
HSP70 Is a Critical Regulator of HSP90 Inhibitor’s Effectiveness in Preventing HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
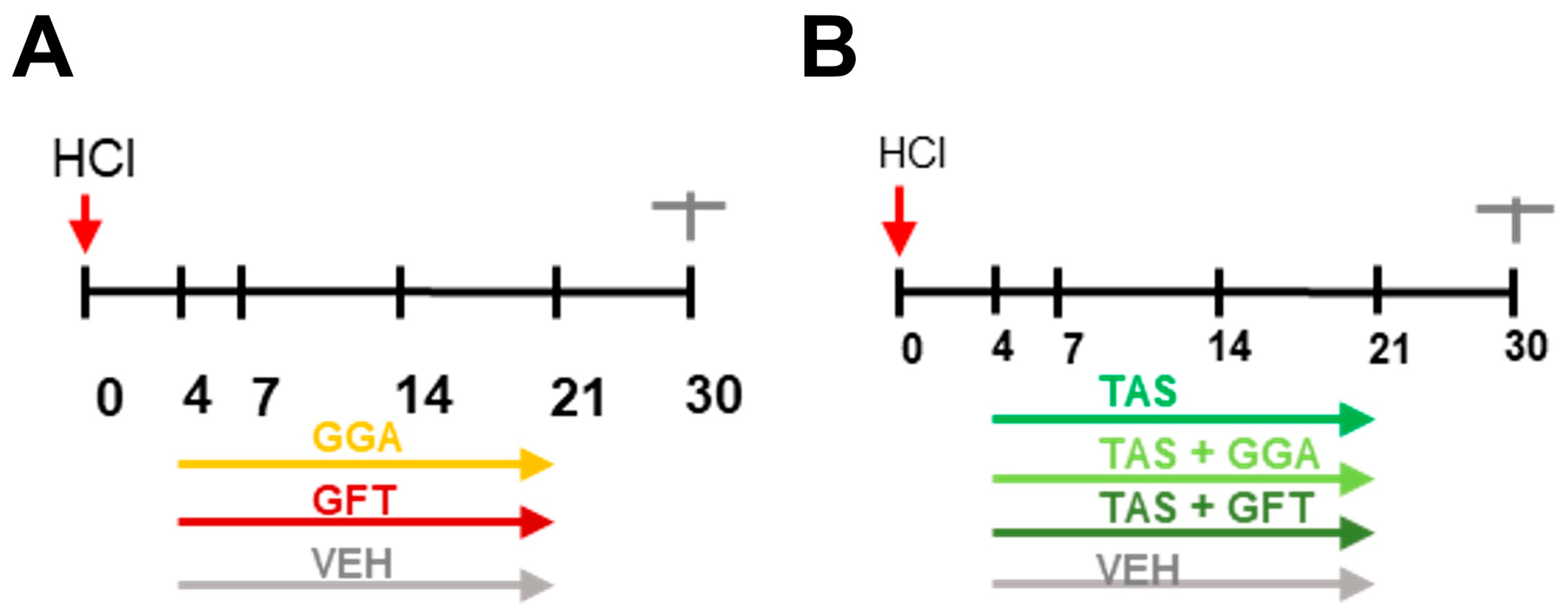
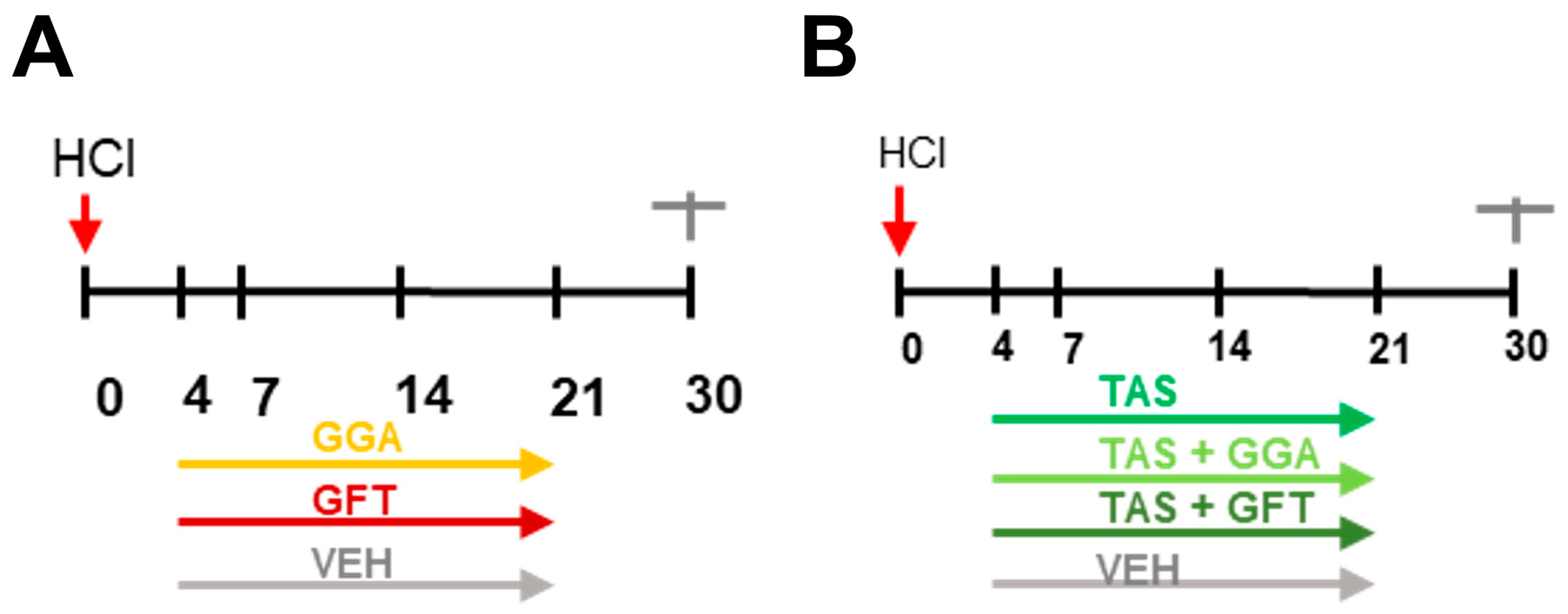
2.1. Timeline of Animal Studies
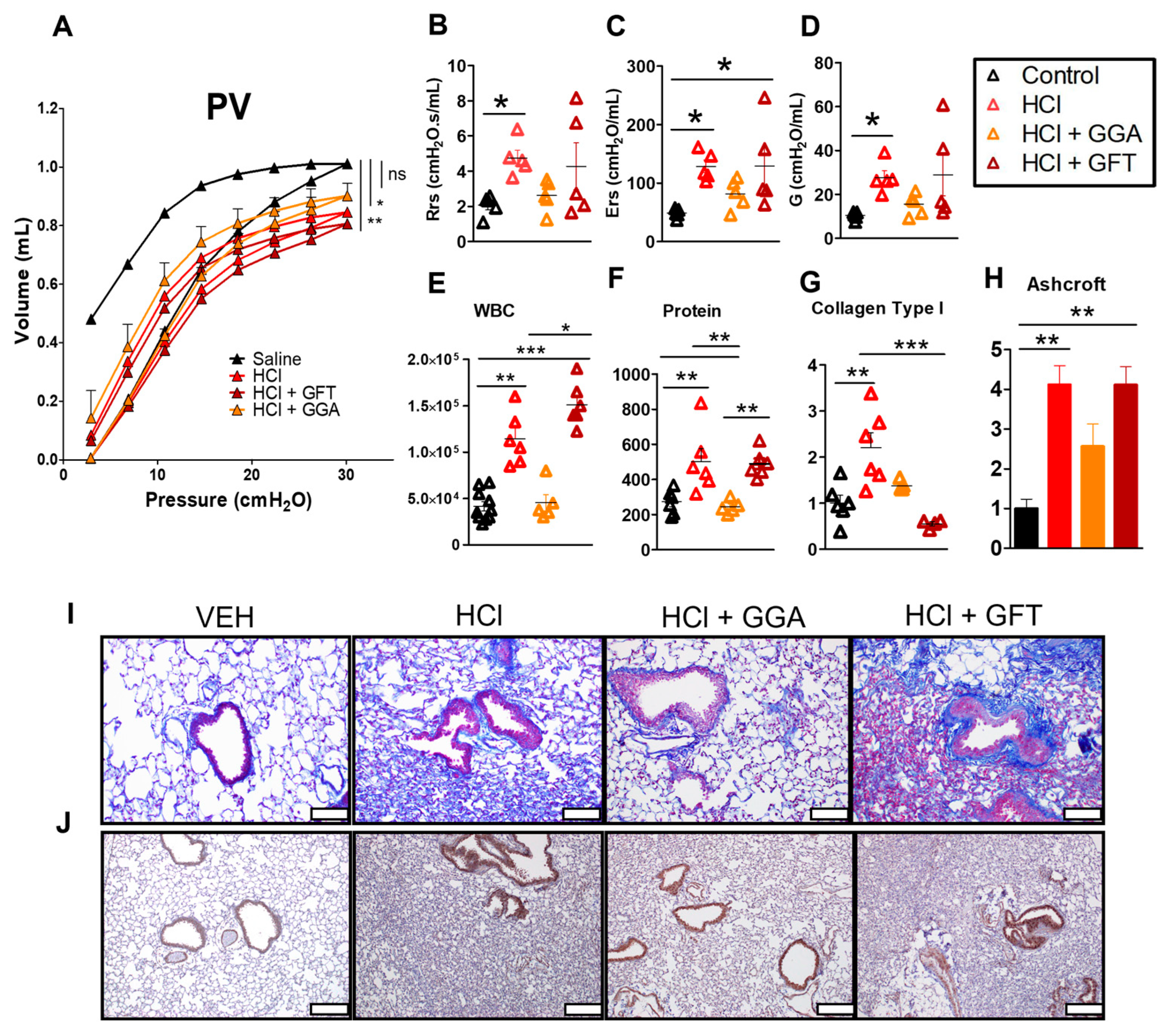
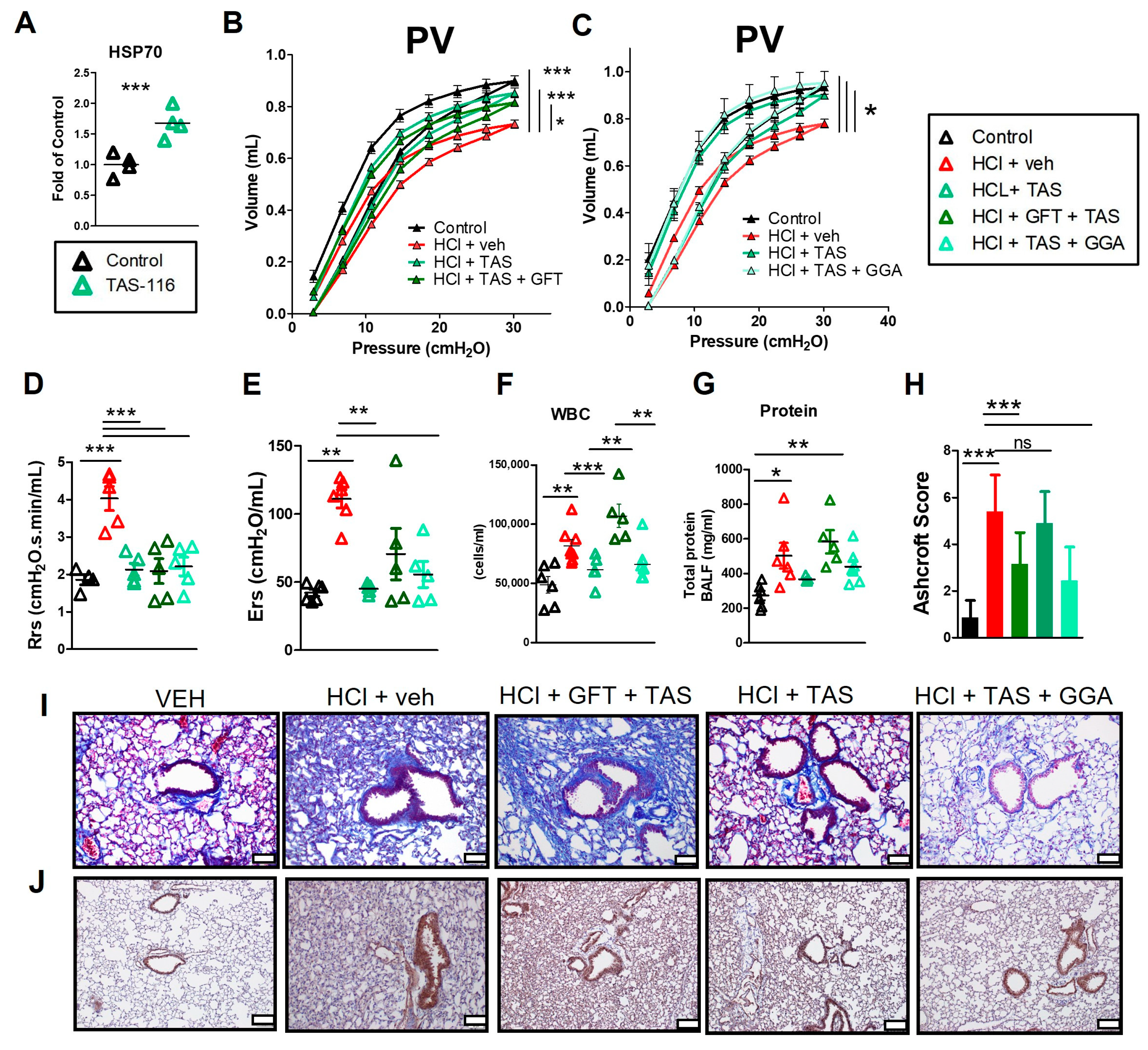
2.2. HSP70 Upregulation Improves HCl-Induced Lung Dysfunction
2.3. HSP70 Upregulation Ameliorates HCl-Induced Persistent Inflammation and Lung Fibrosis
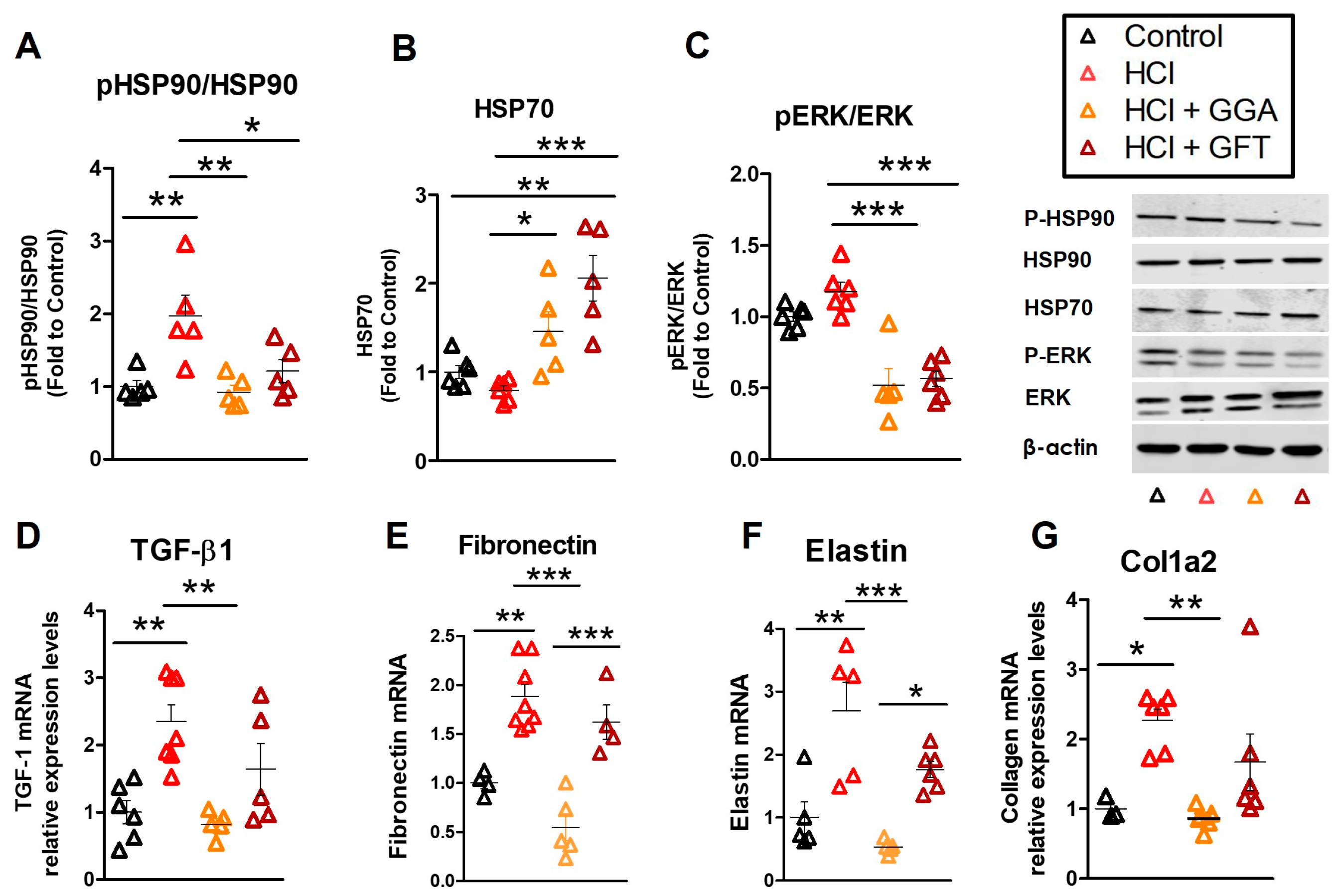
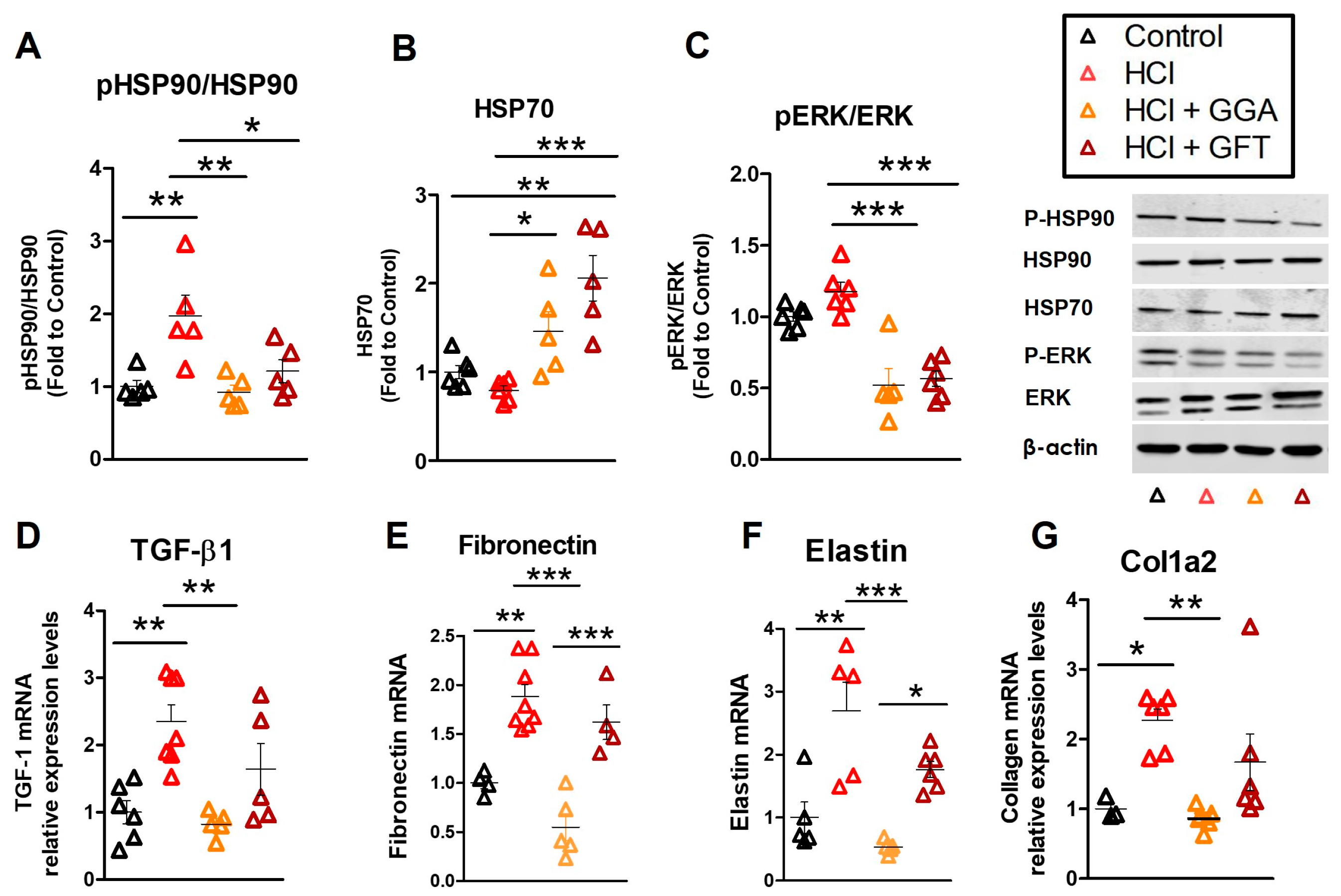
2.4. HCl Exposure Activates Heat Shock Protein 90
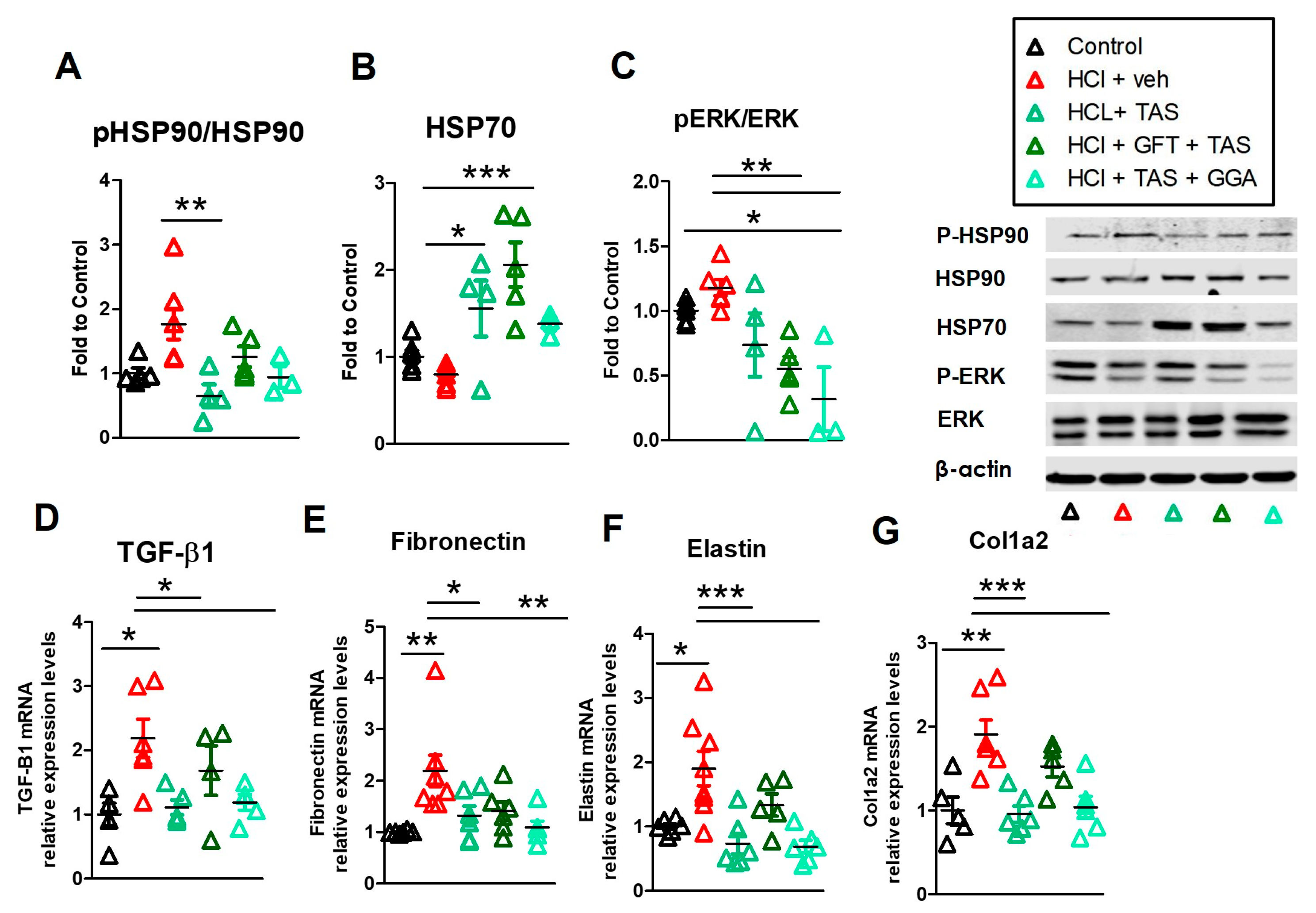
2.5. HSP70 Upregulation Modulates the HCl-Induced Profibrotic Response
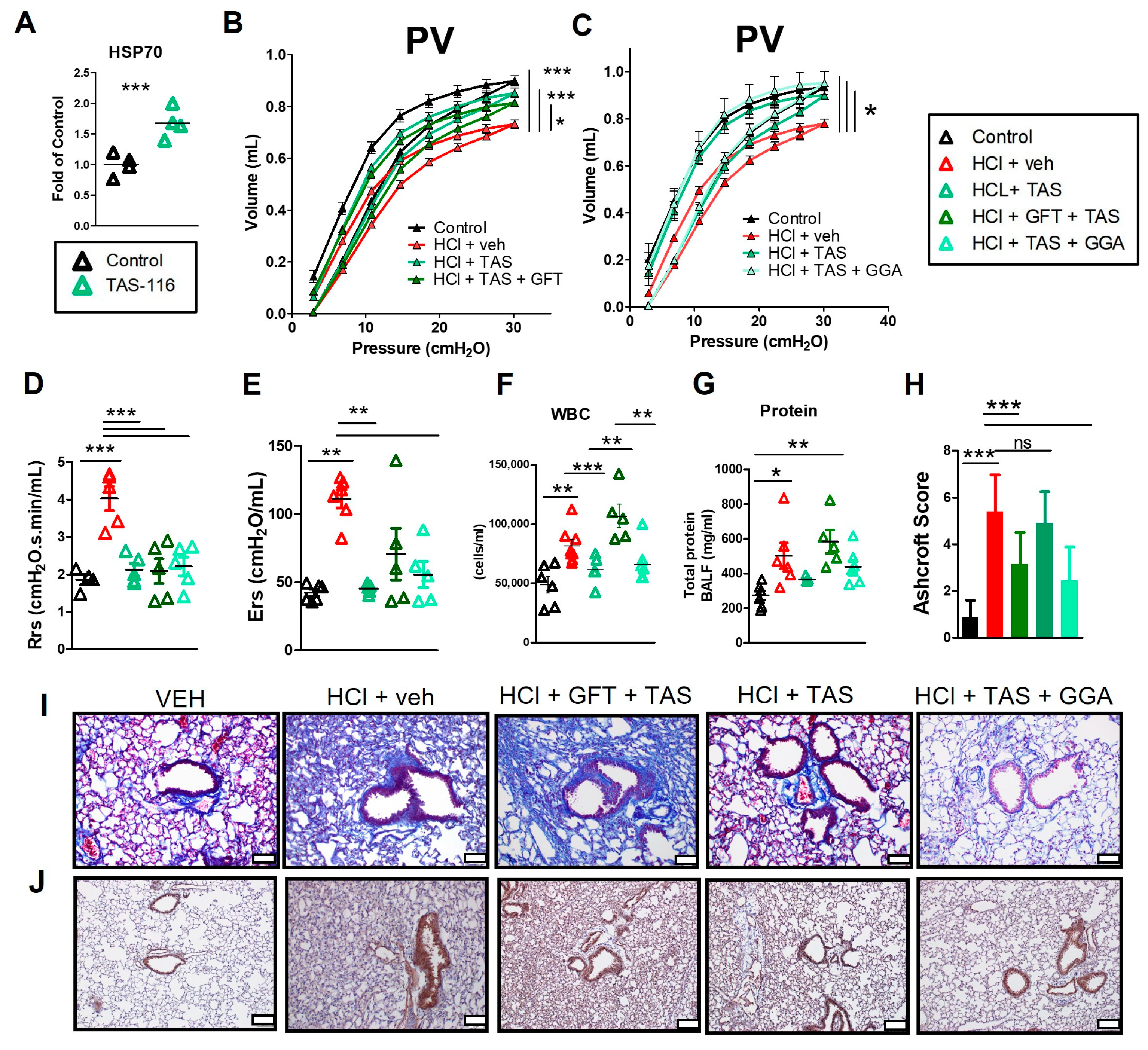
2.6. HSP70 Upregulation Improved the TAS-116 Therapeutic Profile for HCl-Induced Lung Dysfunction
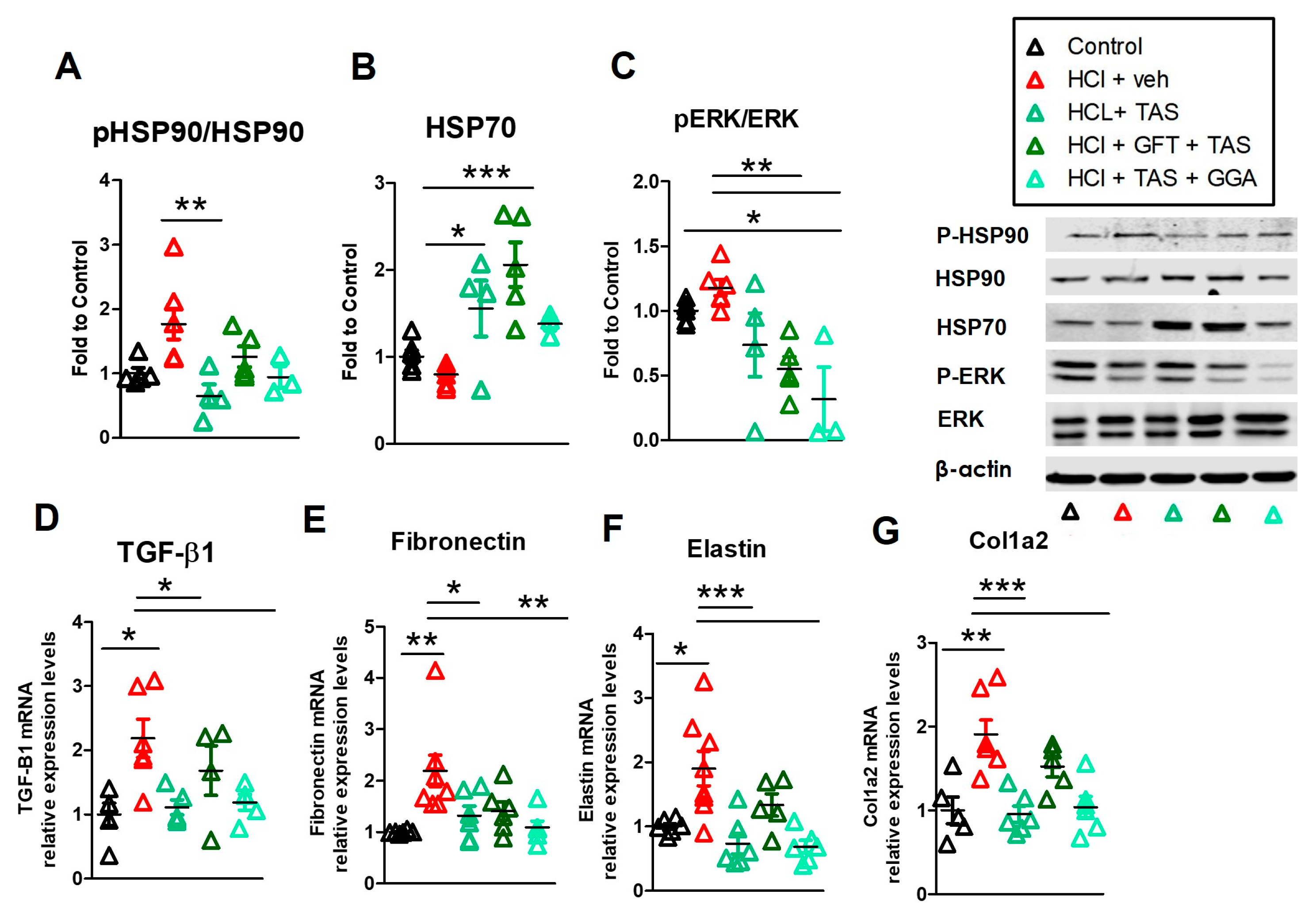
2.7. GFT Impaired HSP90 Inhibitor Amelioration of HCl-Induced Persistent Inflammation and Lung Fibrosis
2.8. TAS-116 Reduced HCl-Induced Activation of Heat Shock Protein 90
2.9. GGA Ameliorates the HCl-Induced Profibrotic Response
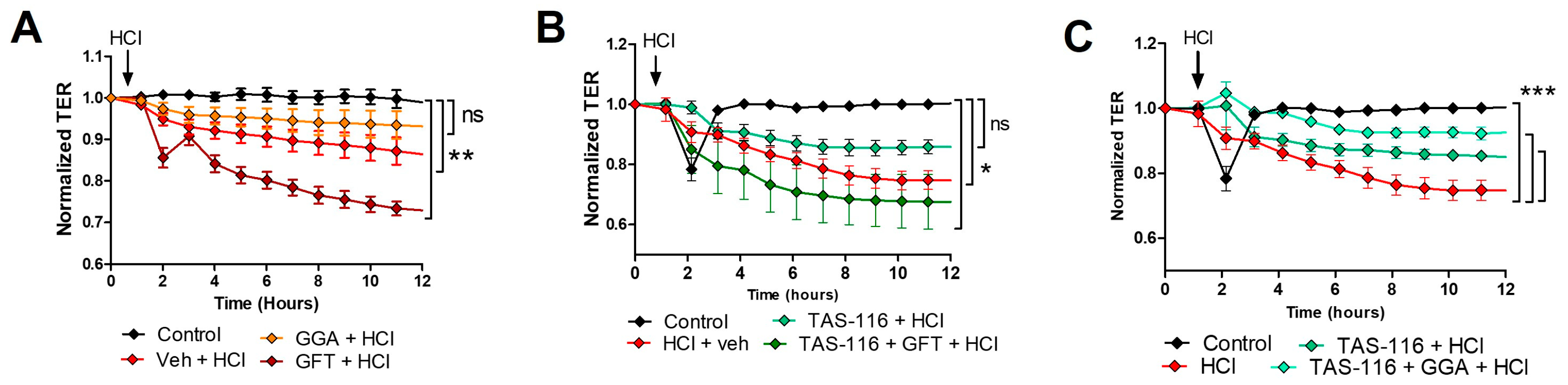
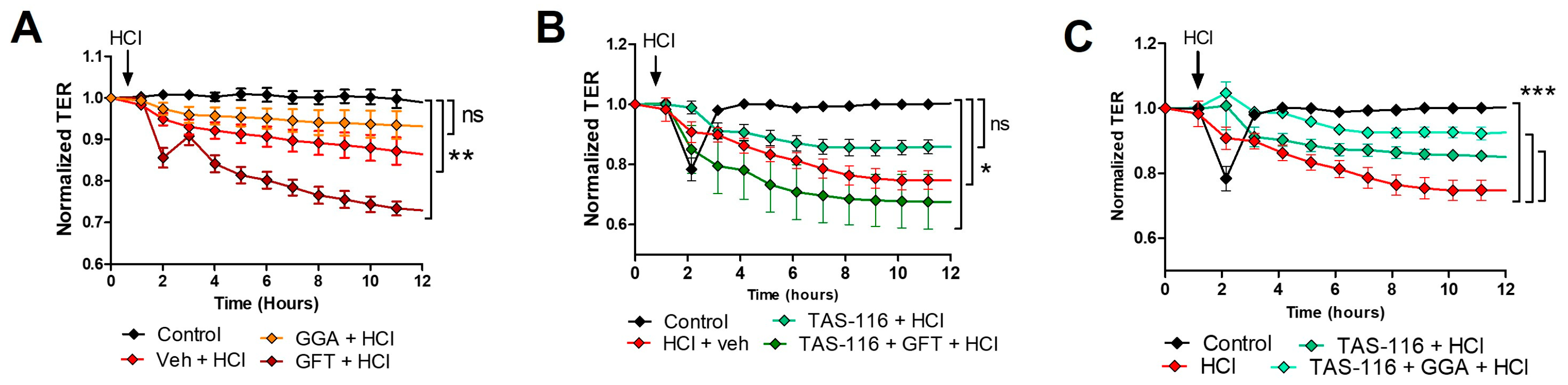
2.10. HSP70 Upregulation Improves TAS-116 Therapeutic Effects on HCl-Induced Hyperpermeability
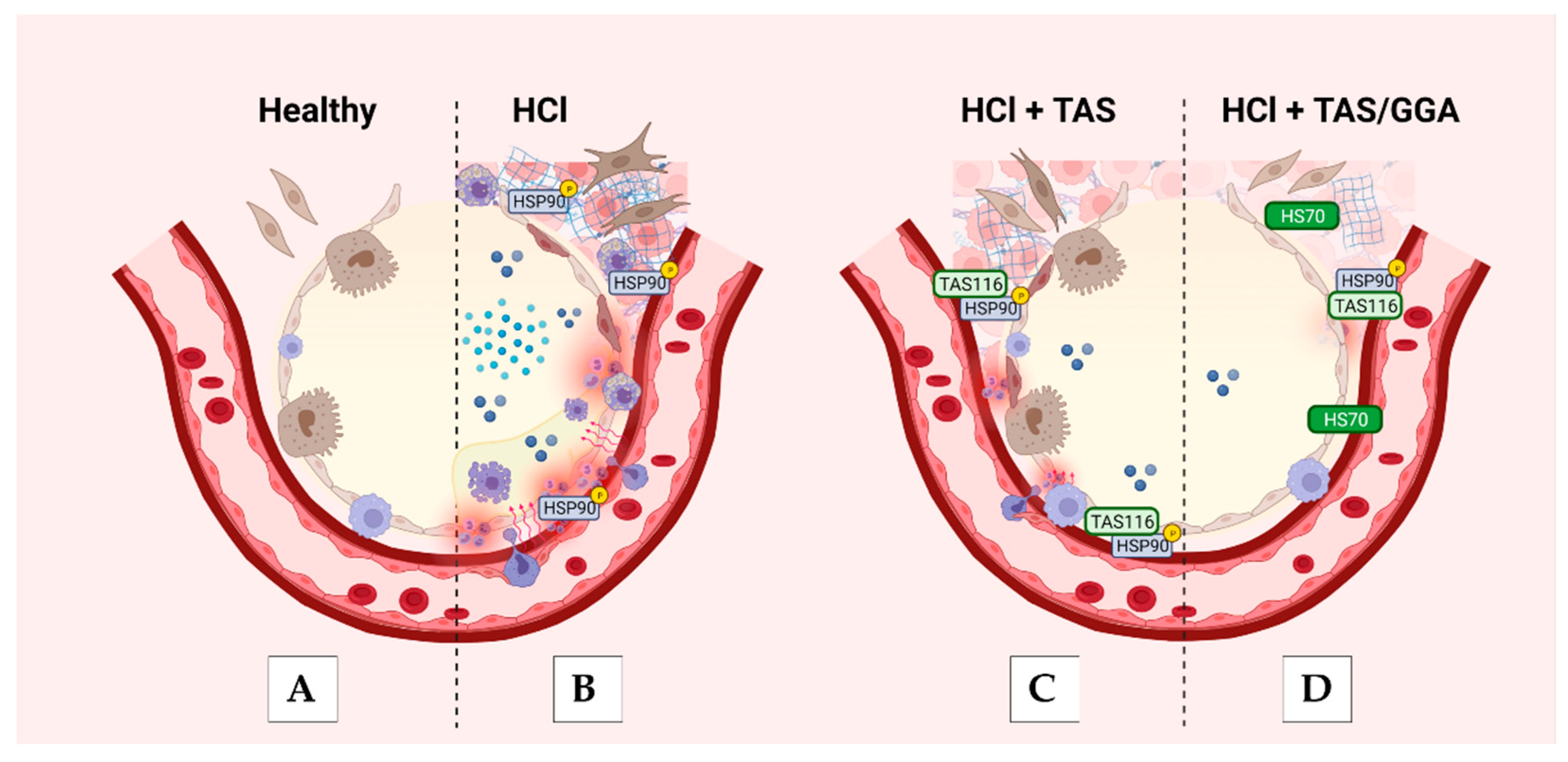
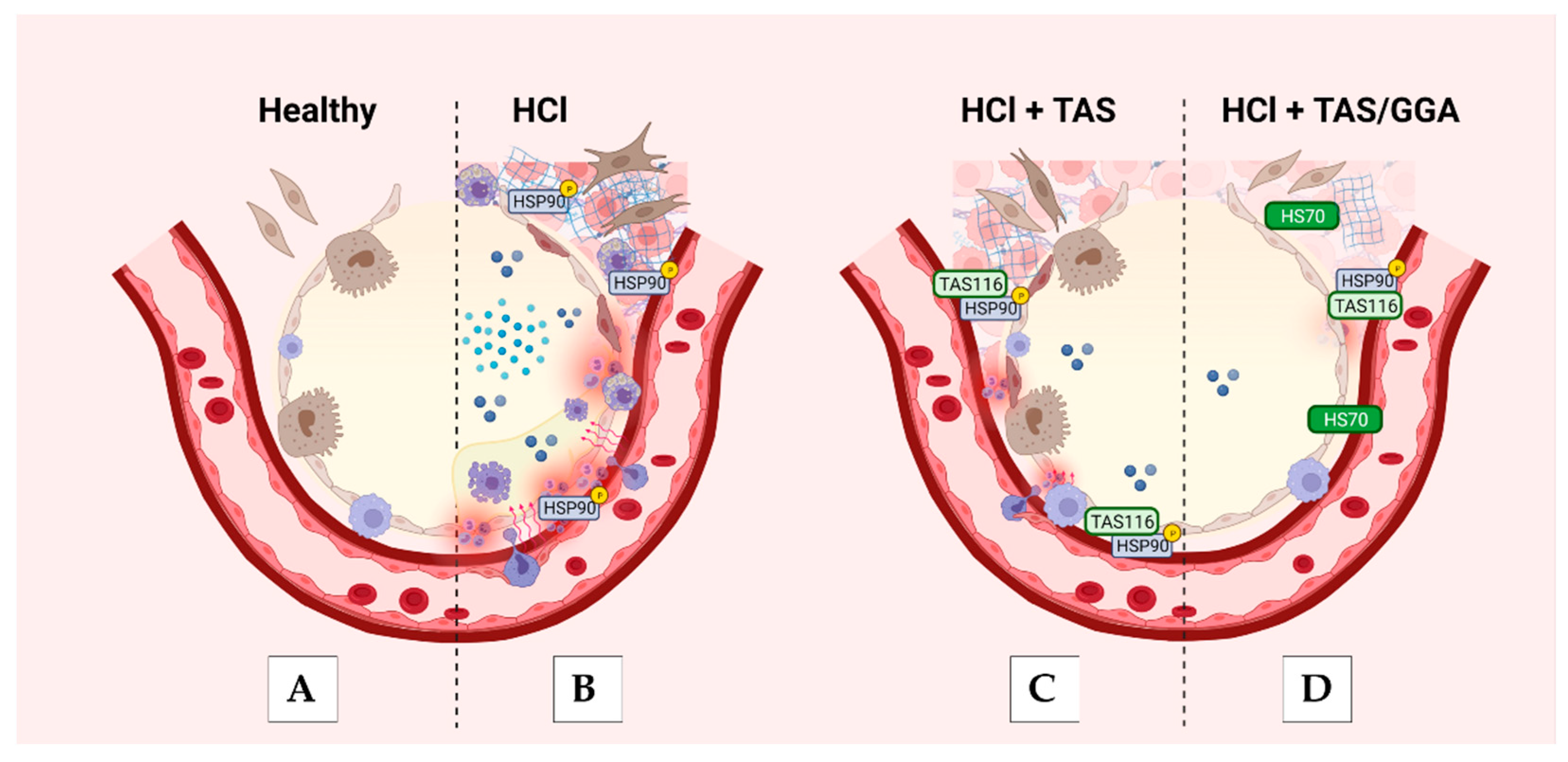
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Procedures
4.3. Bronchoalveolar Lavage Fluid (BALF) Analysis
4.4. Histopathology
4.5. Tissue Collection
4.6. Western Blot Analysis
4.7. Real-Time PCR (qPCR)
