 13, 5073. [Google Scholar] [CrossRef] [PubMed]
- Memon, A.; Bannister, P.; Rogers, I.; Sundin, J.; Al-Ayadhy, B.; James, P.W.; McNally, R.J.Q. Changing epidemiology and age-specific incidence of cutaneous malignant melanoma in England: An analysis of the national cancer registration data by age, gender and anatomical site, 1981–2018. Lancet Reg. Health Eur. 2021, 2, 100024. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, B.K.; Cust, A.E. Sun exposure and skin cancer, and the puzzle of cutaneous melanoma: A perspective on Fears et al. Mathematical models of age and ultraviolet effects on the incidence of skin cancer among whites in the United States. Cancer Epidemiol. 2017, 48, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Picconi, O.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. Eur. J. Cancer 2005, 41, 45–60. [Google Scholar] [CrossRef]
- Kim, J.; Novak, D.; Sachpekidis, C.; Utikal, J.; Larribère, L. STAT3 Relays a Differential Response to Melanoma-Associated NRAS Mutations. Cancers 2020, 12, 119. [Google Scholar] [CrossRef]
- Mo, X.; Preston, S.; Zaidi, M.R. Macroenvironment-gene-microenvironment interactions in ultraviolet radiation-induced melanomagenesis. Adv. Cancer Res. 2019, 144, 1–54. [Google Scholar]
- Georgescu, S.R.; Tampa, M.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Caruntu, A.; Lupu, M.; Matei, C.; Constantin, C.; Neagu, M. Tumour Microenvironment in Skin Carcinogenesis. Adv. Exp. Med. Biol. 2020, 1226, 123–142. [Google Scholar]
- Hernando, B.; Dietzen, M.; Parra, G.; Gil-Barrachina, M.; Pitarch, G.; Mahiques, L.; Valcuende-Cavero, F.; McGranahan, N.; Martinez-Cadenas, C. The effect of age on the acquisition and selection of cancer driver mutations in sun-exposed normal skin. Ann. Oncol. 2021, 32, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.-W.; Chang, C.-H. Nevi, dysplastic nevi, and melanoma: Molecular and immune mechanisms involving the progression. Tzu Chi Med. J. 2022, 34, 1–7. [Google Scholar] [PubMed]
- Damsky, W.E.; Bosenberg, M. Melanocytic nevi and melanoma: Unraveling a complex relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Vega, R.; Chen, C.-F.; Razzak, E.; Vasudeva, P.; Krasieva, T.B.; Shiu, J.; Caldwell, M.G.; Yan, H.; Lowengrub, J.; Ganesan, A.K.; et al. Dynamics of nevus development implicate cell cooperation in the growth arrest of transformed melanocytes. eLife 2020, 9, e61026. [Google Scholar] [CrossRef] [PubMed]
- Shiu, J.; Lander, A.D. When oncogenes do not cause cancer. eLife 2021, 10, e74912. [Google Scholar] [CrossRef]
- McNeal, A.S.; Belote, R.L.; Zeng, H.; Urquijo, M.; Barker, K.; Torres, R.; Curtin, M.; Shain, A.H.; Andtbacka, R.H.; Holmen, S.; et al. BRAF(V600E) induces reversible mitotic arrest in human melanocytes via microrna-mediated suppression of AURKB. eLife 2021, 10, e70385. [Google Scholar] [CrossRef] [PubMed]
- Pissa, M.; Lapins, J.; Sköldmark, C.; Helgadottir, H. Melanoma-specific survival before and after inclusion in a familial melanoma dermatologic surveillance program in CDKN2A mutation carriers and non-carriers. J. Eur. Acad. Dermatology Venereol. 2023, 37, 284–292. [Google Scholar] [CrossRef]
- Sparrow, L.E.; Eldon, M.J.; English, D.R.; Heenan, P.J. p16 and p21WAF1 protein expression in melanocytic tumors by immunohistochemistry. Am. J. Dermatopathol. 1998, 20, 255–261. [Google Scholar] [CrossRef]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef]
