
Saturation Transfer Difference NMR and Molecular Docking Interaction Study of Aralkyl-Thiodigalactosides as Potential Inhibitors of the Human-Galectin-3 Protein

 , , ,
, , ,
Abstract
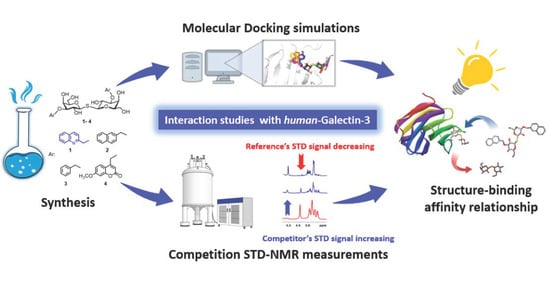
1. Introduction
2. Results and Discussion
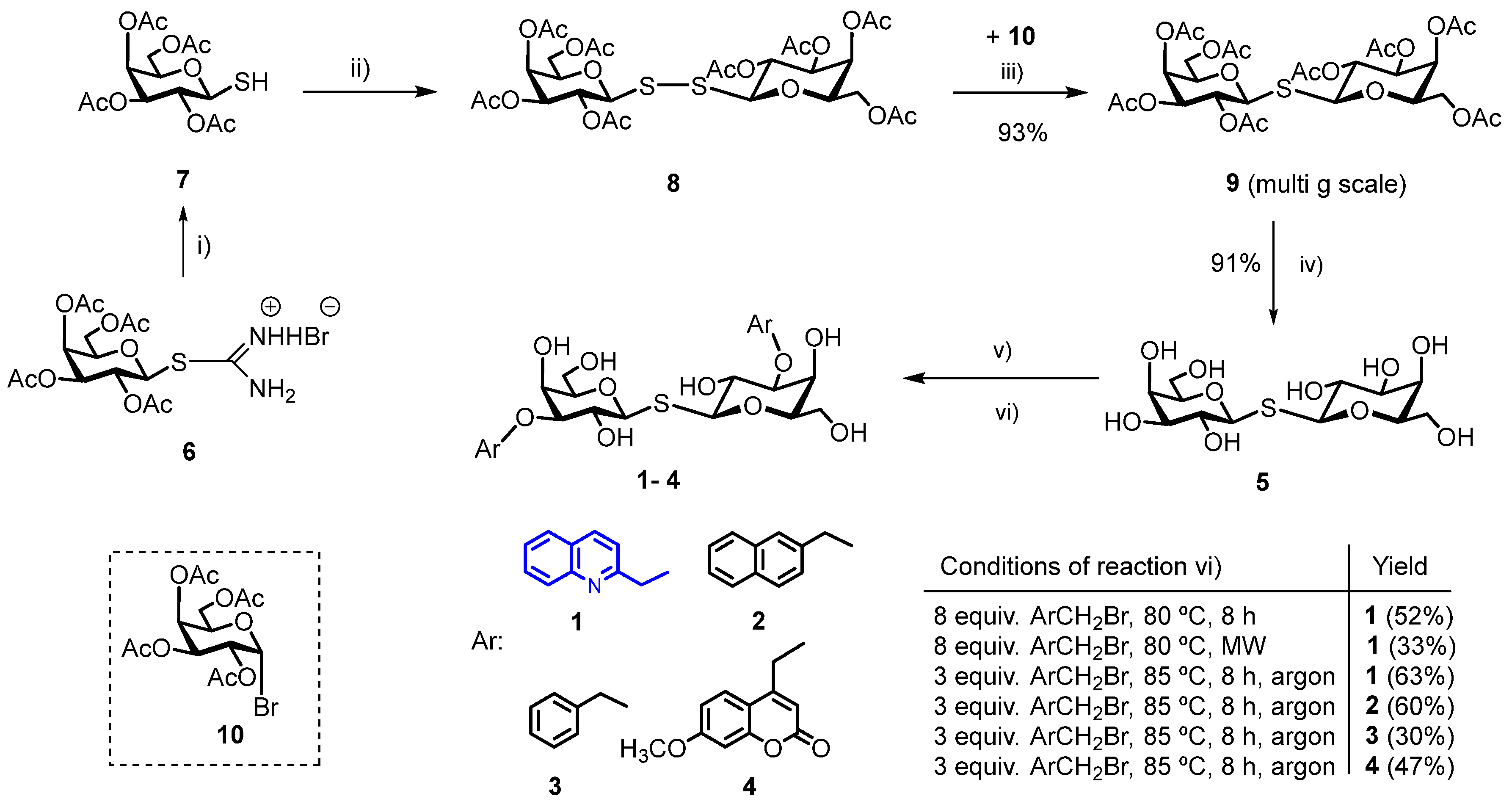
