
Therapeutic Targeting of Hypoxia-Inducible Factors in Cancer

 , ,
, , 
Abstract
1. Introduction
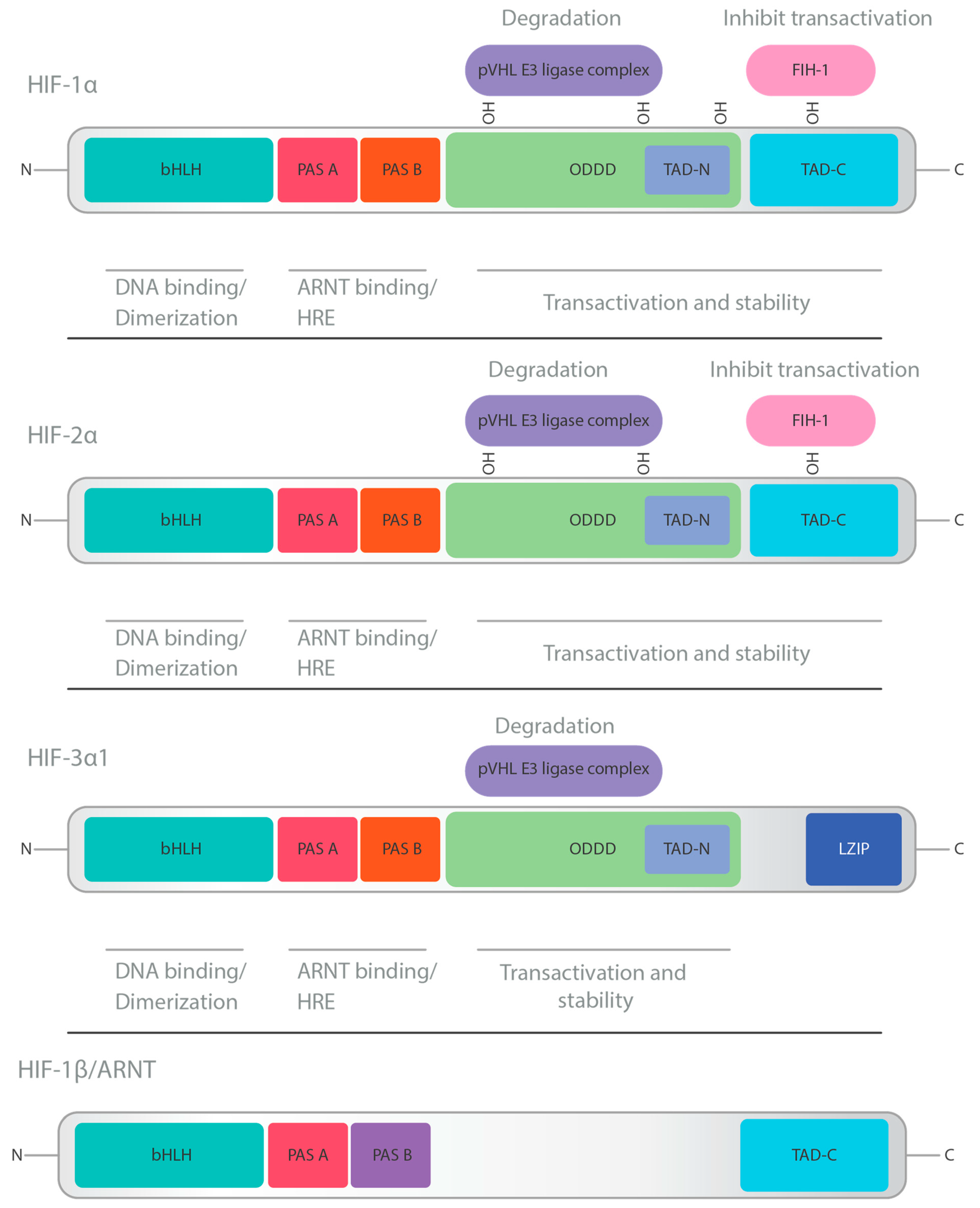
2. HIF Structure and Physiology
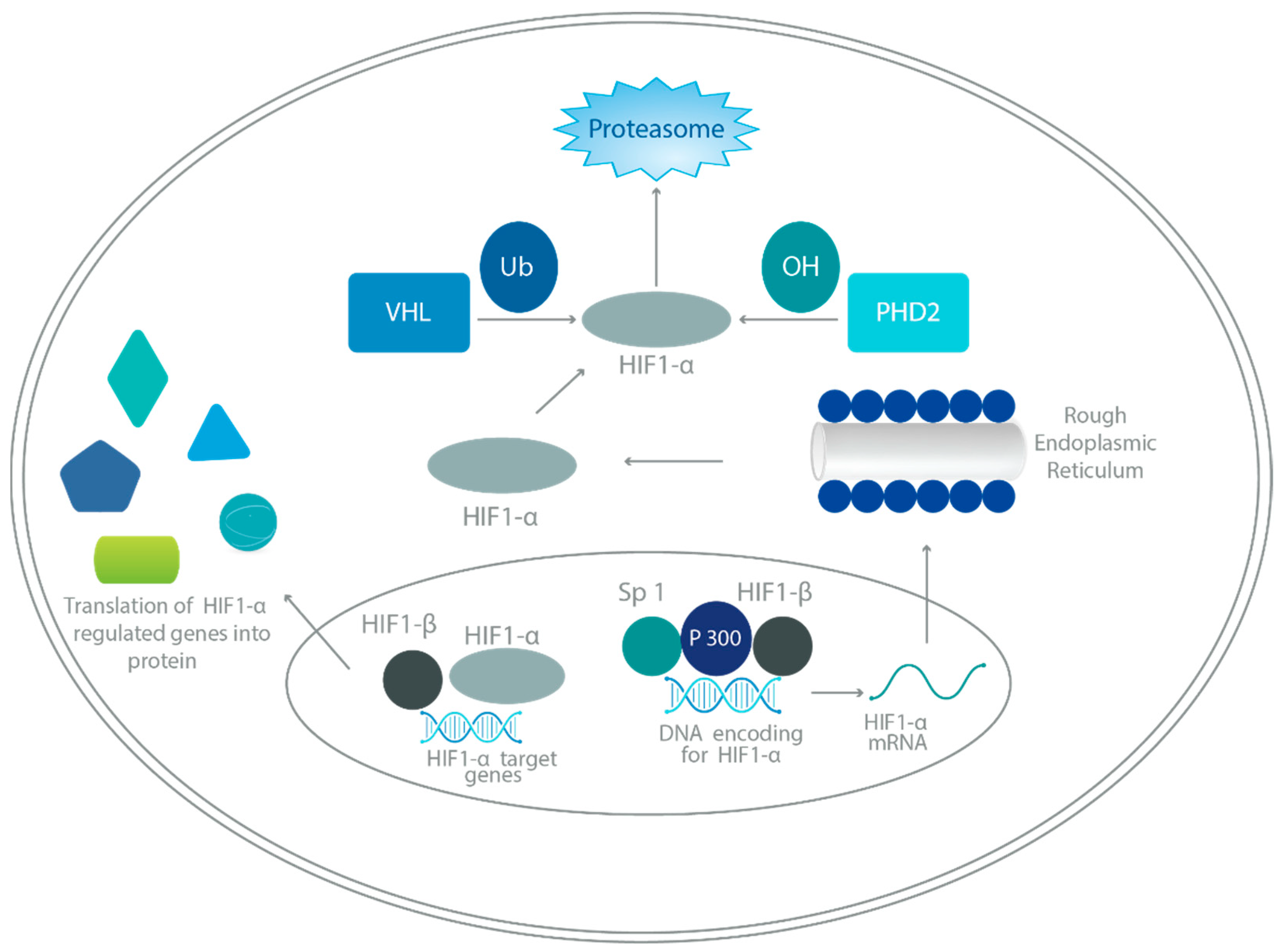
3. HIF Regulation
4. Role of HIFs in Cancer Progression
4.1. Regulation of Signal Transduction Pathways
4.2. Tumor Immune Escape and Immunotherapy Resistance
5. Role of HIFs in Solid Tumors
5.1. Role of HIFs in Liver Cancer
5.2. Role of HIFs in Renal Cell Carcinoma (RCC)
5.3. Role of HIFs in Gastric Cancer
5.4. Role of HIFs in Breast Cancer
6. HIF-Associated Future Cancer Therapies
6.1. Reduction in HIF-1α Protein Synthesis
6.2. HIF-1 Inhibitors Destabilizing HIF-1α
6.3. HIF-1 Dimerization Inhibitor
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, X.; Yu, H. Global burden of cancer. Yale J. Biol. Med. 2006, 79, 85–94. [Google Scholar] [PubMed]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Paredes, F.; Williams, H.C.; San Martin, A. Metabolic adaptation in hypoxia and cancer. Cancer Lett. 2021, 502, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Wicks, E.E.; Semenza, G.L. Hypoxia-inducible factors: Cancer progression and clinical translation. J. Clin. Investig. 2022, 132, e159839. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.W.; Bai, G.H.; Wang, T.L.; Shih, I.M.; Chuang, C.M.; Lo, C.L.; Tsai, M.C.; Chiu, L.Y.; Lin, C.C.; Shen, Y.A. Novel cancer treatment paradigm targeting hypoxia-induced factor in conjunction with current therapies to overcome resistance. J. Exp. Clin. Cancer Res. 2023, 42, 171. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. Faseb J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- Fernandez-Torres, J.; Zamudio-Cuevas, Y.; Martinez-Nava, G.A.; Lopez-Reyes, A.G. Hypoxia-Inducible Factors (HIFs) in the articular cartilage: A systematic review. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2800–2810. [Google Scholar]
- Yang, S.L.; Wu, C.; Xiong, Z.F.; Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-κB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef]
- Masson, N.; Singleton, R.S.; Sekirnik, R.; Trudgian, D.C.; Ambrose, L.J.; Miranda, M.X.; Tian, Y.M.; Kessler, B.M.; Schofield, C.J.; Ratcliffe, P.J. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012, 13, 251–257. [Google Scholar] [CrossRef]
- Dames, S.A.; Martinez-Yamout, M.; De Guzman, R.N.; Dyson, H.J.; Wright, P.E. Structural basis for Hif-1α/CBP recognition in the cellular hypoxic response. Proc. Natl. Acad. Sci. USA 2002, 99, 5271–5276. [Google Scholar] [CrossRef]
- Salati, M.; Caputo, F.; Baldessari, C.; Galassi, B.; Grossi, F.; Dominici, M.; Ghidini, M. IDH Signalling Pathway in Cholangiocarcinoma: From Biological Rationale to Therapeutic Targeting. Cancers 2020, 12, 3310. [Google Scholar] [CrossRef]