4.8. Lung Function Measurements
4.9. Endothelial Barrier Function
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, A.R. Top five chemicals resulting in injuries from acute chemical incidents—Hazardous Substances Emergency Events Surveillance, nine states, 1999–2008. MMWR Suppl. 2015, 64, 39–46. [Google Scholar] [PubMed]
- Zellner, T.; Eyer, F. Choking agents and chlorine gas—History, pathophysiology, clinical effects and treatment. Toxicol. Lett. 2020, 320, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Faria, V.S.; da Silva, S.; Marchini, J.F.M. Reactive airways dysfunction syndrome following inhalation of hydrogen chloride vapor. Autops. Case Rep. 2021, 11, e2021266. [Google Scholar] [CrossRef] [PubMed]
- Gorguner, M.; Aslan, S.; Inandi, T.; Cakir, Z. Reactive airways dysfunction syndrome in housewives due to a bleach-hydrochloric acid mixture. Inhal. Toxicol. 2004, 16, 87–91. [Google Scholar] [CrossRef]
- Deschamps, D.; Soler, P.; Rosenberg, N.; Baud, F.; Gervais, P. Persistent asthma after inhalation of a mixture of sodium hypochlorite and hydrochloric acid. Chest 1994, 105, 1895–1896. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.B.; Sherman, M. Chronic reactive airway disease following acute chlorine gas exposure in an asymptomatic atopic patient. Chest 1991, 100, 855–856. [Google Scholar] [CrossRef] [PubMed]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Acute exposure of mice to hydrochloric acid leads to the development of chronic lung injury and pulmonary fibrosis. Inhal. Toxicol. 2019, 31, 147–160. [Google Scholar] [CrossRef]