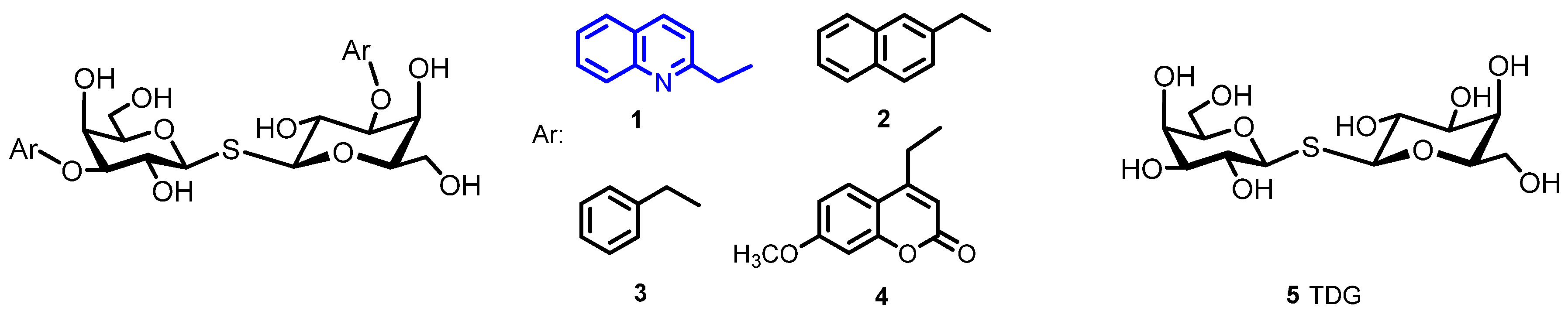
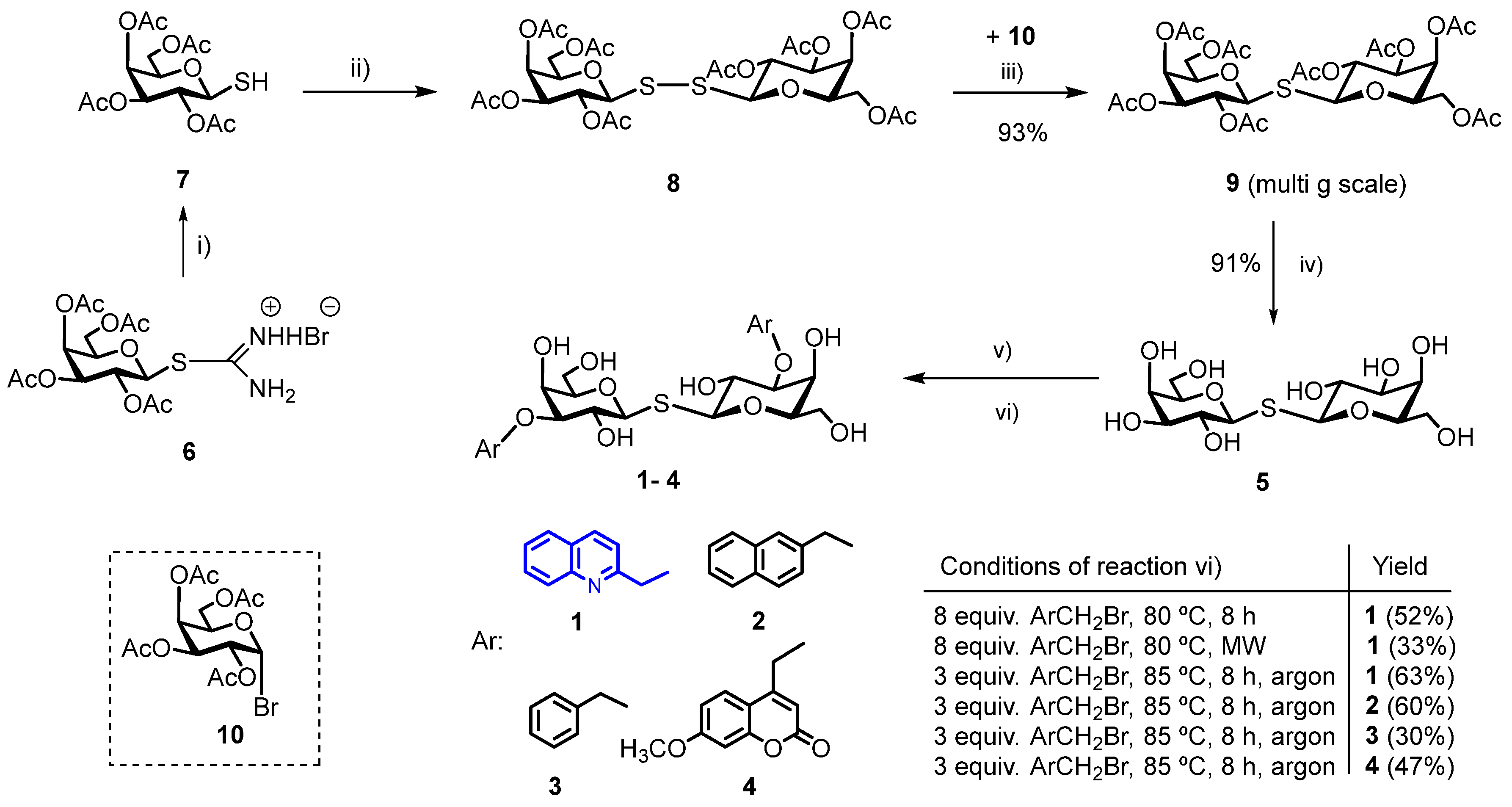
