



DNA Double Strand Break and Response Fluorescent Assays: Choices and Interpretation



Abstract
1. Introduction
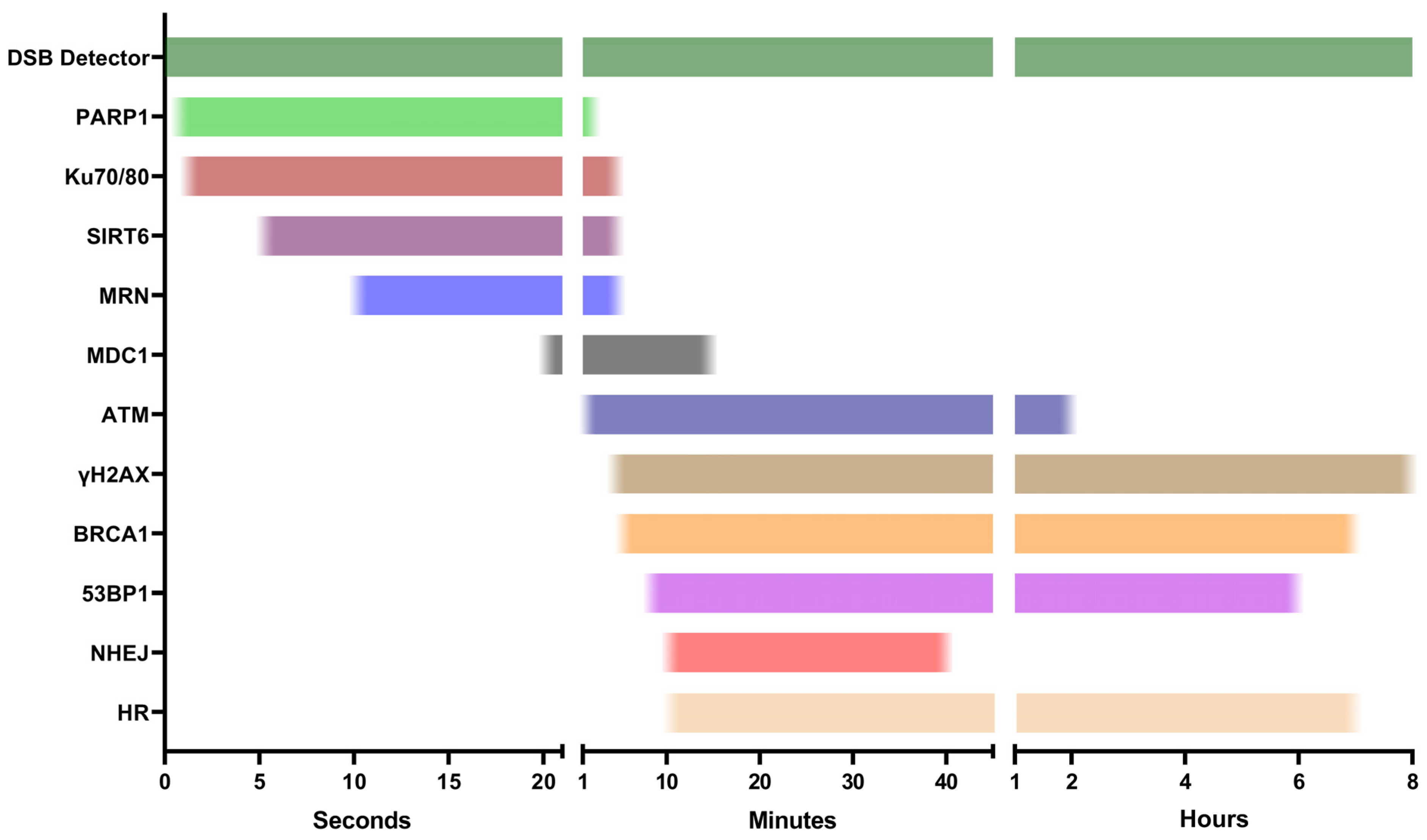
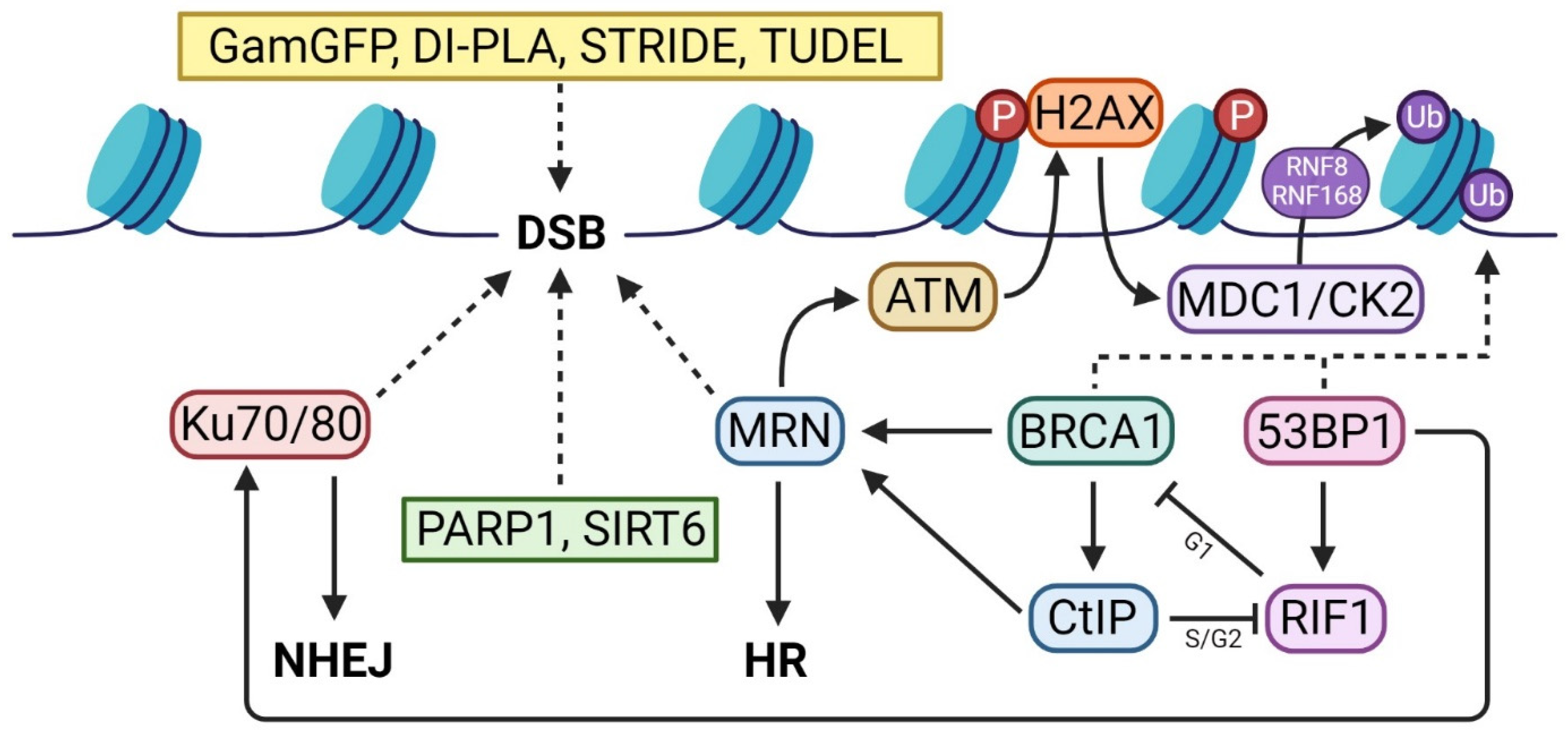
2. DSB Detector Assays
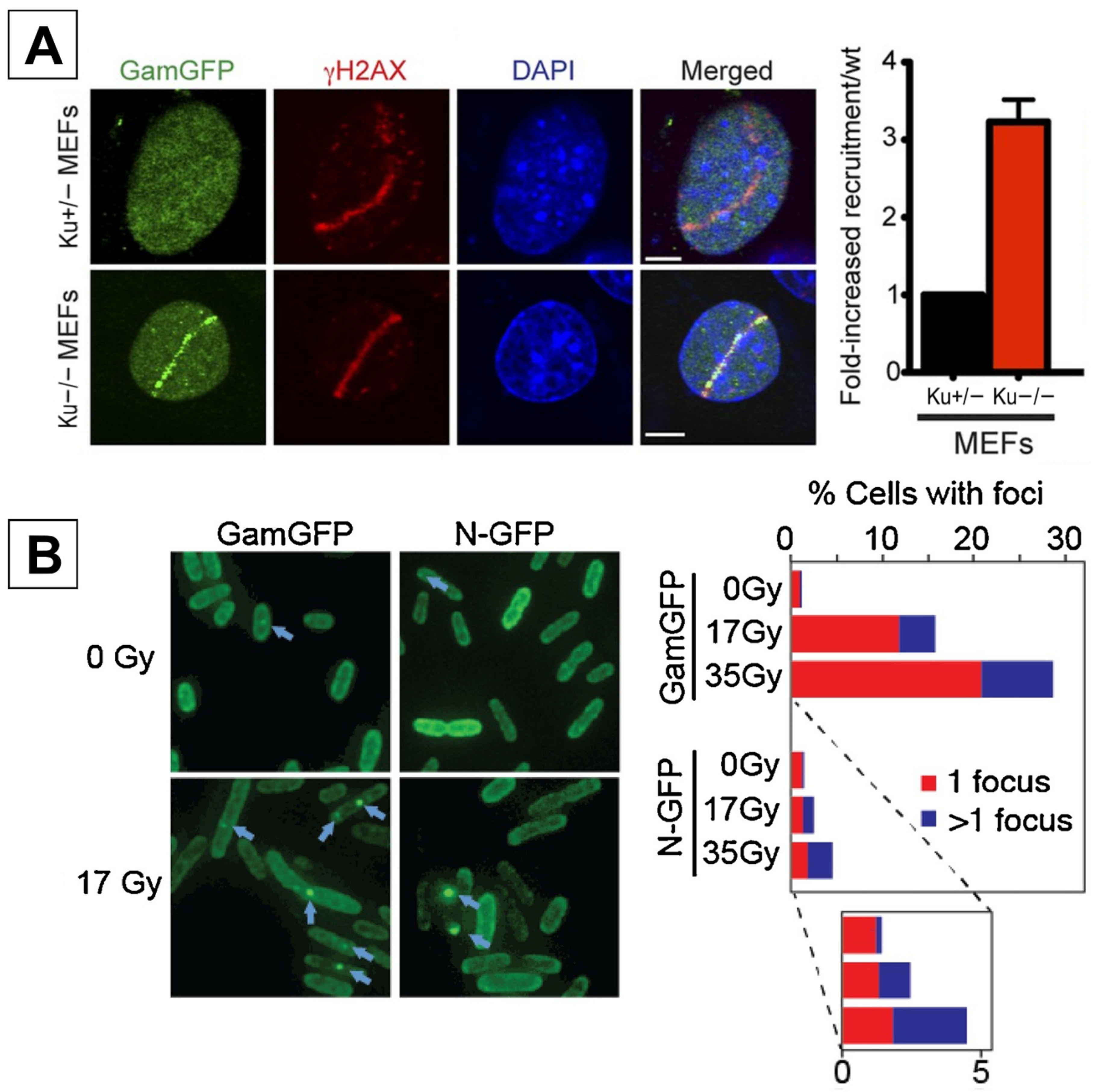
2.1. GamGFP

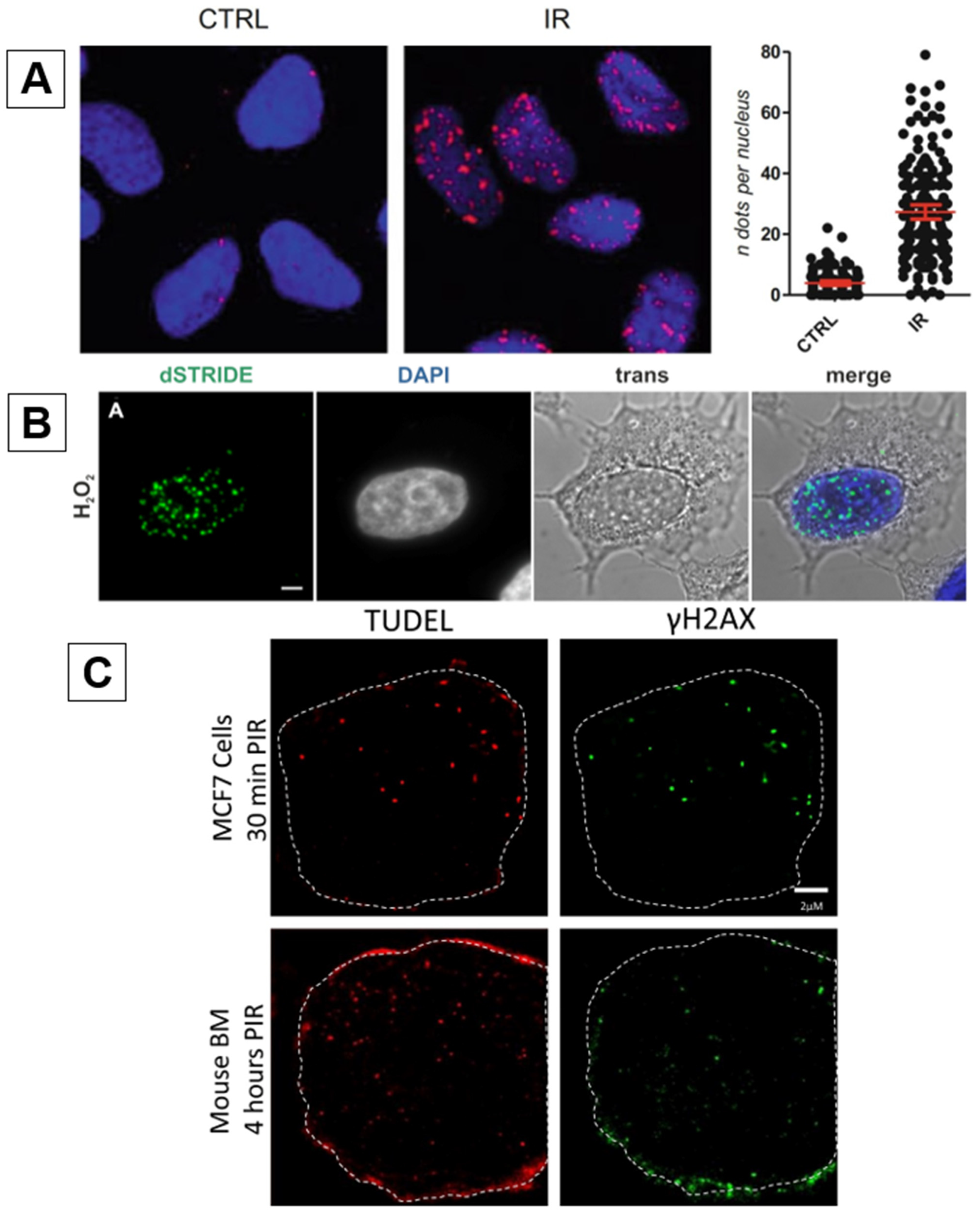
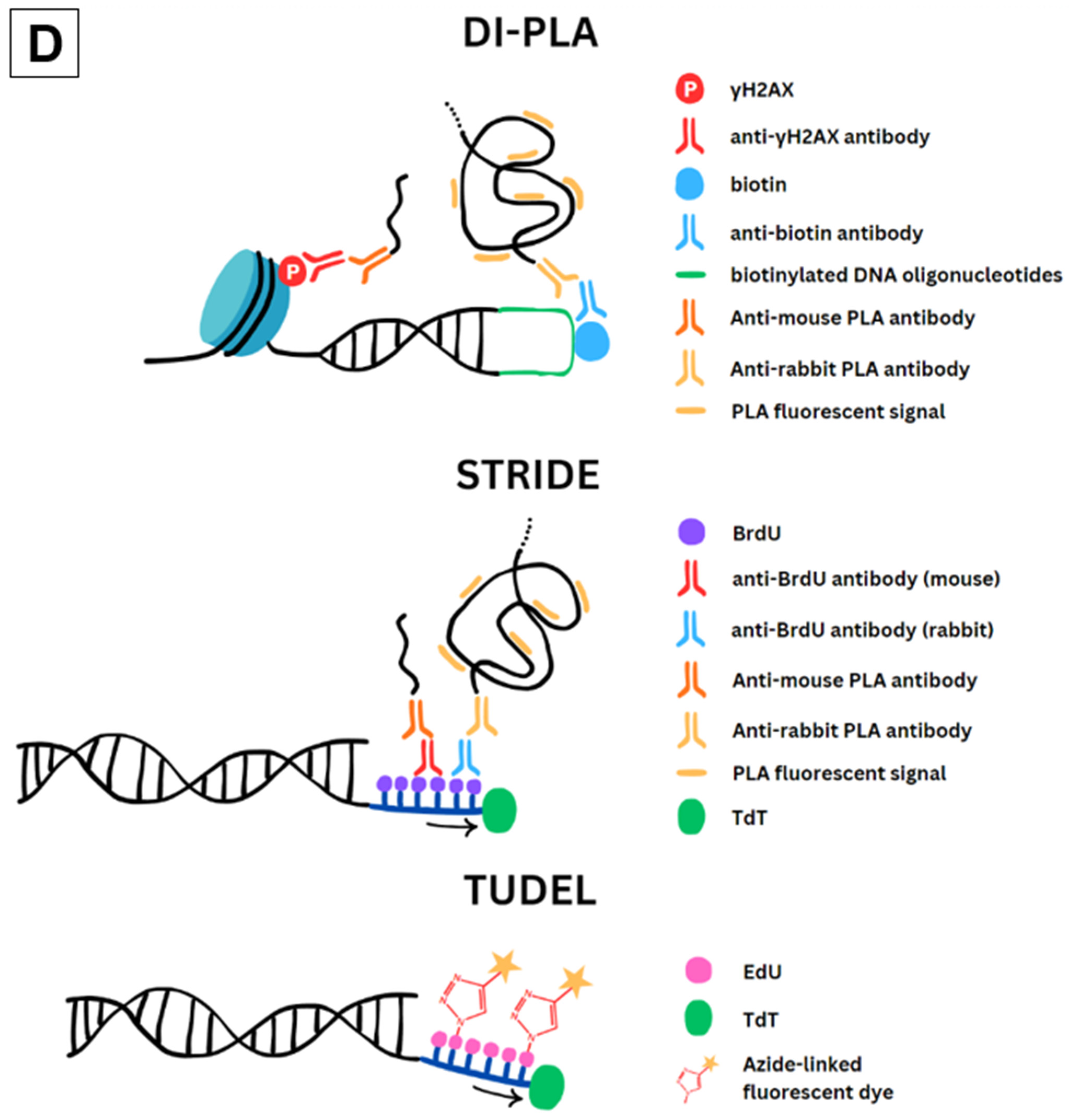
2.2. DI-PLA
2.3. STRIDE
2.4. TUDEL
3. First Responders
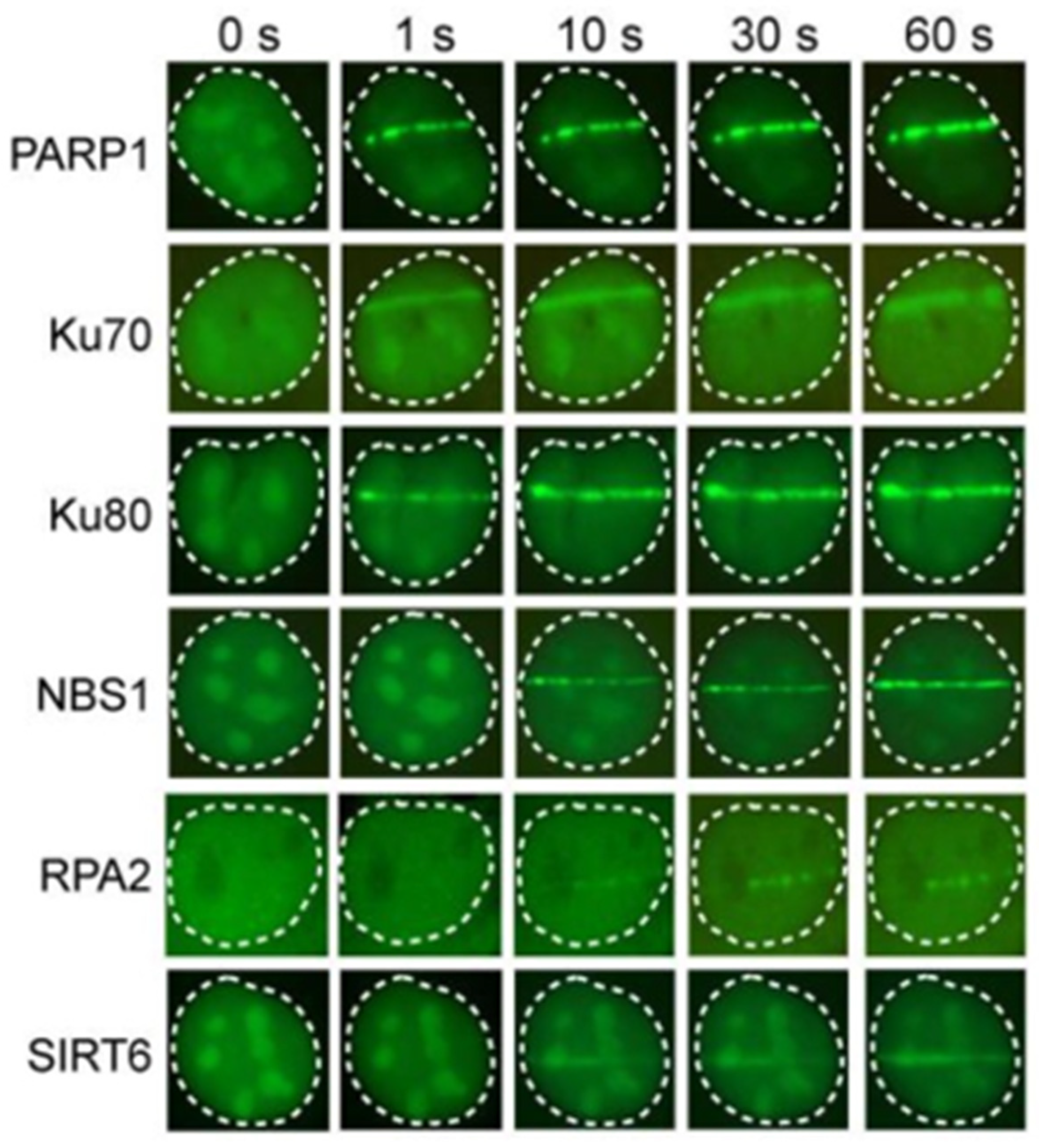
3.1. PARP1
3.2. Ku70/80 and MRN
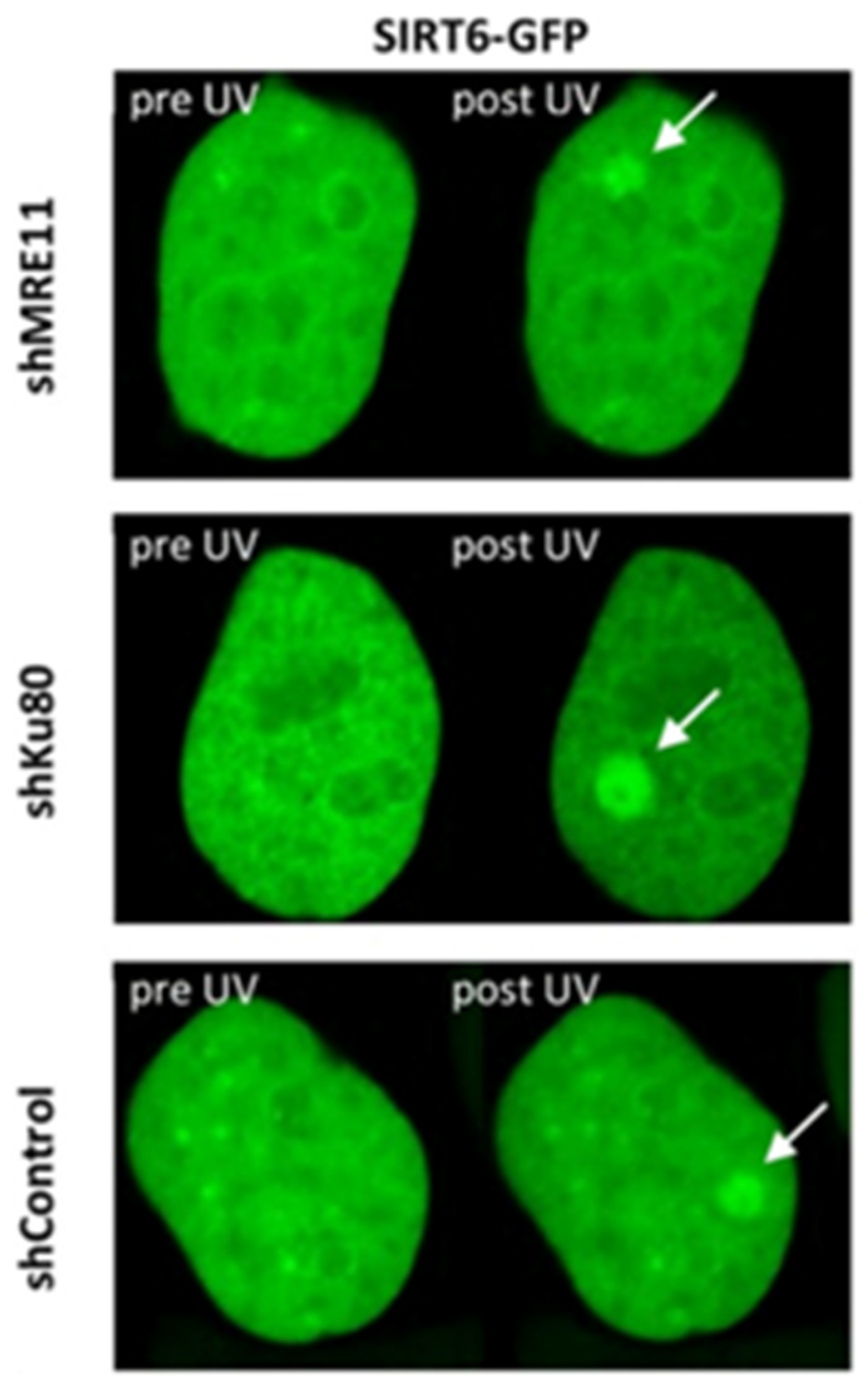
3.3. SIRT6
4. DDR Activators
4.1. ATM
4.2. γH2AX
4.3. MDC1
5. Repair Pathway Specific Assays
BRCA1 and 53BP1
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef]
- Varga, T.; Aplan, P.D. Chromosomal aberrations induced by double strand DNA breaks. DNA Repair 2005, 4, 1038–1046. [Google Scholar] [CrossRef]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Kang, J.-K.; Seo, S.; Jin, Y.W. Health Effects of Radon Exposure. Yonsei Med. J. 2019, 60, 597–603. [Google Scholar] [CrossRef]
- Bhaskaran, R.; Damodaran, R.C.; Kumar, V.A.; Panakal John, J.; Bangaru, D.; Natarajan, C.; Sathiamurthy, B.S.; Mundiyanikal Thomas, J.; Mishra, R. Inhalation Dose and Source Term Studies in a Tribal Area of Wayanad, Kerala, India. J. Environ. Public Health 2017, 2017, 1930787. [Google Scholar] [CrossRef]
- Angelo, G.; Lorena, C.; Marta, G.; Antonella, C. Biochemical Composition and Antioxidant Properties of Lavandula angustifolia Miller Essential Oil are Shielded by Propolis Against UV Radiations. Photochem. Photobiol. 2014, 90, 702–708. [Google Scholar] [CrossRef]
- Durante, M.; Cucinotta, F.A. Physical basis of radiation protection in space travel. Rev. Mod. Phys. 2011, 83, 1245–1281. [Google Scholar] [CrossRef]
- Mishra, B.; Luderer, U. Reproductive hazards of space travel in women and men. Nat. Rev. Endocrinol. 2019, 15, 713–730. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Mol. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX–A novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Atkinson, J.; Bezak, E.; Kempson, I. Imaging DNA double-strand breaks—Are we there yet? Nat. Rev. Mol. Cell Biol. 2022, 23, 579–580. [Google Scholar] [CrossRef]
- Cleaver, J.E.; Feeney, L.; Revet, I. Phosphorylated H2Ax is not an unambiguous marker for DNA double-strand breaks. Cell Cycle 2014, 10, 3223–3224. [Google Scholar] [CrossRef]
- Shee, C.; Cox, B.D.; Gu, F.; Luengas, E.M.; Joshi, M.C.; Chiu, L.Y.; Magnan, D.; Halliday, J.A.; Frisch, R.L.; Gibson, J.L.; et al. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife 2013, 2, e01222. [Google Scholar] [CrossRef]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of apoptosis by TUNEL assay. Methods Mol. Biol. 2012, 887, 41–47. [Google Scholar]
- Hossain, M.A.; Lin, Y.; Yan, S. Single-Strand Break End Resection in Genome Integrity: Mechanism and Regulation by APE2. Int. J. Mol. Sci. 2018, 19, 2389. [Google Scholar] [CrossRef]
- Ahmed, E.A.; Agay, D.; Schrock, G.; Drouet, M.; Meineke, V.; Scherthan, H. Persistent DNA damage after high dose in vivo gamma exposure of minipig skin. PLoS ONE 2012, 7, e39521. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Sestili, P.; Cantoni, O. Osmotically driven radial diffusion of single-stranded DNA fragments on an agarose bed as a convenient measure of DNA strand scission. Free Radic. Biol. Med. 1999, 26, 1019–1026. [Google Scholar] [CrossRef]
- Sestili, P.; Calcabrini, C.; Diaz, A.R.; Fimognari, C.; Stocchi, V. The Fast-Halo Assay for the Detection of DNA Damage. In Fast Detection of DNA Damage: Methods and Protocols; Didenko, V.V., Ed.; Springer: New York, NY, USA, 2017; pp. 75–93. [Google Scholar]