- Cerychova, R.; Pavlinkova, G. HIF-1, Metabolism, and Diabetes in the Embryonic and Adult Heart. Front. Endocrinol. 2018, 9, 460. [Google Scholar] [CrossRef] [PubMed]
- Pandya, N.M.; Dhalla, N.S.; Santani, D.D. Angiogenesis--a new target for future therapy. Vasc. Pharmacol 2006, 44, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Maynard, M.A.; Ohh, M. The role of hypoxia-inducible factors in cancer. Cell. Mol. Life Sci. 2007, 64, 2170–2180. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Liu, L.; Runge, A.; Wang, T.; Yuan, L.; Patel, S.; Iruela-Arispe, L.; Simon, M.C.; Keith, B. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood 2009, 114, 469–477. [Google Scholar] [CrossRef]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef]
- Sang, Q.X. Complex role of matrix metalloproteinases in angiogenesis. Cell Res. 1998, 8, 171–177. [Google Scholar] [CrossRef]
- Ito, K.; Kitajima, Y.; Kai, K.; Matsufuji, S.; Yamada, K.; Egawa, N.; Kitagawa, H.; Okuyama, K.; Tanaka, T.; Noshiro, H. Matrix metalloproteinase-1 expression is regulated by HIF-1-dependent and epigenetic mechanisms and serves a tumor-suppressive role in gastric cancer progression. Int. J. Oncol. 2021, 59, 102. [Google Scholar] [CrossRef]
- Lolmede, K.; Durand de Saint Front, V.; Galitzky, J.; Lafontan, M.; Bouloumie, A. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int. J. Obes. 2003, 27, 1187–1195. [Google Scholar] [CrossRef]
- Vannini, F.; Kashfi, K.; Nath, N. The dual role of iNOS in cancer. Redox Biol. 2015, 6, 334–343. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef]
- Hapke, R.Y.; Haake, S.M. Hypoxia-induced epithelial to mesenchymal transition in cancer. Cancer Lett. 2020, 487, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Bui, B.P.; Nguyen, P.L.; Lee, K.; Cho, J. Hypoxia-Inducible Factor-1: A Novel Therapeutic Target for the Management of Cancer, Drug Resistance, and Cancer-Related Pain. Cancers 2022, 14, 6054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1α Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wong, C.C.; Wei, H.; Gilkes, D.M.; Korangath, P.; Chaturvedi, P.; Schito, L.; Chen, J.; Krishnamachary, B.; Winnard, P.T., Jr.; et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012, 31, 1757–1770. [Google Scholar] [CrossRef]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-inducible factors: Master regulators of hypoxic tumor immune escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhan, H.; Ye, Y.; Yang, J.; Zhang, M.; Li, J.; Zhuang, Y. Current Progress and Future Perspectives of Immune Checkpoint in Cancer and Infectious Diseases. Front. Genet. 2021, 12, 785153. [Google Scholar] [CrossRef] [PubMed]
- Benoit, A.; Vogin, G.; Duhem, C.; Berchem, G.; Janji, B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Cells 2023, 12, 1787. [Google Scholar] [CrossRef]
