
In-Silico Identification of Novel Pharmacological Synergisms: The Trabectedin Case

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Results
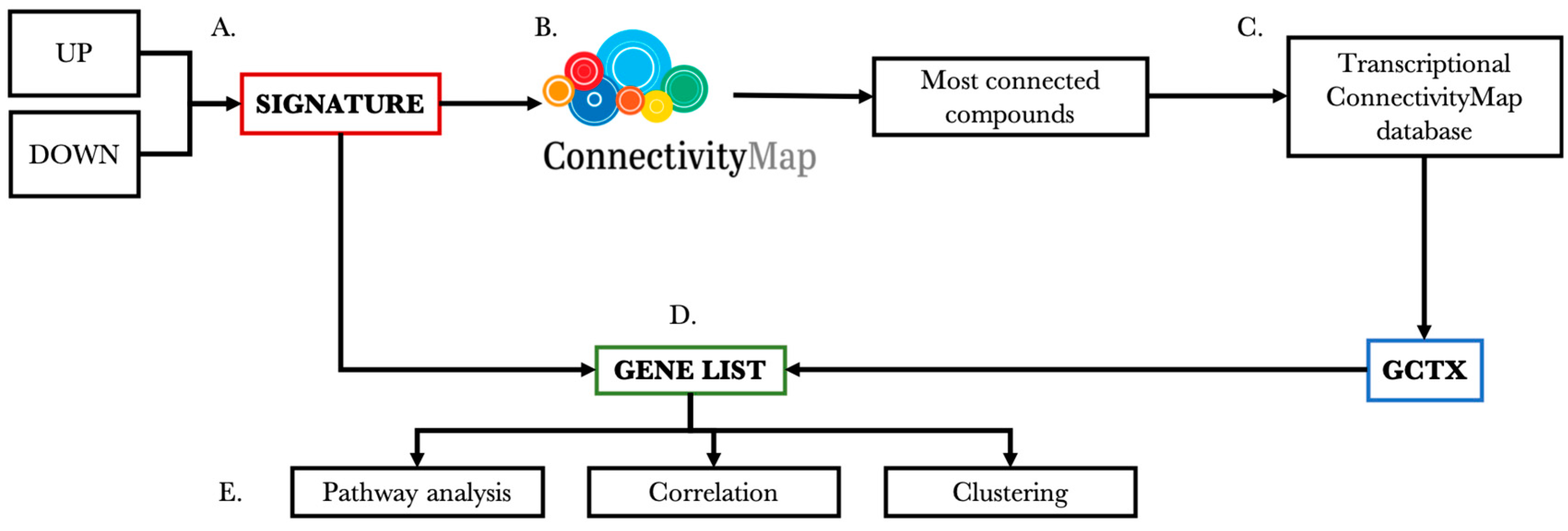
2.1. Workflow for Drug-Specific Gene Selection
2.2. Selection of Transcriptional Datasets
2.3. Extraction of Gene Signatures
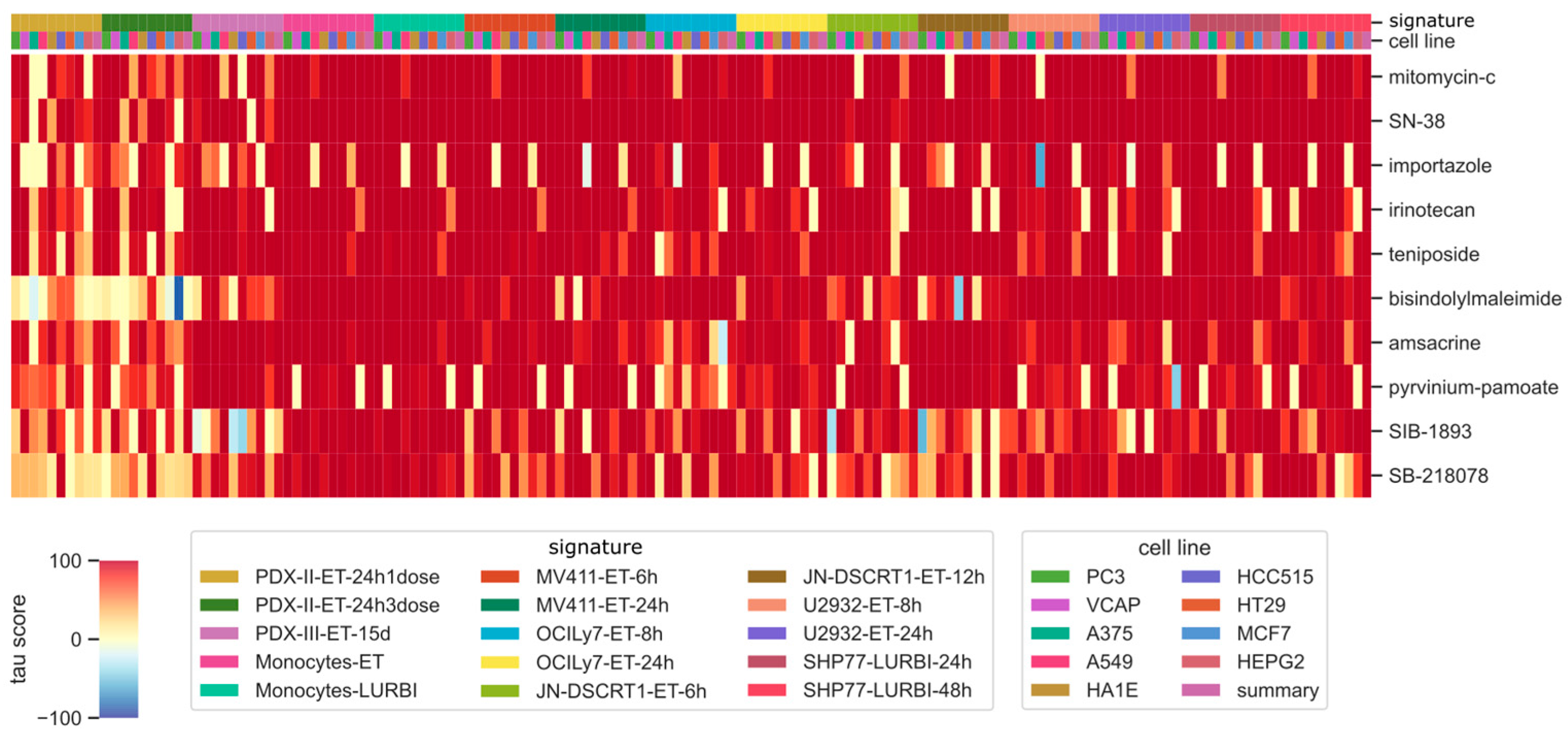
2.4. Identification of Compounds Correlated with the Drug of Interest
2.5. Identification of Genes with the Same Transcriptional Modulation
2.6. Common Pathways
3. Discussion
4. Materials and Methods
4.1. Dataset
- Availability of control samples under untreated conditions;
- Availability of samples treated with either trabectedin or lurbinectedin;
- At least three samples for each condition (either treated or untreated);
- Gene expression data generated by microarray technology.
4.2. Microarray Data Analysis
4.3. Connectivity Map
4.4. Definition of a Gene Signature
4.5. CMap Query
4.6. CMap Query Result Selection
4.7. Identification of Genes with the Same Up- or Down-Regulation Direction
- GSE92742_Broad_LINCS_sig_info.txt, to retrieve information on cell lines and compounds;
- GSE92742_Broad_LINCS_gene_info.txt, to retrieve information on genes;
- GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx, representing the level 5 data z-score vector [19].
- The sig_info file (GSE92742_Broad_LINCS_sig_info.txt) was used to subset the previous gctx file based on the following:
- The pertubagen (i.e., compound) id (pert_id), using the pert_id corresponding to the first 10 results selected as previously reported in section “CMap query result selection”, as reported in Table 3;
- The pertubagen name (pert_iname), using the pert_iname corresponding to the first 10 results selected as previously reported in section “CMap query result selection”, as reported in Table 3;
- The cell lines of human origin of interest (cell_id) associated with the CMap database, as reported in Table 4.