- Solopov, P.; Colunga Biancatelli, R.M.L.; Dimitropoulou, C.; Catravas, J.D. Dietary Phytoestrogens Ameliorate Hydrochloric Acid-Induced Chronic Lung Injury and Pulmonary Fibrosis in Mice. Nutrients 2021, 13, 3599. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress. Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Marinova, M.; Solopov, P.; Dimitropoulou, C.; Colunga Biancatelli, R.M.L.; Catravas, J.D. Post-treatment with a heat shock protein 90 inhibitor prevents chronic lung injury and pulmonary fibrosis, following acute exposure of mice to HCl. Exp. Lung Res. 2020, 46, 203–216. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Dimitropoulou, C.; Gregory, B.; Day, T.; Catravas, J.D. The Heat Shock Protein 90 Inhibitor, AT13387, Protects the Alveolo-Capillary Barrier and Prevents HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis. Cells 2022, 11, 1046. [Google Scholar] [CrossRef] [PubMed]
- Solopov, P.; Biancatelli, R.; Marinova, M.; Dimitropoulou, C.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Ameliorates the Development of Nitrogen Mustard-Induced Pulmonary Fibrosis and Lung Dysfunction in Mice. Int. J. Mol. Sci. 2020, 21, 4740. [Google Scholar] [CrossRef] [PubMed]
- Solopov, P.A.; Colunga Biancatelli, R.M.L.; Dimitropolou, C.; Day, T.; Catravas, J.D. Optimizing antidotal treatment with the oral HSP90 inhibitor TAS-116 against hydrochloric acid-induced pulmonary fibrosis in mice. Front. Pharmacol. 2022, 13, 1034464. [Google Scholar] [CrossRef] [PubMed]
- Serwetnyk, M.A.; Blagg, B.S.J. The disruption of protein-protein interactions with co-chaperones and client substrates as a strategy towards Hsp90 inhibition. Acta Pharm. Sin. B 2021, 11, 1446–1468. [Google Scholar] [CrossRef]
