
The Protein Kinase A Inhibitor KT5720 Prevents Endothelial Dysfunctions Induced by High-Dose Irradiation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
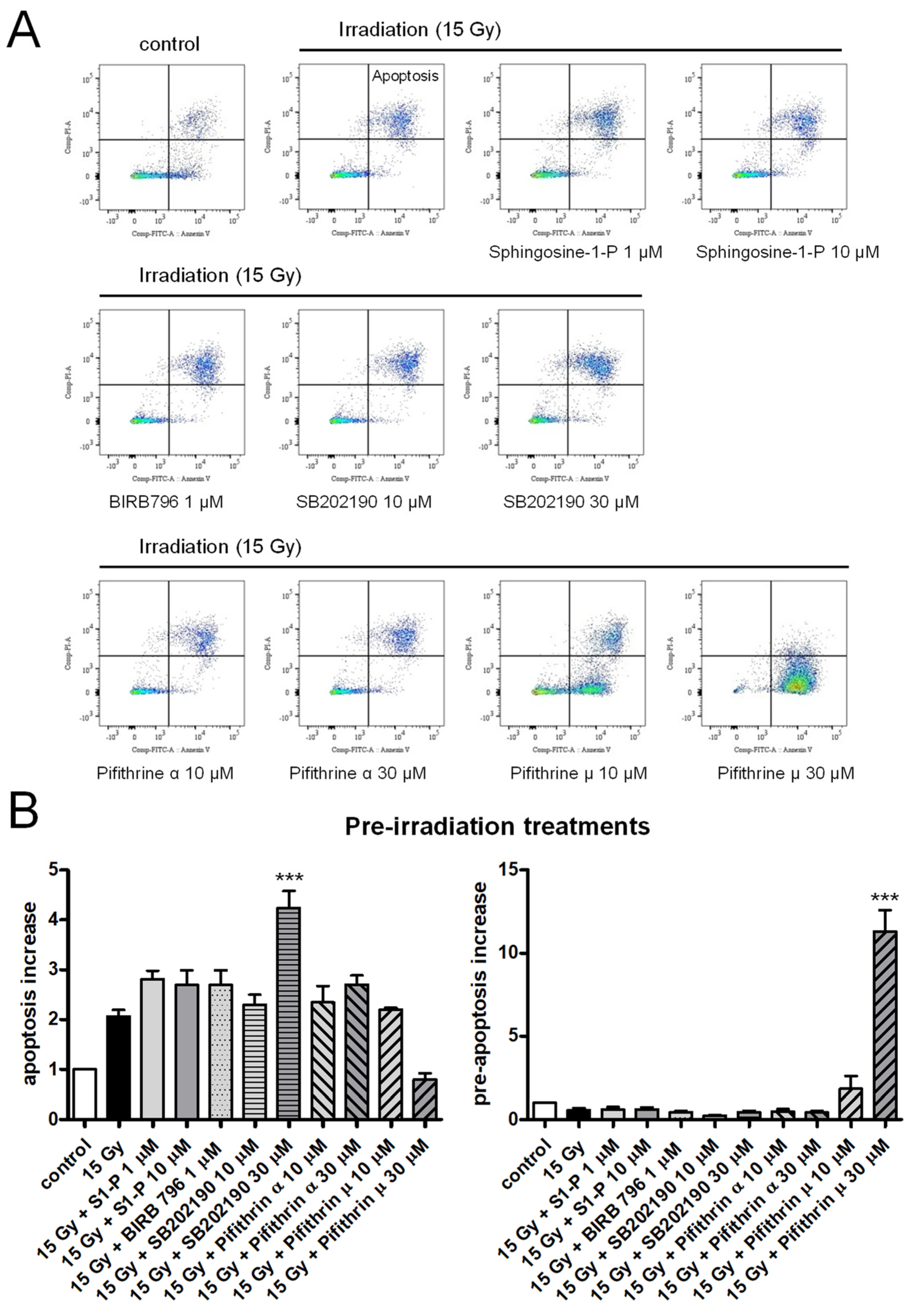
2.1. Effects of Sphingosine-1-P, p38 MAP Kinase, or p53 Inhibitors on Irradiation-Induced Apoptosis of HPMECs
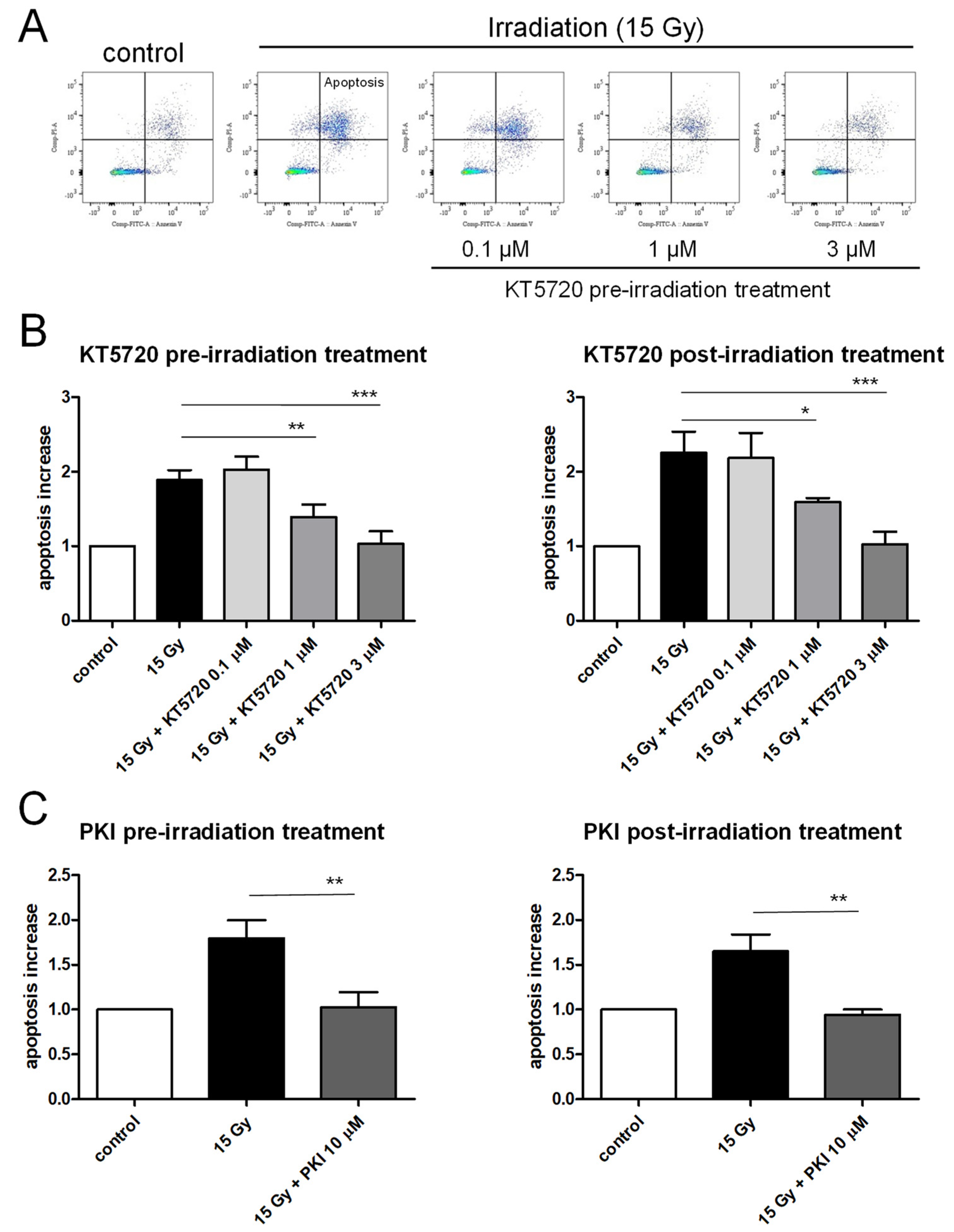
2.2. Both Pre- and Post-Irradiation Treatments with KT5720 Prevent Irradiation-Induced Apoptosis of HPMECs
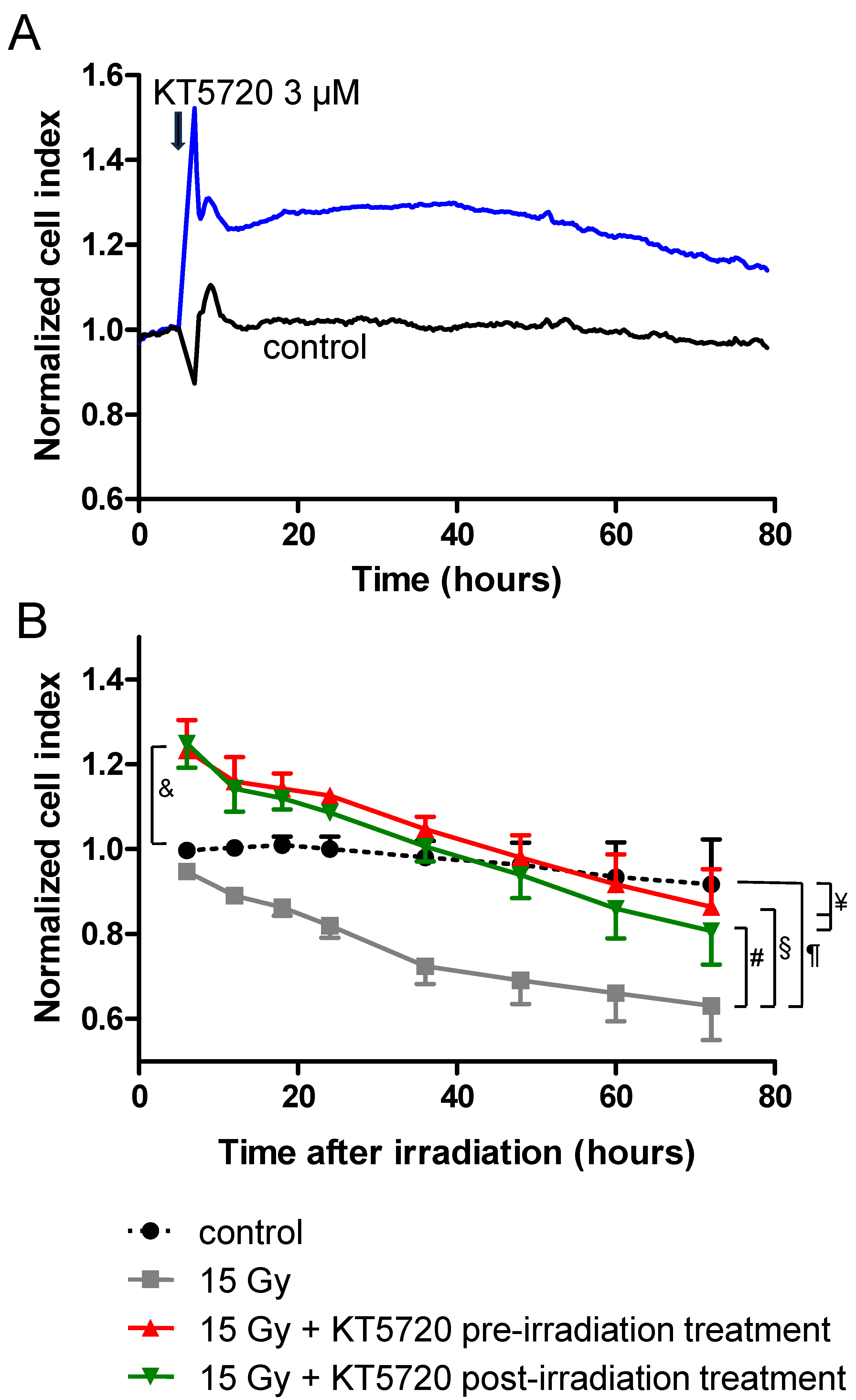
2.3. KT5720 Post-Irradiation Treatment Prevents Irradiation-Induced Alteration of Endothelial Monolayers
2.4. Effects of KT5720 Treatment and Irradiation on Impedance of HPMEC Monolayers
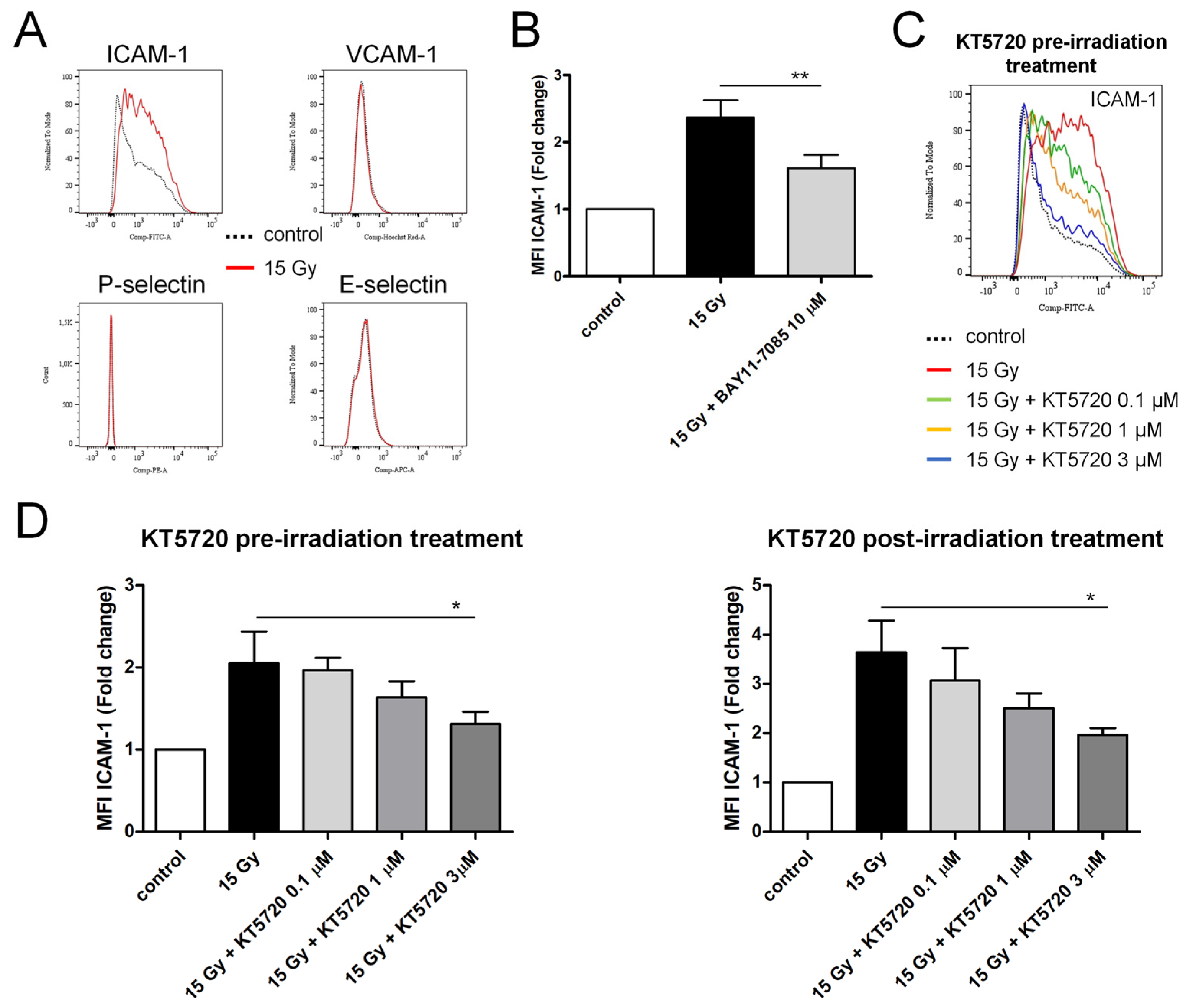
2.5. Both Pre- and Post-Irradiation Treatments with KT5720 Reduce Irradiation-Induced ICAM-1 Overexpression in HPMECs
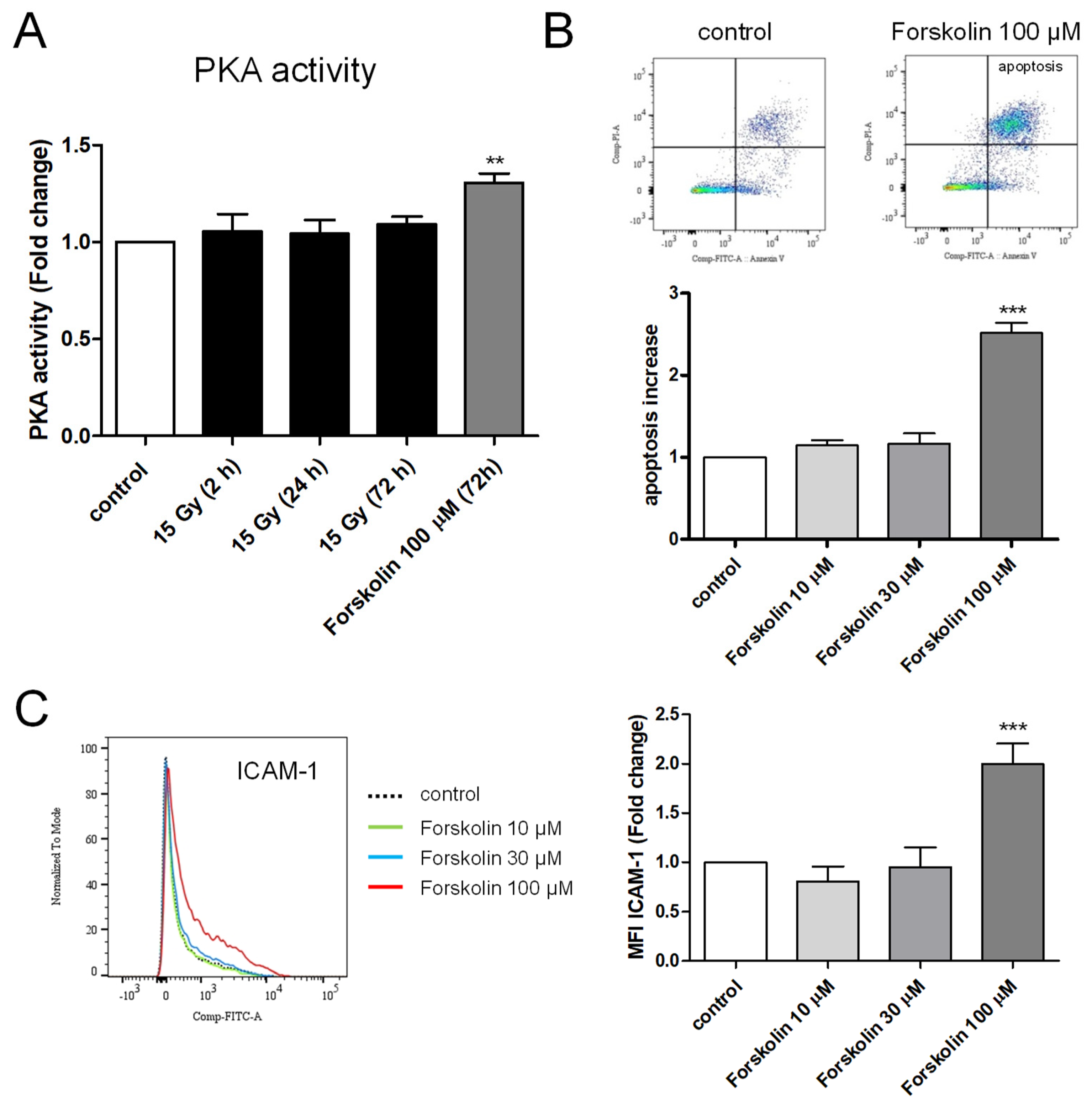
2.6. Measurement of PKA Activity in Irradiated HPMECs
2.7. Effects of Forskolin on Apoptosis and ICAM-1 Membrane Expression in HPMECs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Culture of Microvascular Endothelial Cells
4.3. Irradiation and Treatments
4.4. Immunostainings
4.5. Apoptosis Measurements
4.6. Measurements of Endothelial Cell Surface Expression of Adhesion Molecules
4.7. Measurements of Impedance in HPMEC Monolayers
4.8. Measurements of PKA Activity in HPMECs
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hallahan, D.E.; Virudachalam, S. Intercellular adhesion molecule 1 knockout abrogates radiation induced pulmonary inflammation. Proc. Natl. Acad. Sci. USA 1997, 94, 6432–6437. [Google Scholar] [CrossRef]
- Williams, J.P.; McBride, W.H. After the bomb drops: A new look at radiation-induced multiple organ dysfunction syndrome (MODS). Int. J. Radiat. Biol. 2011, 87, 851–868. [Google Scholar] [CrossRef]