- Sebestyen, A.; Kopper, L.; Danko, T.; Timar, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 27, 1609802. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.H.; Wong, C.C. Hypoxia, Metabolic Reprogramming, and Drug Resistance in Liver Cancer. Cells 2021, 10, 1715. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Wei, L.; Lee, D.; Law, C.T.; Zhang, M.S.; Shen, J.; Chin, D.W.; Zhang, A.; Tsang, F.H.; Wong, C.L.; Ng, I.O.; et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 2019, 10, 4681. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Kaelin, W.G., Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, H. The Roles of VHL-Dependent Ubiquitination in Signaling and Cancer. Front. Oncol. 2012, 2, 35. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, K.; Rizzi, J.P.; Huang, H.; Grina, J.A.; Schlachter, S.T.; Wang, B.; Wehn, P.M.; Yang, H.; Dixon, D.D.; et al. 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2alpha (HIF-2alpha) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J. Med. Chem. 2019, 62, 6876–6893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, T.; Simon, J.; Takada, M.; Saito, R.; Fan, C.; Liu, X.D.; Jonasch, E.; Xie, L.; Chen, X.; et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 2018, 361, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Badgwell, B.D. Current treatment and recent progress in gastric cancer. CA Cancer J. Clin. 2021, 71, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xiao, A.; Liu, C.; Ye, C.; Yin, K.; Lu, M.; Jiao, R.; Chen, X.; Zhang, C.; Liu, M. The HIF-1A/miR-17-5p/PDCD4 axis contributes to the tumor growth and metastasis of gastric cancer. Signal Transduct. Target. Ther. 2020, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Liu, J.; Tan, S.; Liu, W.; Wang, R.; Chen, C. PLGA sustained-release microspheres loaded with an insoluble small-molecule drug: Microfluidic-based preparation, optimization, characterization, and evaluation in vitro and in vivo. Drug Deliv. 2022, 29, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, G.; Yang, X.; Yin, W.; Lv, G.; Wang, S. HIF in Gastric Cancer: Regulation and Therapeutic Target. Molecules 2022, 27, 4893. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta 2016, 1863, 382–391. [Google Scholar] [CrossRef]
- Gilreath, C.; Boerma, M.; Qin, Z.; Hudson, M.K.; Wang, S. The Hypoxic Microenvironment of Breast Cancer Cells Promotes Resistance in Radiation Therapy. Front. Oncol. 2020, 10, 629422. [Google Scholar] [CrossRef]
- Kozal, K.; Krzeslak, A. The Role of Hypoxia-Inducible Factor Isoforms in Breast Cancer and Perspectives on Their Inhibition in Therapy. Cancers 2022, 14, 4518. [Google Scholar] [CrossRef]