- Nedelcu, R.I.; Zurac, S.A.; Brînzea, A.; Cioplea, M.D.; Turcu, G.; Popescu, R.; Popescu, C.M.; Ion, D.A. Morphological features of melanocytic tumors with depigmented halo: Review of the literature and personal results. Rom. J. Morphol. Embryol. 2015, 56, 659–663. [Google Scholar]
- Clark, W.H.J.; Elder, D.E.; Guerry, D., IV; Epstein, M.N.; Greene, M.H.; Van Horn, M. A study of tumor progression: The precursor lesions of superficial spreading and nodular melanoma. Hum. Pathol. 1984, 15, 1147–1165. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.R.; Eliades, P.; Gupta, S.; Tsao, H. Genetics of melanocytic nevi. Pigment Cell Melanoma Res. 2015, 28, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.A.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Davi, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.-M.; Lin, L.L.; Lambie, D.; Flewell-Smith, R.; Jagirdar, K.; Schaider, H.; Sturm, R.A.; Prow, T.W.; Soyer, H.P. BRAF Wild-Type Melanoma in Situ Arising in a BRAF V600E Mutant Dysplastic Nevus. JAMA Dermatol. 2015, 151, 417–421. [Google Scholar] [CrossRef]
- Bezić, J.; Kuret, S.; Vrbičić, B.; Smolić, J.; Borić, I.; Škifić, I.; Ledina, D.; Božić, J. Clinicopathological Characteristics of BRAF V600E Mutated Melanomas in the Dalmatian Region of Croatia. Acta Dermatovenerol. Croat. 2019, 27, 225–230. [Google Scholar] [PubMed]
- Porumb-Andrese, E.; Ursu, R.G.; Ivanov, I.; Caruntu, I.-D.; Porumb, V.; Ferariu, D.; Damian, C.; Ciobanu, D.; Terinte, C.; Iancu, L.S. The BRAF V600E Mutation Detection by quasa Sensitive Real-Time PCR Assay in Northeast Romania Melanoma Patients. Appl. Sci. 2021, 11, 9511. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Si, L.; Kong, Y.; Xu, X.; Flaherty, K.T.; Sheng, X.; Cui, C.; Chi, Z.; Li, S.; Mao, L.; Guo, J. Prevalence of BRAF V600E mutation in Chinese melanoma patients: Large scale analysis of BRAF and NRAS mutations in a 432-case cohort. Eur. J. Cancer 2012, 48, 94–100. [Google Scholar] [CrossRef]
- Hugdahl, E.; Kalvenes, M.B.; Puntervoll, H.E.; Ladstein, R.G.; Akslen, L.A. BRAF-V600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br. J. Cancer 2016, 114, 801–808. [Google Scholar] [CrossRef]
- Edlundh-Rose, E.; Egyhazi, S.; Omholt, K. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: A study based on mutation screening by pyrosequencing. Melanoma Res. 2006, 16, 471–478. [Google Scholar] [CrossRef]
- Zablocka, T.; Nikolajeva, A.; Kreismane, M.; Pjanova, D.; Isajevs, S. Addressing the importance of melanoma tumor-infiltrating lymphocytes in disease progression and clinicopathological characteristics. Mol. Clin. Oncol. 2021, 15, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.; Bowyer, S.E.; White, A.; Grieu-Iacopetta, F.; Trevenen, M.; Iacopetta, B.; Amanuel, B.; Millward, M. FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathology 2015, 47, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Tomei, S.; Bedognetti, D.; De Giorgi, V.; Sommariva, M.; Civini, S.; Reinboth, J.; Al Hashmi, M.; Ascierto, M.L.; Liu, Q.; Ayotte, B.D.; et al. The immune-related role of BRAF in melanoma. Mol. Oncol. 2015, 9, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.J.; Aredo, J.V.; Starrett, J.H.; Stockhammer, P.; van Alderwerelt van Rosenburgh, I.K.; Wurtz, A.; Piper-Valillo, A.J.; Piotrowska, Z.; Falcon, C.; Yu, H.A.; et al. Efficacy of Osimertinib in Patients with Lung Cancer Positive for Uncommon EGFR Exon 19 Deletion Mutations. Clin. Cancer Res. 2023, 29, 2123–2130. [Google Scholar] [CrossRef]
- Yang, G.; Curley, D.; Bosenberg, M.W.; Tsao, H. Loss of xeroderma pigmentosum C (Xpc) enhances melanoma photocarcinogenesis in Ink4a-Arf-deficient mice. Cancer Res. 2007, 67, 5649–5657. [Google Scholar] [CrossRef]