- Fernández, J.L.; Vázquez-Gundín, F.; Rivero, M.T.; Genescá, A.; Gosálvez, J.; Goyanes, V. DBD-fish on neutral comets: Simultaneous analysis of DNA single- and double-strand breaks in individual cells. Exp. Cell Res. 2001, 270, 102–109. [Google Scholar] [CrossRef]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent Kinetics of Recruitment of MRE11 and NBS1 Proteins to Multiple DNA Damage Sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef]
- Yang, G.; Liu, C.; Chen, S.-H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar] [CrossRef]
- Jakob, B.; Taucher-Scholz, G. Live Cell Imaging to Study Real-Time ATM-Mediated Recruitment of DNA Repair Complexes to Sites of Ionizing Radiation-Induced DNA Damage. In ATM Kinase: Methods and Protocols; Kozlov, S.V., Ed.; Springer: New York, NY, USA, 2017; pp. 287–302. [Google Scholar]
- Davis, A.J.; Wang, S.Y.; Chen, D.J.; Chen, B.P.C. Imaging of Fluorescently Tagged ATM Kinase at the Sites of DNA Double Strand Breaks. Methods Mol. Biol. 2017, 1599, 277–285. [Google Scholar]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Wei, L.; Lan, L.; Hong, Z.; Yasui, A.; Ishioka, C.; Chiba, N. Rapid recruitment of BRCA1 to DNA double-strand breaks is dependent on its association with Ku80. Mol. Cell Biol. 2008, 28, 7380–7393. [Google Scholar] [CrossRef]
- Suchánková, J.; Kozubek, S.; Legartová, S.; Sehnalová, P.; Küntziger, T.; Bártová, E. Distinct kinetics of DNA repair protein accumulation at DNA lesions and cell cycle-dependent formation of γH2AX- and NBS1-positive repair foci. Biol. Cell 2015, 107, 440–454. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Lukas, C.; Melander, F.; Bartek, J.; Lukas, J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J. Cell Biol. 2005, 170, 201–211. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Soniat, M.M.; Walker, D.; Jang, S.; Finkelstein, I.J.; Harshey, R.M. Phage Mu Gam protein promotes NHEJ in concert withEscherichia coliligase. Proc. Natl. Acad. Sci. USA 2018, 115, E11614–E11622. [Google Scholar] [CrossRef]
- Abraham, Z.H.; Symonds, N. Purification of overexpressed gam gene protein from bacteriophage Mu by denaturation-renaturation techniques and a study of its DNA-binding properties. Biochem. J. 1990, 269, 679–684. [Google Scholar] [CrossRef]
- Williams, E.S.; Stap, J.; Essers, J.; Ponnaiya, B.; Luijsterburg, M.S.; Krawczyk, P.M.; Ullrich, R.L.; Aten, J.A.; Bailey, S.M. DNA double-strand breaks are not sufficient to initiate recruitment of TRF2. Nat. Genet. 2007, 39, 696–698. [Google Scholar] [CrossRef]
- Horibe, T.; Torisawa, A.; Akiyoshi, R.; Hatta-Ohashi, Y.; Suzuki, H.; Kawakami, K. Transfection efficiency of normal and cancer cell lines and monitoring of promoter activity by single-cell bioluminescence imaging. Luminescence 2014, 29, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Roizman, D.; Pachaimuthu, N.; Blázquez, J.; Rolff, J.; Rodríguez-Rojas, A. Antibiotic-induced recombination in bacteria requires the formation of double-strand breaks. bioRxiv 2022, 2022.03.08.483535. [Google Scholar] [CrossRef]
- Kotlajich, M.V.; Xia, J.; Zhai, Y.; Lin, H.-Y.; Bradley, C.C.; Shen, X.; Mei, Q.; Wang, A.Z.; Lynn, E.J.; Shee, C.; et al. Fluorescent fusions of the N protein of phage Mu label DNA damage in living cells. DNA Repair 2018, 72, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, A.; Beauséjour, C.; d’Adda di Fagagna, F. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell 2017, 16, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, A.; d’Adda di Fagagna, F. DNA Damage In Situ Ligation Followed by Proximity Ligation Assay (DI-PLA). In Cellular Senescence: Methods and Protocols; Demaria, M., Ed.; Springer: New York, NY, USA, 2019; pp. 11–20. [Google Scholar]