- Baselet, B.; Sonveaux, P.; Baatout, S.; Aerts, A. Pathological effects of ionizing radiation: Endothelial activation and dysfunction. Cell. Mol. Life Sci. 2019, 76, 699–728. [Google Scholar] [CrossRef]
- Venkatesulu, B.P.; Mahadevan, L.S.; Aliru, M.L.; Yang, X.; Bodd, M.H.; Singh, P.K.; Yusuf, S.W.; Abe, J.I.; Krishnan, S. Radiation-Induced Endothelial Vascular Injury. A Review of Possible Mechanisms. JACC Basic Transl. Sci. 2018, 3, 563–572. [Google Scholar] [CrossRef]
- Heckmann, M.; Douwes, K.; Peter, R.; Degitz, K. Vascular activation of adhesion molecule mRNA and cell surface expression by ionizing radiation. Exp. Cell Res. 1998, 238, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Quarmby, S.; Kumar, P.; Kumar, S. Radiation-induced normal tissue injury: Role of adhesion molecules in leukocyte-endothelial cell interactions. Int. J. Cancer 1999, 82, 385–395. [Google Scholar] [CrossRef]
- Kouam, P.N.; Rezniczek, G.A.; Adamietz, I.A.; Buhler, H. Ionizing radiation increases the endothelial permeability and the transendothelial migration of tumor cells through ADAM10-activation and subsequent degradation of VE-cadherin. BMC Cancer 2019, 19, 958. [Google Scholar] [CrossRef]
- Wang, H.; Segaran, R.C.; Chan, L.Y.; Aladresi, A.A.M.; Chinnathambi, A.; Alharbi, S.A.; Sethi, G.; Tang, F.R. Gamma Radiation-Induced Disruption of Cellular Junctions in HUVECs Is Mediated through Affecting MAPK/NF-κB Inflammatory Pathways. Oxid. Med. Cell. Longev. 2019, 2019, 1486232. [Google Scholar] [CrossRef]