2.1. Chemical Synthesis
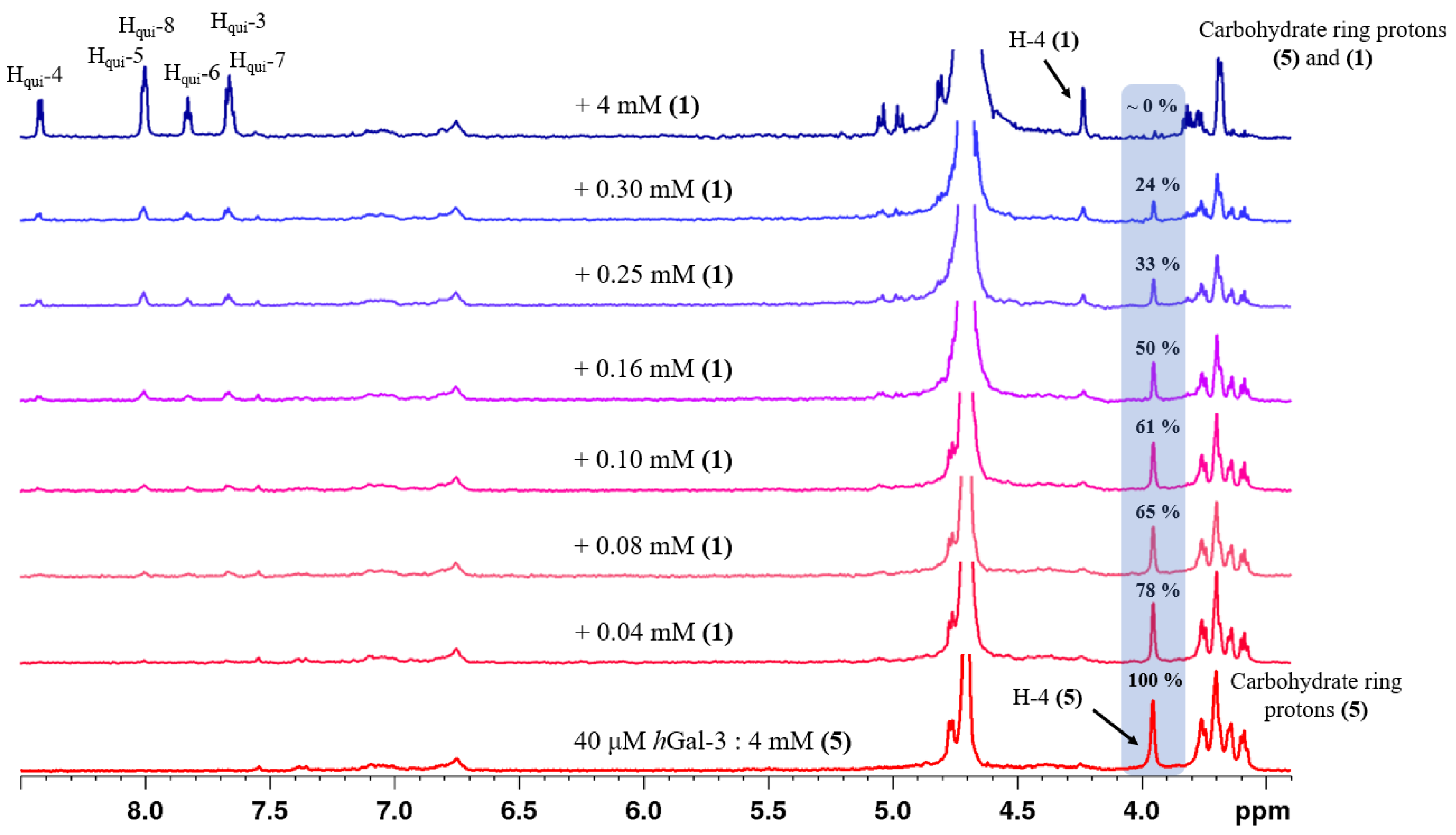
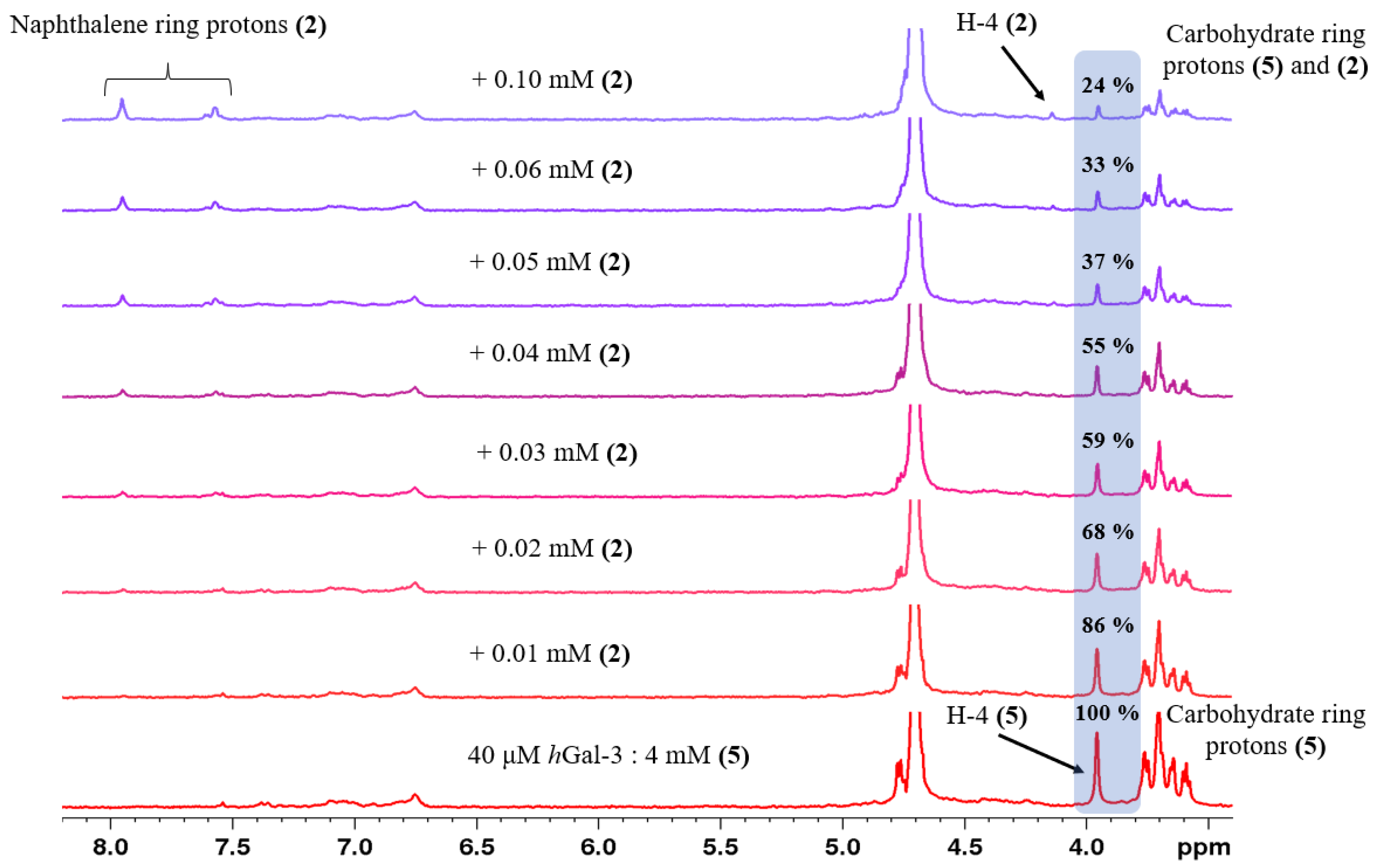