- Kao, E.Y.; Wakeman, K.M.; Wu, Y.; Gross, J.M.; Chen, E.Y.; Ricciotti, R.W.; Liu, Y.J.; Mantilla, J.G. Prevalence and detection of actionable BRAF V600 and NRAS Q61 mutations in malignant peripheral nerve sheath tumor by droplet digital PCR. Hum. Pathol. 2022, 129, 90–97. [Google Scholar] [CrossRef]
- Satzger, I.; Marks, L.; Kerick, M.; Klages, S.; Berking, C.; Herbst, R.; Völker, B.; Schacht, V.; Timmermann, B.; Gutzmer, R. Allele frequencies of BRAFV600 mutations in primary melanomas and matched metastases and their relevance for BRAF inhibitor therapy in metastatic melanoma. Oncotarget 2015, 6, 37895–37905. [Google Scholar] [CrossRef]
- Yang, S.; Xu, J.; Zeng, X. A six-long non-coding RNA signature predicts prognosis in melanoma patients. Int. J. Oncol. 2018, 52, 1178–1188. [Google Scholar] [CrossRef]
- Dobre, E.-G.; Constantin, C.; Neagu, M. Skin Cancer Research Goes Digital: Looking for Biomarkers within the Droplets. J. Pers. Med. 2022, 12, 1136. [Google Scholar] [CrossRef]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: Pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar]
- Kluszczynska, K.; Czyz, M. Extracellular Vesicles-Based Cell-Cell Communication in Melanoma: New Perspectives in Diagnostics and Therapy. Int. J. Mol. Sci. 2023, 24, 965. [Google Scholar] [CrossRef]
- Skopeliti, M.; Voutsas, I.F.; Klimentzou, P.; Tsiatas, M.L.; Beck, A.; Bamias, A.; Moraki, M.; Livaniou, E.; Neagu, M.; Voelter, W.; et al. The immunologically active site of prothymosin alpha is located at the carboxy-terminus of the polypeptide. Evaluation of its in vitro effects in cancer patients. Cancer Immunol. Immunother. 2006, 55, 1247–1257. [Google Scholar] [CrossRef]
- Birmpilis, I.; Karachaliou, C.-E.; Samara, P.; Ioannou, K.; Selemenakis, P.; Kostopoulos, I.V.; Kavrochorianou, N.; Kalbacher, H.; Livaniou, E.; Haralambous, S.; et al. Antitumor Reactive T-Cell Responses Are Enhanced In Vivo by DAMP Prothymosin Alpha and Its C-Terminal Decapeptide. Cancers 2019, 11, 1764. [Google Scholar] [CrossRef]
- LEGE nr. 104 din 27 Martie 2003 Privind Manipularea Cadavrelor Umane şi Prelevarea Organelor şi Tesuturilor de la Cadavre în Vederea Transplantului. Available online: https://legislatie.just.ro/Public/DetaliiDocumentAfis/42803 (accessed on 3 January 2018).
- HOTĂRÂRE nr. 451 din 1 Aprilie 2004 Pentru Aprobarea Normelor Metodologice de Aplicare a Legii nr. 104/2003 Privind Manipularea Cadavrelor Umane şi Prelevarea Organelor şi ţesuturilor de la Cadavre în Vederea Transplantului. Available online: https://legislatie.just.ro/Public/DetaliiDocument/141602 (accessed on 3 January 2018).
- Ross, M.H.; Wojciech, P. An Intro to H&E Staining: Protocol, Best Practices, Steps & More. In Histology: A Text and Atlas: With Correlated Cell and Molecular Biology, 7th ed.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2016; pp. 984–1010. ISBN 978-1451187427. Available online: https://www.leicabiosystems.com (accessed on 3 January 2018).
- Clark, W.H., Jr.; Elder, D.E.; Guerry, D.; Braitman, L.E.; Trock, B.J.; Schultz, D.; Synnestvedt, M.; Halpern, A.C. Model predicting survival in stage I melanoma based on tumor progression. J. Natl. Cancer Inst. 1989, 81, 1893–1904. [Google Scholar] [CrossRef]
- Protocol for the Examination of Biopsy Specimens from Patients with Melanoma of the Skin, Version: 4.3.1.0. Available online: https://documents.cap.org/protocols/Skin.Melanoma.Bx_4.3.1.0.REL_CAPCP.pdf (accessed on 10 July 2022).