- Paris, F.; Fuks, Z.; Kang, A.; Capodieci, P.; Juan, G.; Ehleiter, D.; Haimovitz-Friedman, A.; Cordon-Cardo, C.; Kolesnick, R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 2001, 293, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.L.; Blum, J.M.; Kirsch, D.G. Role of p53 in regulating tissue response to radiation by mechanisms independent of apoptosis. Transl. Cancer Res. 2013, 2, 412–421. [Google Scholar] [PubMed]
- Lee, C.L.; Moding, E.J.; Cuneo, K.C.; Li, Y.; Sullivan, J.M.; Mao, L.; Washington, I.; Jeffords, L.B.; Rodrigues, R.C.; Ma, Y.; et al. p53 functions in endothelial cells to prevent radiation-induced myocardial injury in mice. Sci. Signal. 2012, 5, ra52. [Google Scholar] [CrossRef]
- Lafargue, A.; Degorre, C.; Corre, I.; Alves-Guerra, M.C.; Gaugler, M.H.; Vallette, F.; Pecqueur, C.; Paris, F. Ionizing radiation induces long-term senescence in endothelial cells through mitochondrial respiratory complex II dysfunction and superoxide generation. Free Radic. Biol. Med. 2017, 108, 750–759. [Google Scholar] [CrossRef]
- Bonnaud, S.; Niaudet, C.; Pottier, G.; Gaugler, M.H.; Millour, J.; Barbet, J.; Sabatier, L.; Paris, F. Sphingosine-1-phosphate protects proliferating endothelial cells from ceramide-induced apoptosis but not from DNA damage-induced mitotic death. Cancer Res. 2007, 67, 1803–1811. [Google Scholar] [CrossRef]
- Corre, I.; Guillonneau, M.; Paris, F. Membrane signaling induced by high doses of ionizing radiation in the endothelial compartment. Relevance in radiation toxicity. Int. J. Mol. Sci. 2013, 14, 22678–22696. [Google Scholar] [CrossRef] [PubMed]
- Niaudet, C.; Bonnaud, S.; Guillonneau, M.; Gouard, S.; Gaugler, M.H.; Dutoit, S.; Ripoche, N.; Dubois, N.; Trichet, V.; Corre, I.; et al. Plasma membrane reorganization links acid sphingomyelinase/ceramide to p38 MAPK pathways in endothelial cells apoptosis. Cell. Signal. 2017, 33, 10–21. [Google Scholar] [CrossRef]