- Kordon, M.M.; Zarębski, M.; Solarczyk, K.; Ma, H.; Pederson, T.; Dobrucki, J.W. STRIDE—A fluorescence method for direct, specific in situ detection of individual single- or double-strand DNA breaks in fixed cells. Nucleic Acids Res. 2019, 48, e14. [Google Scholar] [CrossRef]
- Lutze, J.; Warrington, S.E.; Kron, S.J. TdT-dUTP DSB End Labeling (TUDEL), for Specific, Direct In Situ Labeling of DNA Double Strand Breaks. In Biomedical Engineering Technologies; Rasooly, A., Baker, H., Ossandon, M.R., Eds.; Springer: New York, NY, USA, 2022; pp. 299–317. [Google Scholar]
- Darzynkiewicz, Z.; Zhao, H. Detection of DNA strand breaks in apoptotic cells by flow- and image-cytometry. Methods Mol. Biol. 2011, 682, 91–101. [Google Scholar]
- Banér, J.; Nilsson, M.; Mendel-Hartvig, M.; Landegren, U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 1998, 26, 5073–5078. [Google Scholar] [CrossRef]
- Pinto, M.; Pickrell, A.M.; Wang, X.; Bacman, S.R.; Yu, A.; Hida, A.; Dillon, L.M.; Morton, P.D.; Malek, T.R.; Williams, S.L.; et al. Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ. 2017, 24, 288–299. [Google Scholar] [CrossRef]
- Didier, M.; Bursztajn, S.; Adamec, E.; Passani, L.; Nixon, R.A.; Coyle, J.T.; Wei, J.Y.; Berman, S.A. DNA strand breaks induced by sustained glutamate excitotoxicity in primary neuronal cultures. J. Neurosci. 1996, 16, 2238–2250. [Google Scholar] [CrossRef]
- Chen, J.; Jin, K.; Chen, M.; Pei, W.; Kawaguchi, K.; Greenberg, D.A.; Simon, R.P. Early Detection of DNA Strand Breaks in the Brain After Transient Focal Ischemia: Implications for the Role of DNA Damage in Apoptosis and Neuronal Cell Death. J. Neurochem. 1997, 69, 232–245. [Google Scholar] [CrossRef]
- Honda, S.; Sugita, I.; Miki, K.; Saito, I. The Semi-quantitative Comparison of Oxidative Stress Mediated DNA Single and Double Strand Breaks using Terminal Deoxynucleotidyl Transferase Mediated End Labeling Combined with a Slot Blot Technique. Free Radic. Res. 2004, 38, 481–485. [Google Scholar] [CrossRef]
- Leduc, F.; Faucher, D.; Bikond Nkoma, G.; Grégoire, M.-C.; Arguin, M.; Wellinger, R.J.; Boissonneault, G. Genome-Wide Mapping of DNA Strand Breaks. PLoS ONE 2011, 6, e17353. [Google Scholar] [CrossRef] [PubMed]
- Yamadori, I.; Yoshino, T.; Kondo, E.; Cao, L.; Akagi, T.; Matsuo, Y.; Minowada, J. Comparison of Two Methods of Staining Apoptotic Cells of Leukemia Cell Lines: Terminal Deoxynucleotidyl Transferase and DNA Polymerase I Reactions. J. Histochem. Cytochem. 1998, 46, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, P.S. Establishment of a new quantitative method for in situ detection of DNA breaks in human spermatozoa; Oslo Metropolitan University: Oslo, Norway, 2021. [Google Scholar]
- Pandey, N.; Black, B.E. Rapid Detection and Signaling of DNA Damage by PARP-1. Trends Biochem. Sci. 2021, 46, 744–757. [Google Scholar] [CrossRef]
- Izhar, L.; Adamson, B.; Ciccia, A.; Lewis, J.; Pontano-Vaites, L.; Leng, Y.; Liang, A.C.; Westbrook, T.F.; Harper, J.W.; Elledge, S.J. A Systematic Analysis of Factors Localized to Damaged Chromatin Reveals PARP-Dependent Recruitment of Transcription Factors. Cell Rep. 2015, 11, 1486–1500. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef]
- Thorslund, T.; West, S.C. BRCA2: A universal recombinase regulator. Oncogene 2007, 26, 7720–7730. [Google Scholar] [CrossRef] [PubMed]
- Navadgi-Patil, V.M.; Burgers, P.M. A tale of two tails: Activation of DNA damage checkpoint kinase Mec1/ATR by the 9–1–1 clamp and by Dpb11/TopBP1. DNA Repair 2009, 8, 996–1003. [Google Scholar] [CrossRef]