4.8. Correlation and Clustering
4.9. Functional Analysis
4.10. Workflow Implementation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug Repositioning: A Brief Overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of Drug Mode of Action and Drug Repositioning from Transcriptional Responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Galmarini, C.M. A Review of Trabectedin (ET-743): A Unique Mechanism of Action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a Drug Acting on Both Cancer Cells and the Tumour Microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma Treatment in the Era of Molecular Medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef] [PubMed]
- Baruchel, S.; Pappo, A.; Krailo, M.; Baker, K.S.; Wu, B.; Villaluna, D.; Lee-Scott, M.; Adamson, P.C.; Blaney, S.M. A Phase 2 Trial of Trabectedin in Children with Recurrent Rhabdomyosarcoma, Ewing Sarcoma and Non-Rhabdomyosarcoma Soft Tissue Sarcomas: A Report from the Children’s Oncology Group. Eur. J. Cancer 2012, 48, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma after Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Pignata, S.; Scambia, G.; Villanucci, A.; Naglieri, E.; Ibarbia, M.A.; Brusa, F.; Bourgeois, H.; Sorio, R.; Casado, A.; Reichert, D.; et al. A European, Observational, Prospective Trial of Trabectedin Plus Pegylated Liposomal Doxorubicin in Patients with Platinum-Sensitive Ovarian Cancer. Oncologist 2021, 26, e658–e668. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Gadducci, A.; Sehouli, J.; Rulli, E.; Mäenpää, J.; Sessa, C.; Montes, A.; Ottevanger, N.B.; Berger, R.; Vergote, I.; et al. INOVATYON/ENGOT-Ov5 Study: Randomized Phase III International Study Comparing Trabectedin/Pegylated Liposomal Doxorubicin (PLD) Followed by Platinum at Progression vs Carboplatin/PLD in Patients with Recurrent Ovarian Cancer Progressing within 6–12 Months after Last Platinum Line. Br. J. Cancer 2023, 128, 1503–1513. [Google Scholar] [CrossRef]
- Mario Negri Institute for Pharmacological Research. A Phase II Study on Trabectedin in Combination with PPARg Agonist Pioglitazone in Patients with Round Cell Myxoid Liposarcomas or Dedifferentiated G1 and G2 Liposarcomas with Stable Disease after a Monotherapy with Trabectedin. (TRABEPIO); 2021. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04794127 (accessed on 7 February 2024).
- Frapolli, R.; Bello, E.; Ponzo, M.; Craparotta, I.; Mannarino, L.; Ballabio, S.; Marchini, S.; Carrassa, L.; Ubezio, P.; Porcu, L.; et al. Combination of PPARγ Agonist Pioglitazone and Trabectedin Induce Adipocyte Differentiation to Overcome Trabectedin Resistance in Myxoid Liposarcomas. Clin. Cancer Res. 2019, 25, 7565–7575. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Lurbinectedin: First Approval. Drugs 2020, 80, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Santamaría Nuñez, G.; Robles, C.M.G.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.-M. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol. Cancer Ther. 2016, 15, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin Reduces Tumour-Associated Macrophages and the Inflammatory Tumour Microenvironment in Preclinical Models. Br. J. Cancer 2017, 117, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Mannarino, L.; Paracchini, L.; Craparotta, I.; Romano, M.; Marchini, S.; Gatta, R.; Erba, E.; Clivio, L.; Romualdi, C.; D’Incalci, M.; et al. A Systems Biology Approach to Investigate the Mechanism of Action of Trabectedin in a Model of Myelomonocytic Leukemia. Pharmacogenom. J. 2018, 18, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Shrestha, R.L.; Levin, N.; Boilot, V.; Garnett, M.J.; Saez-Rodriguez, J.; Draviam, V.M. A Semi-Supervised Approach for Refining Transcriptional Signatures of Drug Response and Repositioning Predictions. PLoS ONE 2015, 10, e0139446. [Google Scholar] [CrossRef] [PubMed]