- Kumar, P.; Miller, A.I.; Polverini, P.J. p38 MAPK mediates gamma-irradiation-induced endothelial cell apoptosis, and vascular endothelial growth factor protects endothelial cells through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J. Biol. Chem. 2004, 279, 43352–43360. [Google Scholar] [CrossRef]
- Hallahan, D.; Kuchibhotla, J.; Wyble, C. Cell adhesion molecules mediate radiation-induced leukocyte adhesion to the vascular endothelium. Cancer Res. 1996, 56, 5150–5155. [Google Scholar] [PubMed]
- Hallahan, D.E.; Virudachalam, S.; Kuchibhotla, J. Nuclear factor κB dominant negative genetic constructs inhibit X-ray induction of cell adhesion molecules in the vascular endothelium. Cancer Res. 1998, 58, 5484–5488. [Google Scholar]
- Quarmby, S.; Hunter, R.D.; Kumar, S. Irradiation induced expression of CD31, ICAM-1 and VCAM-1 in human microvascular endothelial cells. Anticancer Res. 2000, 20, 3375–3381. [Google Scholar] [PubMed]
- Ma, Z.C.; Hong, Q.; Wang, Y.G.; Tan, H.L.; Xiao, C.R.; Liang, Q.D.; Cai, S.H.; Gao, Y. Ferulic acid attenuates adhesion molecule expression in gamma-radiated human umbilical vascular endothelial cells. Biol. Pharm. Bull. 2010, 33, 752–758. [Google Scholar] [CrossRef]
- Soroush, F.; Tang, Y.; Zaidi, H.M.; Sheffield, J.B.; Kilpatrick, L.E.; Kiani, M.F. PKCδ inhibition as a novel medical countermeasure for radiation-induced vascular damage. FASEB J. 2018, 32, 6436–6444. [Google Scholar] [CrossRef] [PubMed]
- Son, E.W.; Mo, S.J.; Rhee, D.K.; Pyo, S. Inhibition of ICAM-1 expression by garlic component, allicin, in gamma-irradiated human vascular endothelial cells via downregulation of the JNK signaling pathway. Int. Immunopharmacol. 2006, 6, 1788–1795. [Google Scholar] [CrossRef]