- Huang, J.; Gong, Z.; Ghosal, G.; Chen, J. SOSS complexes participate in the maintenance of genomic stability. Mol. Cell 2009, 35, 384–393. [Google Scholar] [CrossRef]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef]
- Sefer, A.; Kallis, E.; Eilert, T.; Röcker, C.; Kolesnikova, O.; Neuhaus, D.; Eustermann, S.; Michaelis, J. Structural dynamics of DNA strand break sensing by PARP-1 at a single-molecule level. Nat. Commun. 2022, 13, 6569. [Google Scholar] [CrossRef] [PubMed]
- Ogino, H.; Nozaki, T.; Gunji, A.; Maeda, M.; Suzuki, H.; Ohta, T.; Murakami, Y.; Nakagama, H.; Sugimura, T.; Masutani, M. Loss of Parp-1 affects gene expression profile in a genome-wide manner in ES cells and liver cells. BMC Genom. 2007, 8, 41. [Google Scholar]
- Virág, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly(ADP-ribose) signaling in cell death. Mol. Asp. Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef]
- Wang, F.; Zhao, M.; Chang, B.; Zhou, Y.; Wu, X.; Ma, M.; Liu, S.; Cao, Y.; Zheng, M.; Dang, Y.; et al. Cytoplasmic PARP1 links the genome instability to the inhibition of antiviral immunity through PARylating cGAS. Mol. Cell 2022, 82, 2032–2049.e7. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, N.S.; Haince, J.-F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-Ribose) (PAR) Binding to Apoptosis-Inducing Factor Is Critical for PAR Polymerase-1-Dependent Cell Death (Parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Haddad, M.; Rhinn, H.; Bloquel, C.; Coqueran, B.; Szabó, C.; Plotkine, M.; Scherman, D.; Margaill, I. Anti-inflammatory effects of PJ34, a poly(ADP-ribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. Br. J. Pharmacol. 2006, 149, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Tang, Y.-D.; Zhan, G.; Su, C.; Zheng, C. The Critical Role of PARPs in Regulating Innate Immune Responses. Front. Immunol. 2021, 12, 712556. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Ratanaphan, A. A DNA Repair BRCA1 Estrogen Receptor and Targeted Therapy in Breast Cancer. Int. J. Med. Sci. 2012, 13, 14898–14916. [Google Scholar] [CrossRef]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef]
- Ryabokon, N.I.; Goncharova, R.I.; Duburs, G.; Hancock, R.; Rzeszowska-Wolny, J. Changes in poly(ADP-ribose) level modulate the kinetics of DNA strand break rejoining. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2008, 637, 173–181. [Google Scholar] [CrossRef]
- Shao, Z.; Lee, B.J.; Rouleau-Turcotte, É.; Langelier, M.F.; Lin, X.; Estes, V.M.; Pascal, J.M.; Zha, S. Clinical PARP inhibitors do not abrogate PARP1 exchange at DNA damage sites in vivo. Nucleic Acids Res. 2020, 48, 9694–9709. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lu, L.Y.; Yang, C.Y.; Wang, S.; Yu, X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 2013, 27, 1752–1768. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Seif El Dahan, M.; Iehl, F.; Fernandez-Varela, P.; Le Du, M.-H.; Ropars, V.; Charbonnier, J.B. The Multifaceted Roles of Ku70/80. Int. J. Med. Sci. 2021, 22, 4134. [Google Scholar] [CrossRef]
- Li, B.; Reddy, S.; Comai, L. Depletion of Ku70/80 reduces the levels of extrachromosomal telomeric circles and inhibits proliferation of ALT cells. Aging (Albany NY) 2011, 3, 395–406. [Google Scholar] [CrossRef]