- Shan, Q.; Ma, F.; Wei, J.; Li, H.; Ma, H.; Sun, P. Physiological Functions of Heat Shock Proteins. Curr. Protein Pept. Sci. 2020, 21, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Gregory, B.; Catravas, J.D. HSP90 Inhibition and Modulation of the Proteome: Therapeutical Implications for Idiopathic Pulmonary Fibrosis (IPF). Int. J. Mol. Sci. 2020, 21, 5286. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Paine-Murrieta, G.D.; Taylor, C.W.; Pulcini, E.J.; Akinaga, S.; Benjamin, I.J.; Whitesell, L. Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin. Cancer Res. 2000, 6, 3312–3318. [Google Scholar] [PubMed]
- Kudryavtsev, V.A.; Khokhlova, A.V.; Mosina, V.A.; Selivanova, E.I.; Kabakov, A.E. Induction of Hsp70 in tumor cells treated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 2017, 12, e0173640. [Google Scholar] [CrossRef]
- Genest, O.; Wickner, S.; Doyle, S.M. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. J. Biol. Chem. 2019, 294, 2109–2120. [Google Scholar] [CrossRef]
- Jacquier-Sarlin, M.R.; Fuller, K.; Dinh-Xuan, A.T.; Richard, M.J.; Polla, B.S. Protective effects of hsp70 in inflammation. Experientia 1994, 50, 1031–1038. [Google Scholar] [CrossRef]
- Evgen’ev, M.B.; Onikienko, S.B.; Chuvakova, L.N.; Garbuz, D.G.; Zatsepina, O.G. The Role of Hsp70 in Adaptation to Adverse Conditions and Its Possible Medical Application. FBL 2023, 28, 25. [Google Scholar] [CrossRef] [PubMed]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Jäättelä, M.; Wissing, D.; Kokholm, K.; Kallunki, T.; Egeblad, M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998, 17, 6124–6134. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Thiele, D.J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Tanaka, K.; Hoshino, T.; Azuma, A.; Mizushima, T. Suppression of expression of heat shock protein 70 by gefitinib and its contribution to pulmonary fibrosis. PLoS ONE 2011, 6, e27296. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-β1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019, 10, 670. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Solopov, P.; Gregory, B.; Catravas, J.D. The HSP90 Inhibitor, AUY-922, Protects and Repairs Human Lung Microvascular Endothelial Cells from Hydrochloric Acid-Induced Endothelial Barrier Dysfunction. Cells 2021, 10, 1489. [Google Scholar] [CrossRef]
- Shah, P.V.; Balani, P.; Lopez, A.R.; Nobleza, C.M.N.; Siddiqui, M.; Khan, S. A Review of Pirfenidone as an Anti-Fibrotic in Idiopathic Pulmonary Fibrosis and Its Probable Role in Other Diseases. Cureus 2021, 13, e12482. [Google Scholar] [CrossRef]
- Lamb, Y.N. Nintedanib: A Review in Fibrotic Interstitial Lung Diseases. Drugs 2021, 81, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Y. TGF-β1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef] [PubMed]
- Halwani, R.; Al-Muhsen, S.; Al-Jahdali, H.; Hamid, Q. Role of Transforming Growth Factor–β in Airway Remodeling in Asthma. Am. J. Respir. Cell Mol. Biol. 2011, 44, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-β: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef]