- Kabacik, S.; Raj, K. Ionising radiation increases permeability of endothelium through ADAM10-mediated cleavage of VE-cadherin. Oncotarget 2017, 8, 82049–82063. [Google Scholar] [CrossRef]
- Young, E.F.; Smilenov, L.B. Impedance-based surveillance of transient permeability changes in coronary endothelial monolayers after exposure to ionizing radiation. Radiat. Res. 2011, 176, 415–424. [Google Scholar] [CrossRef]
- Narayanan, S.A.; Ford, J.; Zawieja, D.C. Impairment of lymphatic endothelial barrier function by X-ray irradiation. Int. J. Radiat. Biol. 2019, 95, 562–570. [Google Scholar] [CrossRef]
- Gabrys, D.; Greco, O.; Patel, G.; Prise, K.M.; Tozer, G.M.; Kanthou, C. Radiation effects on the cytoskeleton of endothelial cells and endothelial monolayer permeability. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Templin, T.; Grabham, P. Short term effects of gamma radiation on endothelial barrier function: Uncoupling of PECAM-1. Microvasc. Res. 2013, 86, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kase, H.; Iwahashi, K.; Nakanishi, S.; Matsuda, Y.; Yamada, K.; Takahashi, M.; Murakata, C.; Sato, A.; Kaneko, M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res. Commun. 1987, 142, 436–440. [Google Scholar] [CrossRef]
- Boittin, F.X.; Beddek, K.; Thery, H.; Pateux, J.; Cosler, G.; Riccobono, D.; Drouet, M.; Bobe, R. The immunosuppressant drug Cyclosporin A aggravates irradiation effects in endothelial cells. Biochem. Biophys. Res. Commun. 2022, 602, 127–134. [Google Scholar] [CrossRef]
- Strom, E.; Sathe, S.; Komarov, P.G.; Chernova, O.B.; Pavlovska, I.; Shyshynova, I.; Bosykh, D.A.; Burdelya, L.G.; Macklis, R.M.; Skaliter, R.; et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat. Chem. Biol. 2006, 2, 474–479. [Google Scholar] [CrossRef]