- Qiu, S.; Huang, J. MRN complex is an essential effector of DNA damage repair. J. Zhejiang Univ. Sci. B 2021, 22, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Deriano, L.; Stracker, T.H.; Baker, A.; Petrini, J.H.J.; Roth, D.B. Roles for NBS1 in Alternative Nonhomologous End-Joining of V(D)J Recombination Intermediates. Mol. Cell 2009, 34, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Dinkelmann, M.; Spehalski, E.; Stoneham, T.; Buis, J.; Wu, Y.; Sekiguchi, J.M.; Ferguson, D.O. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 2009, 16, 808–813. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Zhou, G.; Lajeunesse, M.; Le, N.; Stawicki, B.N.; Corcino, Y.L.; Berkner, K.L.; Runge, K.W. Nonhomologous End-Joining with Minimal Sequence Loss Is Promoted by the Mre11-Rad50-Nbs1-Ctp1 Complex in Schizosaccharomyces pombe. Genetics 2017, 206, 481–496. [Google Scholar] [CrossRef][Green Version]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 Dimers Coordinate DNA End Bridging and Nuclease Processing in Double-Strand-Break Repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.; Iwasaki, H.; Russell, P.; et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef]
- Wu, Y.; Xiao, S.; Zhu, X.D. MRE11-RAD50-NBS1 and ATM function as co-mediators of TRF1 in telomere length control. Nat. Struct. Mol. Biol. 2007, 14, 832–840. [Google Scholar] [CrossRef]
- Khayat, F.; Cannavo, E.; Alshmery, M.; Foster, W.R.; Chahwan, C.; Maddalena, M.; Smith, C.; Oliver, A.W.; Watson, A.T.; Carr, A.M.; et al. Inhibition of MRN activity by a telomere protein motif. Nat. Commun. 2021, 12, 3856. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.; Pavletich, N.P. Structure of the human ATM kinase and mechanism of Nbs1 binding. eLife 2022, 11, e74218. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Petrini, J.H. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell Biol. 2001, 21, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Britton, S.; Coates, J.; Jackson, S.P. A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J. Cell Biol. 2013, 202, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Dmitrieva, N.I.; Malide, D.; Burg, M.B. Mre11 is expressed in mammalian mitochondria where it binds to mitochondrial DNA. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R632–R640. [Google Scholar] [CrossRef] [PubMed]
- Coffey, G.; Lakshmipathy, U.; Campbell, C. Mammalian mitochondrial extracts possess DNA end-binding activity. Nucleic Acids Res. 1999, 27, 3348–3354. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity—Critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Bosch-Presegué, L.; Vaquero, A. Sirtuins in stress response: Guardians of the genome. Oncogene 2014, 33, 3764–3775. [Google Scholar] [CrossRef]
- Li, X.; Liu, L.; Li, T.; Liu, M.; Wang, Y.; Ma, H.; Mu, N.; Wang, H. SIRT6 in Senescence and Aging-Related Cardiovascular Diseases. Front. Cell Dev. Biol. 2021, 9, 641315. [Google Scholar] [CrossRef]
- Tian, X.; Firsanov, D.; Zhang, Z.; Cheng, Y.; Luo, L.; Tombline, G.; Tan, R.; Simon, M.; Henderson, S.; Steffan, J.; et al. SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 2019, 177, 622–638.e22. [Google Scholar] [CrossRef]
- Toiber, D.; Erdel, F.; Bouazoune, K.; Silberman, D.M.; Zhong, L.; Mulligan, P.; Sebastian, C.; Cosentino, C.; Martinez-Pastor, B.; Giacosa, S.; et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 2013, 51, 454–468. [Google Scholar] [CrossRef]
- Onn, L.; Portillo, M.; Ilic, S.; Cleitman, G.; Stein, D.; Kaluski, S.; Shirat, I.; Slobodnik, Z.; Einav, M.; Erdel, F.; et al. SIRT6 is a DNA double-strand break sensor. eLife 2020, 9, e51636. [Google Scholar] [CrossRef]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Jackson, S.P. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep. 2008, 9, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Löbrich, M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc. Natl. Acad. Sci. USA 2003, 100, 5057–5062. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.M.; Rezaeian, A.H. ATM: Main Features, Signaling Pathways, and Its Diverse Roles in DNA Damage Response, Tumor Suppression, and Cancer Development. Genes 2021, 12, 845. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Blevins, C.; Wani, G.; Wani, A.A. ATR- and ATM-Mediated DNA Damage Response Is Dependent on Excision Repair Assembly during G1 but Not in S Phase of Cell Cycle. PLoS ONE 2016, 11, e0159344. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, R.; Kastan, M.B. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Huang, X.; Halicka, H.D.; Zhao, H.; Traganos, F.; Albino, A.P.; Dai, W.; Darzynkiewicz, Z. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A 2007, 71, 648–661. [Google Scholar] [CrossRef] [PubMed]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.A.; van Ijcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.J.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. ATM’s Role in the Repair of DNA Double-Strand Breaks. Genes 2021, 12, 1370. [Google Scholar] [CrossRef]
- Savic, V.; Yin, B.; Maas, N.L.; Bredemeyer, A.L.; Carpenter, A.C.; Helmink, B.A.; Yang-Iott, K.S.; Sleckman, B.P.; Bassing, C.H. Formation of Dynamic γ-H2AX Domains along Broken DNA Strands Is Distinctly Regulated by ATM and MDC1 and Dependent upon H2AX Densities in Chromatin. Mol. Cell 2009, 34, 298–310. [Google Scholar] [CrossRef]
- Shroff, R.; Arbel-Eden, A.; Pilch, D.; Ira, G.; Bonner, W.M.; Petrini, J.H.; Haber, J.E.; Lichten, M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 2004, 14, 1703–1711. [Google Scholar] [CrossRef]
- Adams, B.R.; Golding, S.E.; Rao, R.R.; Valerie, K. Dynamic Dependence on ATR and ATM for Double-Strand Break Repair in Human Embryonic Stem Cells and Neural Descendants. PLoS ONE 2010, 5, e10001. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Matsuda, R.; Matsuda, Y.; Yanagisawa, S.-y.; Ikura, M.; Ikura, T.; Matsuda, T. An Easy-to-use Genotoxicity Assay Using EGFP-MDC1-expressing Human Cells. Genes Environ. 2014, 36, 17–28. [Google Scholar] [CrossRef]
- Feng, L.; Li, N.; Li, Y.; Wang, J.; Gao, M.; Wang, W.; Chen, J. Cell cycle-dependent inhibition of 53BP1 signaling by BRCA1. Cell Discov. 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Petersen, S.; Romanienko, P.J.; Fernandez-Capetillo, O.; Chen, H.T.; Sedelnikova, O.A.; Reina-San-Martin, B.; Coppola, V.; Meffre, E.; Difilippantonio, M.J.; et al. Genomic instability in mice lacking histone H2AX. Science 2002, 296, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Rybak, P.; Hoang, A.; Bujnowicz, L.; Bernas, T.; Berniak, K.; Zarębski, M.; Darzynkiewicz, Z.; Dobrucki, J. Low level phosphorylation of histone H2AX on serine 139 (γH2AX) is not associated with DNA double-strand breaks. Oncotarget 2016, 7, 49574–49587. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Voss, K.-O.; Tobias, F.; Jakob, B.; Durante, M.; Taucher-Scholz, G. Clustered DNA damage induces pan-nuclear H2AX phosphorylation mediated by ATM and DNA–PK. Nucleic Acids Res. 2013, 41, 6109–6118. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.-Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.-D.; Guan, H.; Zhang, S.-M.; Tang, Y.; Rang, W.-Q.; Zhou, P.-K. γH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef]
- Bouquet, F.; Muller, C.; Salles, B. The Loss of gammaH2AX Signal is a Marker of DNA Double Strand Breaks Repair Only at Low Levels of DNA Damage. Cell Cycle 2006, 5, 1116–1122. [Google Scholar] [CrossRef]
- Bouquet, F.; Pal, A.; Pilones, K.A.; Demaria, S.; Hann, B.; Akhurst, R.J.; Babb, J.S.; Lonning, S.M.; DeWyngaert, J.K.; Formenti, S.C.; et al. TGFβ1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin. Cancer Res. 2011, 17, 6754–6765. [Google Scholar] [CrossRef]