- Fang, W.; Liao, C.; Shi, R.; Simon, J.M.; Ptacek, T.S.; Zurlo, G.; Ye, Y.; Han, L.; Fan, C.; Bao, L.; et al. ZHX2 promotes HIF1alpha oncogenic signaling in triple-negative breast cancer. Elife 2021, 10, e70412. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xing, C.; Deng, Y.; Ye, C.; Peng, H. HIF-1alpha signaling: Essential roles in tumorigenesis and implications in targeted therapies. Genes Dis. 2024, 11, 234–251. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1alpha), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Saluja, A. Minnelide, a novel drug for pancreatic and liver cancer. Pancreatology 2015, 15, S39–S43. [Google Scholar] [CrossRef]
- McGinn, O.; Gupta, V.K.; Dauer, P.; Arora, N.; Sharma, N.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. Inhibition of hypoxic response decreases stemness and reduces tumorigenic signaling due to impaired assembly of HIF1 transcription complex in pancreatic cancer. Sci. Rep. 2017, 7, 7872. [Google Scholar] [CrossRef]
- Skorupan, N.; Ahmad, M.I.; Steinberg, S.M.; Trepel, J.B.; Cridebring, D.; Han, H.; Von Hoff, D.D.; Alewine, C. A phase II trial of the super-enhancer inhibitor Minnelide in advanced refractory adenosquamous carcinoma of the pancreas. Future Oncol. 2022, 18, 2475–2481. [Google Scholar] [CrossRef]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef]
- Norris, R.E.; Shusterman, S.; Gore, L.; Muscal, J.A.; Macy, M.E.; Fox, E.; Berkowitz, N.; Buchbinder, A.; Bagatell, R. Phase 1 evaluation of EZN-2208, a polyethylene glycol conjugate of SN38, in children adolescents and young adults with relapsed or refractory solid tumors. Pediatr. Blood Cancer 2014, 61, 1792–1797. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Andreu-Vieyra, C.V.; Berenson, J.R. The potential of panobinostat as a treatment option in patients with relapsed and refractory multiple myeloma. Ther. Adv. Hematol. 2014, 5, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Bubna, A.K. Vorinostat-An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Park, S.R.; Rapisarda, A.; Fer, N.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; et al. Weekly EZN-2208 (PEGylated SN-38) in combination with bevacizumab in patients with refractory solid tumors. Invest. New Drugs 2014, 32, 340–346. [Google Scholar] [CrossRef]
- Garrett, C.R.; Bekaii-Saab, T.S.; Ryan, T.; Fisher, G.A.; Clive, S.; Kavan, P.; Shacham-Shmueli, E.; Buchbinder, A.; Goldberg, R.M. Randomized phase 2 study of pegylated SN-38 (EZN-2208) or irinotecan plus cetuximab in patients with advanced colorectal cancer. Cancer 2013, 119, 4223–4230. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.E.; Boothman, D.A.; Fattah, F.J.; Dong, Y.; Zhu, H.; Skelton, R.A.; Priddy, L.L.; Vo, P.; Dowell, J.E.; Sarode, V.; et al. Phase 1 study of romidepsin plus erlotinib in advanced non-small cell lung cancer. Lung Cancer 2015, 90, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Haigentz, M., Jr.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.L.; Mina, R.; Jakubowiak, A.J.; Zimmerman, T.L.; Wolf, J.J.; Lewis, C.; Gleason, C.; Sharp, C.; Martin, T.; Heffner, L.T.; et al. Combining carfilzomib and panobinostat to treat relapsed/refractory multiple myeloma: Results of a Multiple Myeloma Research Consortium Phase I Study. Blood Cancer J. 2019, 9, 3. [Google Scholar] [CrossRef]