- Lussier, Y.A.; Chen, J.L. The Emergence of Genome-Based Drug Repositioning. Sci. Transl. Med. 2011, 3, 96ps35. [Google Scholar] [CrossRef]
- Lamb, J. The Connectivity Map: A New Tool for Biomedical Research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update–from Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique Features of Trabectedin Mechanism of Action. Cancer Chemother. Pharmacol. 2016, 77, 663–671. [Google Scholar] [CrossRef]
- Acikgoz, E.; Guven, U.; Duzagac, F.; Uslu, R.; Kara, M.; Soner, B.C.; Oktem, G. Enhanced G2/M Arrest, Caspase Related Apoptosis and Reduced E-Cadherin Dependent Intercellular Adhesion by Trabectedin in Prostate Cancer Stem Cells. PLoS ONE 2015, 10, e0141090. [Google Scholar] [CrossRef]
- Pignochino, Y.; Capozzi, F.; D’Ambrosio, L.; Dell’Aglio, C.; Basiricò, M.; Canta, M.; Lorenzato, A.; Vignolo Lutati, F.; Aliberti, S.; Palesandro, E.; et al. PARP1 Expression Drives the Synergistic Antitumor Activity of Trabectedin and PARP1 Inhibitors in Sarcoma Preclinical Models. Mol. Cancer 2017, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cruzado, L.; Tornin, J.; Rodriguez, A.; Santos, L.; Allonca, E.; Fernandez-Garcia, M.T.; Astudillo, A.; Garcia-Pedrero, J.M.; Rodriguez, R. Trabectedin and Campthotecin Synergistically Eliminate Cancer Stem Cells in Cell-of-Origin Sarcoma Models. Neoplasia 2017, 19, 460–470. [Google Scholar] [CrossRef] [PubMed]
- van Raalte, D.H.; Li, M.; Pritchard, P.H.; Wasan, K.M. Peroxisome Proliferator-Activated Receptor (PPAR)-Alpha: A Pharmacological Target with a Promising Future. Pharm. Res. 2004, 21, 1531–1538. [Google Scholar] [CrossRef]
- Riccardi, A.; Meco, D.; Ubezio, P.; Mazzarella, G.; Marabese, M.; Faircloth, G.T.; Jimeno, J.; D’Incalci, M.; Riccardi, R. Combination of Trabectedin and Irinotecan Is Highly Effective in a Human Rhabdomyosarcoma Xenograft. Anticancer Drugs 2005, 16, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- CONNECTOPEDIA [Clue.Io]. Available online: https://clue.io/connectopedia/ (accessed on 26 September 2022).
- Enache, O.M.; Lahr, D.L.; Natoli, T.E.; Litichevskiy, L.; Wadden, D.; Flynn, C.; Gould, J.; Asiedu, J.K.; Narayan, R.; Subramanian, A. The GCTx Format and cmap{Py, R, M, J} Packages: Resources for Optimized Storage and Integrated Traversal of Annotated Dense Matrices. Bioinformatics 2019, 35, 1427–1429. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.; Granger, B.E. IPython: A System for Interactive Scientific Computing. Comput. Sci. Eng. 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Waskom, M.L. Seaborn: Statistical Data Visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- The Pandas Development Team. Pandas-Dev/Pandas. Available online: https://zenodo.org/records/10537285 (accessed on 7 February 2024).
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human. R Package Version 3.8.2. 2019. Available online: https://doi.org/10.18129/B9.bioc.org.Hs.eg.db (accessed on 7 February 2024).