- Pei, Q.; Ni, W.; Yuan, Y.; Yuan, J.; Zhang, X.; Yao, M. HSP70 Ameliorates Septic Lung Injury via Inhibition of Apoptosis by Interacting with KANK2. Biomolecules 2022, 12, 410. [Google Scholar] [CrossRef] [PubMed]
- Aschkenasy, G.; Bromberg, Z.; Raj, N.; Deutschman, C.S.; Weiss, Y.G. Enhanced Hsp70 expression protects against acute lung injury by modulating apoptotic pathways. PLoS ONE 2011, 6, e26956. [Google Scholar] [CrossRef]
- Bellaye, P.S.; Shimbori, C.; Yanagihara, T.; Carlson, D.A.; Hughes, P.; Upagupta, C.; Sato, S.; Wheildon, N.; Haystead, T.; Ask, K.; et al. Synergistic role of HSP90α and HSP90β to promote myofibroblast persistence in lung fibrosis. Eur. Respir. J. 2018, 51, 1700386. [Google Scholar] [CrossRef]
- Štorkánová, H.; Oreská, S.; Špiritović, M.; Heřmánková, B.; Bubová, K.; Komarc, M.; Pavelka, K.; Vencovský, J.; Distler, J.H.W.; Šenolt, L.; et al. Plasma Hsp90 levels in patients with systemic sclerosis and relation to lung and skin involvement: A cross-sectional and longitudinal study. Sci. Rep. 2021, 11, 1. [Google Scholar] [CrossRef]
- Chakraborty, A.; Edkins, A.L. HSP90 as a regulator of extracellular matrix dynamics. Biochem. Soc. Trans. 2021, 49, 2611–2625. [Google Scholar] [CrossRef]
- Chakraborty, A.; Boel, N.M.; Edkins, A.L. HSP90 Interacts with the Fibronectin N-terminal Domains and Increases Matrix Formation. Cells 2020, 9, 272. [Google Scholar] [CrossRef] [PubMed]