- Goldberg, M.; Stucki, M.; Falck, J.; D’Amours, D.; Rahman, D.; Pappin, D.; Bartek, J.; Jackson, S.P. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 2003, 421, 952–956. [Google Scholar] [CrossRef]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef]
- Zhang, F.; Bick, G.; Park, J.Y.; Andreassen, P.R. MDC1 and RNF8 function in a pathway that directs BRCA1-dependent localization of PALB2 required for homologous recombination. J. Cell Sci. 2012, 125, 6049–6057. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, J.; Ma, Z.; Treszezamsky, A.; Powell, S.N. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat. Struct. Mol. Biol. 2005, 12, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Park, S.-J.; Hariharasudhan, G.; Kim, M.-J.; Jung, S.M.; Jeong, S.-Y.; Chang, I.-Y.; Kim, C.; Kim, E.; Yu, J.; et al. ID3 regulates the MDC1-mediated DNA damage response in order to maintain genome stability. Nat. Commun. 2017, 8, 903. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Adhikari, S.; Butler, B.E.; Pandita, T.K.; Mitra, S.; Hegde, M.L. A Perspective on Chromosomal Double Strand Break Markers in Mammalian Cells. Jacobs J. Radiat. Oncol. 2014, 1, 3. [Google Scholar]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.-L.; Wu, X. Cell Cycle-dependent Complex Formation of BRCA1·CtIP·MRN Is Important for DNA Double-strand Break Repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.-T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Dimitrova, N.; Chen, Y.-C.M.; Spector, D.L.; de Lange, T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008, 456, 524–528. [Google Scholar] [CrossRef]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef]
- Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40–63. [Google Scholar] [CrossRef]
- Walsh, C.S. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Polato, F.; Callen, E.; Wong, N.; Faryabi, R.; Bunting, S.; Chen, H.-T.; Kozak, M.; Kruhlak, M.J.; Reczek, C.R.; Lee, W.-H.; et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014, 211, 1027–1036. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, L.-Y. BRCA1 and homologous recombination: Implications from mouse embryonic development. Cell Biosci. 2020, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534. [Google Scholar] [CrossRef]
- Nakamura, K.; Saredi, G.; Becker, J.R.; Foster, B.M.; Nguyen, N.V.; Beyer, T.E.; Cesa, L.C.; Faull, P.A.; Lukauskas, S.; Frimurer, T.; et al. H4K20me0 recognition by BRCA1–BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019, 21, 311–318. [Google Scholar] [CrossRef]
- Michelena, J.; Pellegrino, S.; Spegg, V.; Altmeyer, M. Replicated chromatin curtails 53BP1 recruitment in BRCA1-proficient and BRCA1-deficient cells. Life Sci. Alliance 2021, 4, e202101023. [Google Scholar] [CrossRef]
- Paiano, J.; Zolnerowich, N.; Wu, W.; Pavani, R.; Wang, C.; Li, H.; Zheng, L.; Shen, B.; Sleckman, B.P.; Chen, B.R.; et al. Role of 53BP1 in end protection and DNA synthesis at DNA breaks. Genes Dev. 2021, 35, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. Roles for 53BP1 in the repair of radiation-induced DNA double strand breaks. DNA Repair 2020, 93, 102915. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Lei, K.-H.; Biak Sang, P.; Shiva, O.; Chastain, M.; Chi, P.; Chai, W. Human CST complex protects stalled replication forks by directly blocking MRE11 degradation of nascent-strand DNA. EMBO J. 2021, 40, e103654. [Google Scholar] [CrossRef] [PubMed]
- Altieri, A.; Dell’Aquila, M.; Pentimalli, F.; Giordano, A.; Luigi, A. SMART (Single Molecule Analysis of Resection Tracks) Technique for Assessing DNA end-Resection in Response to DNA Damage. Bio Protoc. 2020, 10, e3701. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDR Stage | Marker | Complexity | Uses | Challenges |