- IHC_SOP_v3.0. Available online: https://www.protocols.io/view/immunohistochemistry-novolink-polymer-detection-sy-yxmvmkwdog3p/v1 (accessed on 3 January 2023).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total (n = 15) | ||
|---|---|---|
| n | % | |
| Age (years) | ||
| Mean ± SD | 39.8 ± 18.32 | |
| Median [Q1–Q3] | 34 [25; 55] | |
| Gender | ||
| Male | 7 | (46.67%) |
| Female | 8 | (53.33%) |
| Site | ||
| Trunk | 8 | (53.33%) |
| Head and neck | 7 | (46.67%) |
| Nevus subtype | ||
| Intradermal | 8 | (53.33%) |
| Junctional | 3 | (20.00%) |
| Mixed | 4 | (26.67%) |
| BRAF mutation | ||
| BRAF V600 | 13 | (86.66%) |
| NRAS mutation | ||
| NRAS Q61 | 1 | (6.67%) |
| NRAS G12/G13 | 6 | (40.00%) |
| BRAF/NRAS co-mutant | 5 | (33.3%) |
| Total (n = 22) | ||
|---|---|---|
| n | % | |
| Age (years) | ||
| Mean ± SD | 64.5 ± 13.10 | |
| Median [Q1–Q3] | 65 [58; 72] | |
| Gender | ||
| Male | 13 | (59%) |
| Female | 9 | (41%) |
| Site | ||
| Trunk | 13 | (59.09%) |
| Head and neck | 4 | (18.18%) |
| Upper limbs | 5 | (22.73%) |
| Histological type | ||
| SSM | 15 | (68.2%) |
| NM | 7 | (31.8%) |
| Clark level | ||
| III | 3 | (13.64%) |
| IV | 19 | (86.36%) |
| Breslow index (mm) | ||
| Mean ± SD | 2.82 ± 2.07 | |
| Median [Q1–Q3] | 2.4 [1.3; 3.6] | |
| <2 | 10 | (45.46%) |
| 2–4 | 8 | (36.36%) |
| 4+ | 4 | (18.18%) |
| Mitotic index (mitoses/mm2) | ||
| Mean ± SD | 5.63 ± 7.42 | |
| Median [Q1–Q3] | 3 [0; 6] | |
| 0–1 | 7 | (31.8%) |
| 1+ | 15 | (68.2%) |
| Ulceration | ||
| Yes | 12 | (54.55%) |
| No | 10 | (45.45%) |
| TIL score | ||
| Non-brisk | 5 | (22.73%) |
| Brisk | 8 | (36.36%) |
| Absent | 9 | (40.91%) |
| Regression | ||
| Absent | 17 | (77.27%) |
| Present | 5 | (22.73%) |
| Perivascular invasion | 6 | (27.3%) |
| Nevus-associated melanoma | 5 | (22.73%) |
| BRAF mutation | 12 | (54.55%) |
| NRAS mutation | 11 | (50.00%) |
| BRAF/NRAS co-mutant | 8 | (36.36%) |
| BRAF/NRAS WT | 7 | (31.8%) |
| BRAF Mutation | ||||
|---|---|---|---|---|
| Variable | BRAF V600 | BRAF WT | p Value | |
| Number (%) | 12 (54.55%) | 10 (45.45%) | ||
| Age (years) | ||||
| Median (range) | 63 (41–83) | 68.5 (44–87) | 0.138 a | |
| Gender | ||||
| Male | 5 (41.67%) | 8 (80%) | 0.099 b | |
| Female | 7 (58.33%) | 2 (20%) | ||
| Breslow (mm) | ||||
| Median (range) | 3.35 (1.1–7.5) | 1.5 (0–7) | 0.029 a,* | |
| Clark level | ||||
| III | 0 (0%) | 3 (30%) | 0.07 b | |
| IV | 12 (100%) | 7 (70%) | ||
| Mitotic index | ||||
| Median (range) | 3.5 (1–15) | 0.1 (0–25) | 0.11 a | |
| Site | ||||
| Trunk | 7 (58.34%) | 6 (60%) | 0.34 b | |
| Limbs | 4 (33.33%) | 1 (10%) | ||
| Head and neck | 1 (8.33%) | 3 (30%) | ||
| Histological type | ||||
| NM | 3 (25%) | 4 (40%) | 0.65 b | |
| SSM | 9 (75%) | 6 (60%) | ||
| TILs ¶ | ||||
| Present | 10 (83.33%) | 3 (30%) | 0.027 b,* | |
| Absent | 2 (16.67%) | 7 (70%) | ||
| Ulceration | ||||
| Yes | 10 (83.33%) | 4 (40%) | 0.07 b | |
| No | 2 (16.67%) | 6 (60%) | ||
| NRAS Mutation | ||||
|---|---|---|---|---|