- García, R.; Merino, D.; Gómez, J.M.; Nistal, J.F.; Hurlé, M.A.; Cortajarena, A.L.; Villar, A.V. Extracellular heat shock protein 90 binding to TGFβ receptor I participates in TGFβ-mediated collagen production in myocardial fibroblasts. Cell. Signal. 2016, 28, 1563–1579. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K. Cdc37 as a co-chaperone to Hsp90. Subcell. Biochem. 2015, 78, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; An, Y.S.; Kim, M.R.; Kim, Y.A.; Lee, J.K.; Hwang, C.S.; Chung, E.; Park, I.C.; Yi, J.Y. Heat Shock Protein 90 Regulates Subcellular Localization of Smads in Mv1Lu Cells. J. Cell Biochem. 2016, 117, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Roque, W. Heat shock proteins in pulmonary fibrosis: Pawns of cell homeostasis. Am. J. Physiol.-Cell Physiol. 2022, 322, C1105–C1109. [Google Scholar] [CrossRef] [PubMed]
- Barabutis, N. Heat shock protein 90 inhibition in the inflamed lungs. Cell Stress. Chaperones 2020, 25, 195–197. [Google Scholar] [CrossRef]
- Dubey, A.; Prajapati, K.S.; Swamy, M.; Pachauri, V. Heat shock proteins: A therapeutic target worth to consider. Vet. World 2015, 8, 46–51. [Google Scholar] [CrossRef]
- Schilling, D.; Garrido, C.; Combs, S.E.; Multhoff, G. The Hsp70 inhibiting peptide aptamer A17 potentiates radiosensitization of tumor cells by Hsp90 inhibition. Cancer Lett. 2017, 390, 146–152. [Google Scholar] [CrossRef]
- Kühnel, A.; Schilling, D.; Combs, S.E.; Haller, B.; Schwab, M.; Multhoff, G. Radiosensitization of HSF-1 Knockdown Lung Cancer Cells by Low Concentrations of Hsp90 Inhibitor NVP-AUY922. Cells 2019, 8, 1166. [Google Scholar] [CrossRef]
- Martin, T.R.; Hagimoto, N.; Nakamura, M.; Matute-Bello, G. Apoptosis and epithelial injury in the lungs. Proc. Am. Thorac. Soc. 2005, 2, 214–220. [Google Scholar] [CrossRef]