- Zhu, J.; Singh, M.; Selivanova, G.; Peuget, S. Pifithrin-α alters p53 post-translational modifications pattern and differentially inhibits p53 target genes. Sci. Rep. 2020, 10, 1049. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Yan, D.; Xiao, J.; Feng, H.; Chang, M.X. The Zebrafish Antiapoptotic Protein BIRC2 Promotes Edwardsiella piscicida Infection by Inhibiting Caspases and Accumulating p53 in a p53 Transcription-Dependent and -Independent Manner. Front. Immunol. 2021, 12, 781680. [Google Scholar] [CrossRef] [PubMed]
- Neganova, I.; Chichagova, V.; Armstrong, L.; Lako, M. A critical role for p38MAPK signalling pathway during reprogramming of human fibroblasts to iPSCs. Sci. Rep. 2017, 7, 41693. [Google Scholar] [CrossRef] [PubMed]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Marquez, R.; Cuenda, A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005, 280, 19472–19479. [Google Scholar] [CrossRef]
- Blackwood, B.P.; Wood, D.R.; Yuan, C.; Nicolas, J.; De Plaen, I.G.; Farrow, K.N.; Chou, P.; Turner, J.R.; Hunter, C.J. A Role for cAMP and Protein Kinase A in Experimental Necrotizing Enterocolitis. Am. J. Pathol. 2017, 187, 401–417. [Google Scholar] [CrossRef]
- Glass, D.B.; Cheng, H.C.; Mende-Mueller, L.; Reed, J.; Walsh, D.A. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J. Biol. Chem. 1989, 264, 8802–8810. [Google Scholar] [CrossRef]
- Bischoff, I.; Hornburger, M.C.; Mayer, B.A.; Beyerle, A.; Wegener, J.; Furst, R. Pitfalls in assessing microvascular endothelial barrier function: Impedance-based devices versus the classic macromolecular tracer assay. Sci. Rep. 2016, 6, 23671. [Google Scholar] [CrossRef]