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Begley, C.G.; Ashton, M.; Baell, J.; Bettess, M.; Brown, M.P.; Carter, B.; Charman, W.N.; Davis, C.; Fisher, S.; Frazer, I.; et al. Drug Repurposing: Misconceptions, Challenges, and Opportunities for Academic Researchers. Sci. Transl. Med. 2021, 13, eabd5524. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Dataset ID | Brief Description | Origin | Available at | Accession ID |
|---|---|---|---|---|
| MV411 | Gene expression profiling of a cell line model of myelomonocytic leukemia | Cell line | ArrayExpress | E-MTAB-2978 |
| JN-DSCRT1 | Desmoplastic small round cell sarcoma | Cell line | ArrayExpress | E-MTAB-4532 |
| PDX-II | Myxoid liposarcoma—type II | Patient-derived xenograft | ArrayExpress | E-MTAB-8632 |
| PDX-III | Myxoid liposarcoma type III | Patient-derived xenograft | ArrayExpress | E-MTAB-8632 |
| OCILy7 | Lymphoma DLBCL subtype germinal center B cell (GBC) | Cell line | Gene Expression Omnibus | GSE104197 |
| U2932 | Lymphoma DLBCL subtype activated B cell-like (ABC) | Cell line | Gene Expression Omnibus | GSE104197 |
| Monocytes | Immune system cells from donors | Blood cells | ArrayExpress | E-MTAB-5366 |
| SHP-77 | Small cell lung cancer | Cell line | in-house | - |
| Dataset ID | Annotation Package | Source |
|---|---|---|
| MV4-11 | HsAgilentDesign026652 | Agilent, Santa Clara, CA, USA |
| JNDSCRT1 | HsAgilentDesign039494 | Agilent, Santa Clara, CA, USA |
| PDX-II, PDX-III | HsAgilentDesign026652 | Agilent, Santa Clara, CA, USA |
| OCILy7, U2932 | IlluminaHumanv4 | Illumina, San Diego, CA, USA |
| Monocytes | HsAgilentDesign039494 | Agilent, Santa Clara, CA, USA |
| SHP-77 | HsAgilentDesign039494 | Agilent, Santa Clara, CA, USA |
| pert_id | part_iname |
|---|---|
| BRD-A02481876 | importazole |
| BRD-A06352508 | SB-218078 |
| BRD-A35588707 | teniposide |
| BRD-A36630025 | SN-38 |
| BRD-A48237631 | mitomycin-c |
| BRD-K08547377 | irinotecan |
| BRD-K31342827 | bisindolylmaleimide |
| BRD-K67439147 | SIB-1893 |
| BRD-K98490050 | amsacrine |
| BRD-M86331534 | pyrvinium-pamoate |
| cell_id | Brief Description |
|---|---|
| A375 | malignant melanoma |
| A549 | non-small cell lung carcinoma |
| HA1E | kidney epithelial immortalized (non-cancer cell line) |
| HCC515 | non-small cell lung adenocarcinoma |
| HEPG2 | hepatocellular carcinoma |
| HT29 | colorectal adenocarcinoma |
| MCF7 | breast adenocarcinoma |
| PC3 | prostate adenocarcinoma |
| VCAP | metastatic prostate cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mannarino, L.; Ravasio, N.; D’Incalci, M.; Marchini, S.; Masseroli, M. In-Silico Identification of Novel Pharmacological Synergisms: The Trabectedin Case. Int. J. Mol. Sci. 2024, 25, 2059. https://doi.org/10.3390/ijms25042059
Mannarino L, Ravasio N, D’Incalci M, Marchini S, Masseroli M. In-Silico Identification of Novel Pharmacological Synergisms: The Trabectedin Case. International Journal of Molecular Sciences. 2024; 25(4):2059. https://doi.org/10.3390/ijms25042059
Chicago/Turabian StyleMannarino, Laura, Nicholas Ravasio, Maurizio D’Incalci, Sergio Marchini, and Marco Masseroli. 2024. "In-Silico Identification of Novel Pharmacological Synergisms: The Trabectedin Case" International Journal of Molecular Sciences 25, no. 4: 2059. https://doi.org/10.3390/ijms25042059
APA StyleMannarino, L., Ravasio, N., D’Incalci, M., Marchini, S., & Masseroli, M. (2024). In-Silico Identification of Novel Pharmacological Synergisms: The Trabectedin Case. International Journal of Molecular Sciences, 25(4), 2059. https://doi.org/10.3390/ijms25042059