- Drakopanagiotakis, F.; Xifteri, A.; Polychronopoulos, V.; Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 2008, 32, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colunga Biancatelli, R.M.L.; Solopov, P.A.; Day, T.; Gregory, B.; Osei-nkansah, M.; Dimitropoulou, C.; Catravas, J.D. HSP70 Is a Critical Regulator of HSP90 Inhibitor’s Effectiveness in Preventing HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis. Int. J. Mol. Sci. 2024, 25, 1920. https://doi.org/10.3390/ijms25031920
Colunga Biancatelli RML, Solopov PA, Day T, Gregory B, Osei-nkansah M, Dimitropoulou C, Catravas JD. HSP70 Is a Critical Regulator of HSP90 Inhibitor’s Effectiveness in Preventing HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis. International Journal of Molecular Sciences. 2024; 25(3):1920. https://doi.org/10.3390/ijms25031920
Chicago/Turabian StyleColunga Biancatelli, Ruben M. L., Pavel A. Solopov, Tierney Day, Betsy Gregory, Michael Osei-nkansah, Christiana Dimitropoulou, and John D. Catravas. 2024. "HSP70 Is a Critical Regulator of HSP90 Inhibitor’s Effectiveness in Preventing HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis" International Journal of Molecular Sciences 25, no. 3: 1920. https://doi.org/10.3390/ijms25031920
APA StyleColunga Biancatelli, R. M. L., Solopov, P. A., Day, T., Gregory, B., Osei-nkansah, M., Dimitropoulou, C., & Catravas, J. D. (2024). HSP70 Is a Critical Regulator of HSP90 Inhibitor’s Effectiveness in Preventing HCl-Induced Chronic Lung Injury and Pulmonary Fibrosis. International Journal of Molecular Sciences, 25(3), 1920. https://doi.org/10.3390/ijms25031920