| Variable | NRAS G12/G13 | NRAS WT | p Value | |
| Number (%) | 11 (50%) | 11 (50%) | ||
| Age (years) | ||||
| Median (range) | 68 (42–83) | 63 (41–87) | 0.35 a | |
| Gender | ||||
| Male | 6 (54.55%) | 7 (63.64%) | 1.00 b | |
| Female | 5 (45.45%) | 4 (36.36%) | ||
| Breslow (mm) | ||||
| Median (range) | 3.6 (1.7–7.5) | 1.3 (0–6.25) | 0.01 a* | |
| Clark level | ||||
| III | 0 (0%) | 3 (27.27%) | 0.21 b | |
| IV | 11 (100%) | 8 (72.73%) | ||
| Mitotic index | ||||
| Median (range) | 4 (1–25) | 0.1 (0–15) | 0.04 a* | |
| Site | ||||
| Trunk | 7 (63.64%) | 6 (54.55%) | 0.85 b | |
| Limbs | 2 (18.18%) | 3 (27.27%) | ||
| Head and neck | 2 (18.18%) | 2 (18.18%) | ||
| Histological type | ||||
| NM | 2 (18.18%) | 5 (45.45%) | 0.36 b | |
| SSM | 9 (81.82%) | 6 (54.55%) | ||
| TILs ¶ | ||||
| Present | 8 (72.73%) | 5 (45.45%) | 0.38 b | |
| Absent | 3 (27.27%) | 6 (54.55%) | ||
| Ulceration | ||||
| Yes | 6 (54.55%) | 6 (54.55%) | 1.00 b | |
| No | 5 (45.45%) | 5 (45.45%) | ||
| Antibody (Mouse) | Clone | Company | Working Dilution | Pre-Treatment |
|---|---|---|---|---|
| CD3 | LN10 | Leica | RTU * | HIER *, buffer citrate, and pH 6 |
| CD4 | 4B12 | Leica | 1:100 | HIER, EDTA, and pH 9 |
| CD8 | 4B11 | Leica | 1:50 | HIER, EDTA, and pH 9 |
| CD20 | L26 | Leica | 1:150 | HIER, buffer citrate, and pH 6 |
| Antibody (Mouse) | Clone | Company | Working Dilution | Pre-Treatment |
|---|---|---|---|---|
| P16 | 6H12 | Leica | RTU | HIER, EDTA and pH 9 |
| BCL2 | BCL-2/100D5 | Leica | RTU | HIER, EDTA and pH 9 |
| P21 | 4D10 | Leica | 1:20 | HIER, buffer citrate and pH 6 |
| CycD1 | EP12 | Leica | RTU | HIER, EDTA and pH 9 |
| P53 | DO7 | Leica | RTU | HIER, EDTA and pH 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobre, E.-G.; Nichita, L.; Popp, C.; Zurac, S.; Neagu, M. Assessment of RAS-RAF-MAPK Pathway Mutation Status in Healthy Skin, Benign Nevi, and Cutaneous Melanomas: Pilot Study Using Droplet Digital PCR. Int. J. Mol. Sci. 2024, 25, 2308. https://doi.org/10.3390/ijms25042308
Dobre E-G, Nichita L, Popp C, Zurac S, Neagu M. Assessment of RAS-RAF-MAPK Pathway Mutation Status in Healthy Skin, Benign Nevi, and Cutaneous Melanomas: Pilot Study Using Droplet Digital PCR. International Journal of Molecular Sciences. 2024; 25(4):2308. https://doi.org/10.3390/ijms25042308
Chicago/Turabian StyleDobre, Elena-Georgiana, Luciana Nichita, Cristiana Popp, Sabina Zurac, and Monica Neagu. 2024. "Assessment of RAS-RAF-MAPK Pathway Mutation Status in Healthy Skin, Benign Nevi, and Cutaneous Melanomas: Pilot Study Using Droplet Digital PCR" International Journal of Molecular Sciences 25, no. 4: 2308. https://doi.org/10.3390/ijms25042308
APA StyleDobre, E.-G., Nichita, L., Popp, C., Zurac, S., & Neagu, M. (2024). Assessment of RAS-RAF-MAPK Pathway Mutation Status in Healthy Skin, Benign Nevi, and Cutaneous Melanomas: Pilot Study Using Droplet Digital PCR. International Journal of Molecular Sciences, 25(4), 2308. https://doi.org/10.3390/ijms25042308