- Hu, X.; Janssen, W.E.; Moscinski, L.C.; Bryington, M.; Dangsupa, A.; Rezai-Zadeh, N.; Babbin, B.A.; Zuckerman, K.S. An IκBα inhibitor causes leukemia cell death through a p38 MAP kinase-dependent, NF-κB-independent mechanism. Cancer Res. 2001, 61, 6290–6296. [Google Scholar]
- Alasbahi, R.H.; Melzig, M.F. Forskolin and derivatives as tools for studying the role of cAMP. Pharmazie 2012, 67, 5–13. [Google Scholar] [PubMed]
- Bonnaud, S.; Niaudet, C.; Legoux, F.; Corre, I.; Delpon, G.; Saulquin, X.; Fuks, Z.; Gaugler, M.H.; Kolesnick, R.; Paris, F. Sphingosine-1-phosphate activates the AKT pathway to protect small intestines from radiation-induced endothelial apoptosis. Cancer Res. 2010, 70, 9905–9915. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Kelly, J.; Vemavarapu, L.; Thompson, W.J.; Strada, S.J. Activation and induction of cyclic AMP phosphodiesterase (PDE4) in rat pulmonary microvascular endothelial cells. Biochem. Pharmacol. 2004, 68, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yu, C.; Yang, F.; Paganini-Hill, A.; Fisher, M.J. Phosphodiesterase inhibitor modulation of brain microvascular endothelial cell barrier properties. J. Neurol. Sci. 2012, 320, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S.; Zhou, H.; Makhlouf, G.M. PKA-dependent activation of PDE3A and PDE4 and inhibition of adenylyl cyclase V/VI in smooth muscle. Am. J. Physiol. Cell. Physiol. 2002, 282, C508–C517. [Google Scholar] [CrossRef] [PubMed]
- Spindler, V.; Peter, D.; Harms, G.S.; Asan, E.; Waschke, J. Ultrastructural analysis reveals cAMP-dependent enhancement of microvascular endothelial barrier functions via Rac1-mediated reorganization of intercellular junctions. Am. J. Pathol. 