- Wood, P.J.; Strong, R.; McArthur, G.A.; Michael, M.; Algar, E.; Muscat, A.; Rigby, L.; Ferguson, M.; Ashley, D.M. A phase I study of panobinostat in pediatric patients with refractory solid tumors, including CNS tumors. Cancer Chemother. Pharmacol. 2018, 82, 493–503. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Gunther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: A multicentre, randomised, double-blind phase 3 trial. Lancet Oncol 2014, 15, 1195–1206. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Miller, C.R.; Sarkaria, J.N.; Jaeckle, K.; Buckner, J.C.; Ligon, K.L.; Ballman, K.V.; Moore, D.F., Jr.; Nebozhyn, M.; et al. Phase I/II trial of vorinostat combined with temozolomide and radiation therapy for newly diagnosed glioblastoma: Results of Alliance N0874/ABTC 02. Neuro Oncol. 2018, 20, 546–556. [Google Scholar] [CrossRef]
- Haas, N.B.; Quirt, I.; Hotte, S.; McWhirter, E.; Polintan, R.; Litwin, S.; Adams, P.D.; McBryan, T.; Wang, L.; Martin, L.P.; et al. Phase II trial of vorinostat in advanced melanoma. Invest. New Drugs 2014, 32, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.M.; Kindler, H.L.; Calvert, H.; Manegold, C.; Tsao, A.S.; Fennell, D.; Ohman, R.; Plummer, R.; Eberhardt, W.E.; Fukuoka, K.; et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol 2015, 16, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Hillman, D.W.; Vaishampayan, U.; Sheng, S.; Sarkar, F.; Harper, F.; Gaskins, M.; Pitot, H.C.; Tan, W.; Ivy, S.P.; et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 2008, 14, 7940–7946. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Chanan-Khan, A.A.; Lonial, S.; Krishnan, A.Y.; Carroll, M.P.; Alsina, M.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: Results of a phase 1/2 study. Br. J. Haematol. 2011, 153, 729–740. [Google Scholar] [CrossRef]
- Hanrahan, E.O.; Kies, M.S.; Glisson, B.S.; Khuri, F.R.; Feng, L.; Tran, H.T.; Ginsberg, L.E.; Truong, M.T.; Hong, W.K.; Kim, E.S. A phase II study of Lonafarnib (SCH66336) in patients with chemorefractory, advanced squamous cell carcinoma of the head and neck. Am. J. Clin. Oncol. 2009, 32, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kemeny, N.; Kelsen, D.P.; Ilson, D.; O’Reilly, E.; Zaknoen, S.; Baum, C.; Statkevich, P.; Hollywood, E.; Zhu, Y.; et al. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann. Oncol. 2002, 13, 1067–1071. [Google Scholar] [CrossRef]