|---|---|---|---|---|
| DSB Detector Assays | GamGFP | ★★★☆☆ | Binds specifically to DSBs. Simple protocol compared to other DSB assays. | Competition with endogenous Ku in mammalian cells. Optimizing transfection efficiency. |
| DNA damage In situ ligation followed by Proximity Ligation Assay (DI-PLA) | ★★★★★ | Identifies DDR proteins proximal to DSB sites. | May not target all DSBs due to changes in DDR activity. | |
| SensiTive Recognition of Individual DNA Ends (STRIDE) | ★★★★★ | Highly sensitive. Detects single CRISPR/Cas9 nicks. | TdT enzyme may result in non-specificity (SSBs, cytosolic DNA, mitochondrial DNA) | |
| TdT-UTP DSB End Labeling (TUDEL) | ★★★★☆ | High sensitivity. | ||
| First Responders | Poly (ADP-ribose) Polymerase-1 (PARP1) | ★★★☆☆ | Active 0.5 s post-insult. Recruits multiple DDR factors. | Can reside at intracellular locations distinct from DSBs. |
| Ku70/80 | ★★☆☆☆ | Active 1 s post-insult. Involved in canonical-NHEJ (c-NHEJ). | Displaced from DSBs during S/G2 phases and HR. | |
| Sirtun 6 (SIRT6) | ★★★☆☆ | Active 5 s post-insult. Binds to DSBs independently of other DDR factors. | Binds to SSBs. FRET to identify dimer pairs at DSB sites. | |
| MRN | ★★☆☆☆ | Active 10 s post-insult. Involved in HR. | Involved in c-NHEJ and alternative-NHEJ (a-NHEJ), role not yet established. | |
| DDR Activators | Ataxia-Telangiectasia Mutated (ATM) | ★★☆☆☆ | Key DDR regulator. Phosphorylates H2AX. | Activity is cell-cycle-dependent. Non-DSB damage can activate ATM, do not form discrete foci. |
| γH2AX | ★★☆☆☆ | Well-established DDR marker. Activated to promote DSB repair molecules. | Foci do not always indicate the presence or proximity of DSBs. Activation kinetics vary. | |
| Mediator of DNA Damage Checkpoint protein 1 (MDC1) | ★★☆☆☆ | Enhances signaling of damage. Regulates multiple molecules across most stages of repair. | Damage kinetics can be inconsistent across cell types due to regulating multiple proteins. | |
| Repair Pathway Specific Assays | Breast Cancer 1 (BRCA1) | ★★★☆☆ | Regulates HR initiation. | BRCA1 and 53BP1 antagonism. Repair kinetics altered in different cell types (e.g., cancers). |
| p53 Binding Protein 1 (53BP1) | ★★★☆☆ | Regulates NHEJ initiation. |
| Technique | T4 Pol Blunting | Incorporation Reagent | DSB End Conjugate | Antibody Pair | Signal Amplification | Fluorescent Signal |
|---|---|---|---|---|---|---|
| DI-PLA | Yes | T4 Ligase | Biotin linker | Biotin + proximal DDR marker | Yes | Proximity Ligation Assay |
| STRIDE | No | TdT | BrdU | BrdU + BrdU | Yes | Proximity Ligation Assay |
| TUDEL | Yes | TdT | EdU | None | No | Azide-linked Fluorophore |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atkinson, J.; Bezak, E.; Le, H.; Kempson, I. DNA Double Strand Break and Response Fluorescent Assays: Choices and Interpretation. Int. J. Mol. Sci. 2024, 25, 2227. https://doi.org/10.3390/ijms25042227
Atkinson J, Bezak E, Le H, Kempson I. DNA Double Strand Break and Response Fluorescent Assays: Choices and Interpretation. International Journal of Molecular Sciences. 2024; 25(4):2227. https://doi.org/10.3390/ijms25042227
Chicago/Turabian StyleAtkinson, Jake, Eva Bezak, Hien Le, and Ivan Kempson. 2024. "DNA Double Strand Break and Response Fluorescent Assays: Choices and Interpretation" International Journal of Molecular Sciences 25, no. 4: 2227. https://doi.org/10.3390/ijms25042227
APA StyleAtkinson, J., Bezak, E., Le, H., & Kempson, I. (2024). DNA Double Strand Break and Response Fluorescent Assays: Choices and Interpretation. International Journal of Molecular Sciences, 25(4), 2227. https://doi.org/10.3390/ijms25042227