2011, 178, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Radeva, M.Y.; Kugelmann, D.; Spindler, V.; Waschke, J. PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS ONE 2014, 9, e106733. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.K.; Reszka, A.A.; Abraham, I. KT5720 and U-98017 inhibit MAPK and alter the cytoskeleton and cell morphology. J. Cell. Physiol. 1998, 176, 525–536. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Dos, S.M.; Paget, V.; Trompier, F.; Gruel, G.; Milliat, F. Dosimetry for Cell Irradiation using Orthovoltage (40-300 kV) X-Ray Facilities. J. Vis. Exp. 2021, 168, e61645. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boittin, F.-X.; Guitard, N.; Toth, M.; Riccobono, D.; Théry, H.; Bobe, R. The Protein Kinase A Inhibitor KT5720 Prevents Endothelial Dysfunctions Induced by High-Dose Irradiation. Int. J. Mol. Sci. 2024, 25, 2269. https://doi.org/10.3390/ijms25042269
Boittin F-X, Guitard N, Toth M, Riccobono D, Théry H, Bobe R. The Protein Kinase A Inhibitor KT5720 Prevents Endothelial Dysfunctions Induced by High-Dose Irradiation. International Journal of Molecular Sciences. 2024; 25(4):2269. https://doi.org/10.3390/ijms25042269
Chicago/Turabian StyleBoittin, François-Xavier, Nathalie Guitard, Maeliss Toth, Diane Riccobono, Hélène Théry, and Régis Bobe. 2024. "The Protein Kinase A Inhibitor KT5720 Prevents Endothelial Dysfunctions Induced by High-Dose Irradiation" International Journal of Molecular Sciences 25, no. 4: 2269. https://doi.org/10.3390/ijms25042269
APA StyleBoittin, F.-X., Guitard, N., Toth, M., Riccobono, D., Théry, H., & Bobe, R. (2024). The Protein Kinase A Inhibitor KT5720 Prevents Endothelial Dysfunctions Induced by High-Dose Irradiation. International Journal of Molecular Sciences, 25(4), 2269. https://doi.org/10.3390/ijms25042269