- Kim, E.S.; Kies, M.S.; Fossella, F.V.; Glisson, B.S.; Zaknoen, S.; Statkevich, P.; Munden, R.F.; Summey, C.; Pisters, K.M.; Papadimitrakopoulou, V.; et al. Phase II study of the farnesyltransferase inhibitor lonafarnib with paclitaxel in patients with taxane-refractory/resistant nonsmall cell lung carcinoma. Cancer 2005, 104, 561–569. [Google Scholar] [CrossRef]
- Brown, T.D.; Goodman, P.J.; Fleming, T.R.; Taylor, S.A.; Macdonald, J.S. Phase II trial of echinomycin in advanced colorectal cancer. A Southwest Oncology Group study. Invest. New Drugs 1991, 9, 113–114. [Google Scholar] [CrossRef]
- Marshall, M.E.; Wolf, M.K.; Crawford, E.D.; Taylor, S.; Blumenstein, B.; Flanigan, R.; Meyers, F.J.; Hynes, H.E.; Barlogie, B.; Eisenberger, M. Phase II trial of echinomycin for the treatment of advanced renal cell carcinoma. A Southwest Oncology Group study. Invest. New Drugs 1993, 11, 207–209. [Google Scholar] [CrossRef]
- Muss, H.B.; Blessing, J.A.; Baker, V.V.; Barnhill, D.R.; Adelson, M.D. Echinomycin (NSC 526417) in advanced ovarian cancer. A phase II trial of the Gynecologic Oncology Group. Am. J. Clin. Oncol. 1990, 13, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Schilsky, R.L.; Faraggi, D.; Korzun, A.; Vogelzang, N.; Ellerton, J.; Wood, W.; Henderson, I.C. Phase II study of echinomycin in patients with advanced breast cancer: A report of Cancer and Leukemia Group B protocol 8641. Invest. New Drugs 1991, 9, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Richardson, P.G.; Lacy, M.Q.; Dispenzieri, A.; Greipp, P.R.; Witzig, T.E.; Schlossman, R.; Sidor, C.F.; Anderson, K.C.; Gertz, M.A. Novel therapy with 2-methoxyestradiol for the treatment of relapsed and plateau phase multiple myeloma. Clin. Cancer Res. 2007, 13, 6162–6167. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.R.; Hahn, N.M.; Pili, R.; Oh, W.K.; Hammers, H.; Sweeney, C.; Kim, K.; Perlman, S.; Arnott, J.; Sidor, C.; et al. A phase II study of 2-methoxyestradiol (2ME2) NanoCrystal(R) dispersion (NCD) in patients with taxane-refractory, metastatic castrate-resistant prostate cancer (CRPC). Invest. New Drugs 2011, 29, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Schilder, J.; Sutton, G.; Perkins, S.; Breen, T.; Quon, C.; Sidor, C. Activity of 2 methoxyestradiol (Panzem NCD) in advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Gynecol. Oncol. 2009, 115, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Chan, J.A.; Meyerhardt, J.A.; Zhu, A.X.; Abrams, T.A.; Blaszkowsky, L.S.; Regan, E.; Sidor, C.; Fuchs, C.S. A prospective phase II study of 2-methoxyestradiol administered in combination with bevacizumab in patients with metastatic carcinoid tumors. Cancer Chemother. Pharmacol. 2011, 68, 293–300. [Google Scholar] [CrossRef]
- Salman, S.; Meyers, D.J.; Wicks, E.E.; Lee, S.N.; Datan, E.; Thomas, A.M.; Anders, N.M.; Hwang, Y.; Lyu, Y.; Yang, Y.; et al. HIF inhibitor 32-134D eradicates murine hepatocellular carcinoma in combination with anti-PD1 therapy. J. Clin. Investig. 2022, 132, e156774. [Google Scholar] [CrossRef]
- Piorecka, K.; Kurjata, J.; Stanczyk, W.A. Acriflavine, an Acridine Derivative for Biomedical Application: Current State of the Art. J. Med. Chem. 2022, 65, 11415–11432. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Mechanism of HIF-1 Inhibition | HIF-1 Inhibitor | Cancer Type | Reference |
|---|---|---|---|
| Decreasing HIF-1α mRNA expression | EZN-2208 (PEG-SN38) | Refractory solid tumors Metastatic colorectal cancer | [59,64,65] |
| Decreasing HIF-1α protein synthesis | EZN-2968 | Refractory solid tumors | [54] |
| Decreasing HIF-1α stabilization | FK228 (Romidepsin) | Non-small cell lung carcinoma (NSCLC) Head and neck cancer | [66,67] |
| LBH589 (Panobinostat) | Multiple myeloma (MM) Refractory solid tumors Refractory MM | [68,69,70] | |
| Vorinostat | Glioblastoma Advanced melanoma Mesothelioma | [71,72,73] | |
| 17-allylamino-17-demethoxygeldanamycin (17-AAG) (Tanespimycin) | Prostate cancer Refractory MM | [74,75] | |
| SCH66336 (Lonafarnib) | Squamous cell carcinoma of the head and neck (SCCHN) Lung carcinoma Colorectal cancer | [76,77,78] | |
| Decreasing HIF-1/ DNA binding | Echinomycin | Ovarian cancer Breast cancer Renal cell carcinoma Colorectal cancer | [79,80,81,82] |
| Decreasing HIF-1α protein synthesis and transcriptional activity | 2-methoxyestradiol (2ME2/Panzem) | MM Prostate cancer Ovarian cancer Carcinoid tumors | [83,84,85,86] |
| Decreasing HIF-1α and HIF-2α protein synthesis and transcriptional activity | 32-134D | Hepatocellular carcinoma | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musleh Ud Din, S.; Streit, S.G.; Huynh, B.T.; Hana, C.; Abraham, A.-N.; Hussein, A. Therapeutic Targeting of Hypoxia-Inducible Factors in Cancer. Int. J. Mol. Sci. 2024, 25, 2060. https://doi.org/10.3390/ijms25042060
Musleh Ud Din S, Streit SG, Huynh BT, Hana C, Abraham A-N, Hussein A. Therapeutic Targeting of Hypoxia-Inducible Factors in Cancer. International Journal of Molecular Sciences. 2024; 25(4):2060. https://doi.org/10.3390/ijms25042060
Chicago/Turabian StyleMusleh Ud Din, Saba, Spencer G. Streit, Bao Tran Huynh, Caroline Hana, Anna-Ninny Abraham, and Atif Hussein. 2024. "Therapeutic Targeting of Hypoxia-Inducible Factors in Cancer" International Journal of Molecular Sciences 25, no. 4: 2060. https://doi.org/10.3390/ijms25042060
APA StyleMusleh Ud Din, S., Streit, S. G., Huynh, B. T., Hana, C., Abraham, A.-N., & Hussein, A. (2024). Therapeutic Targeting of Hypoxia-Inducible Factors in Cancer. International Journal of Molecular Sciences, 25(4), 2060. https://doi.org/10.3390/ijms25042060