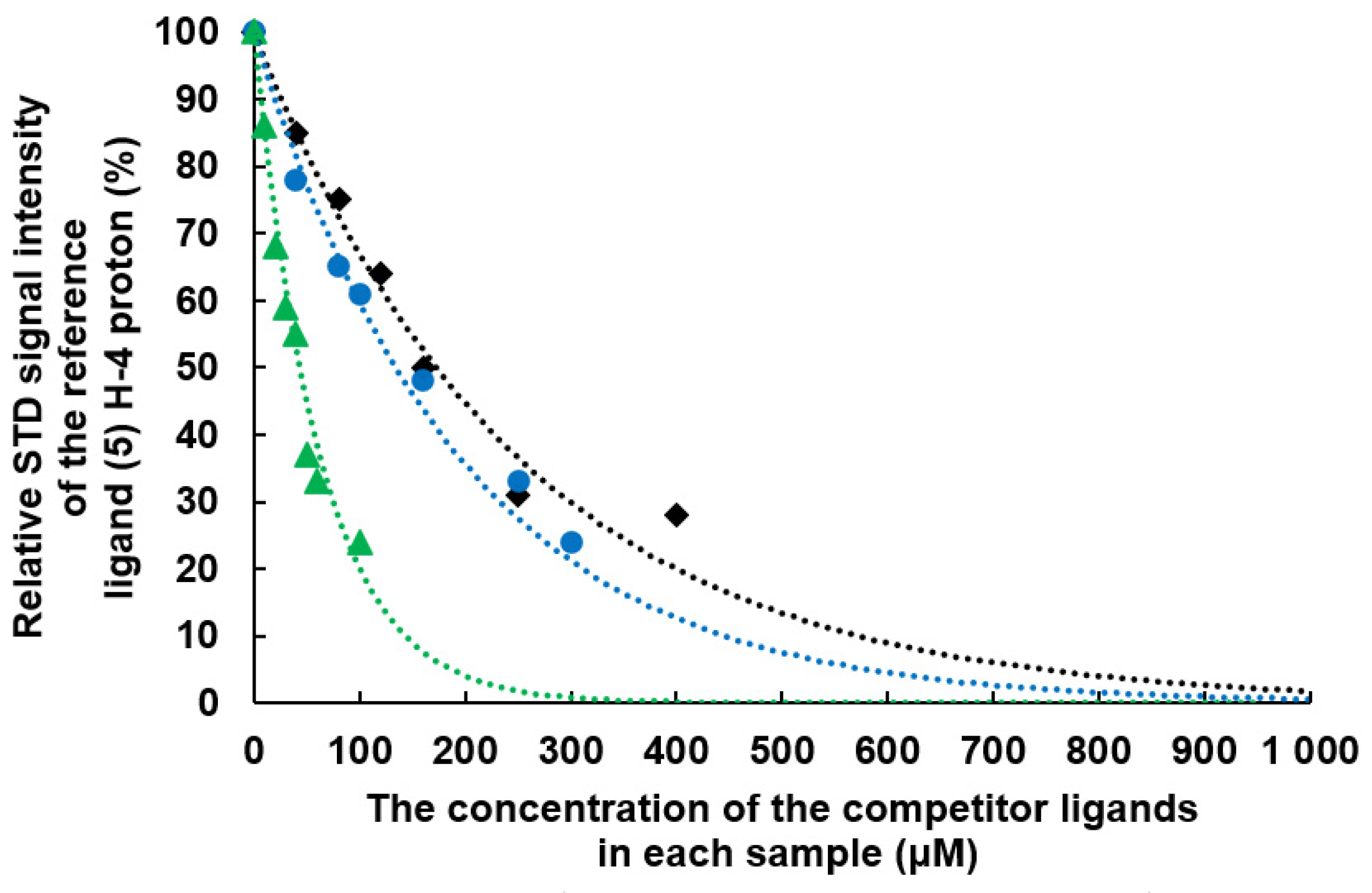
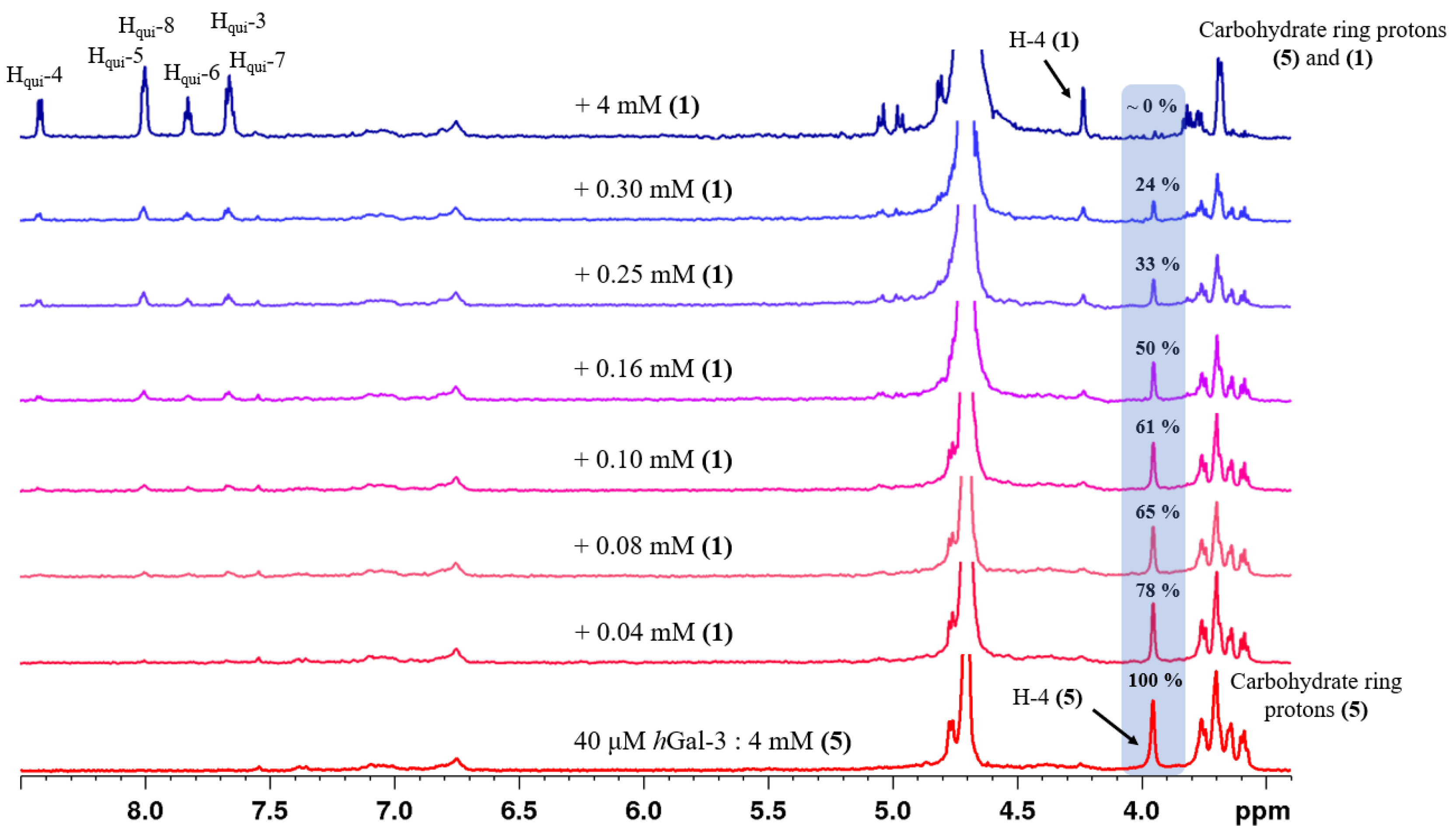
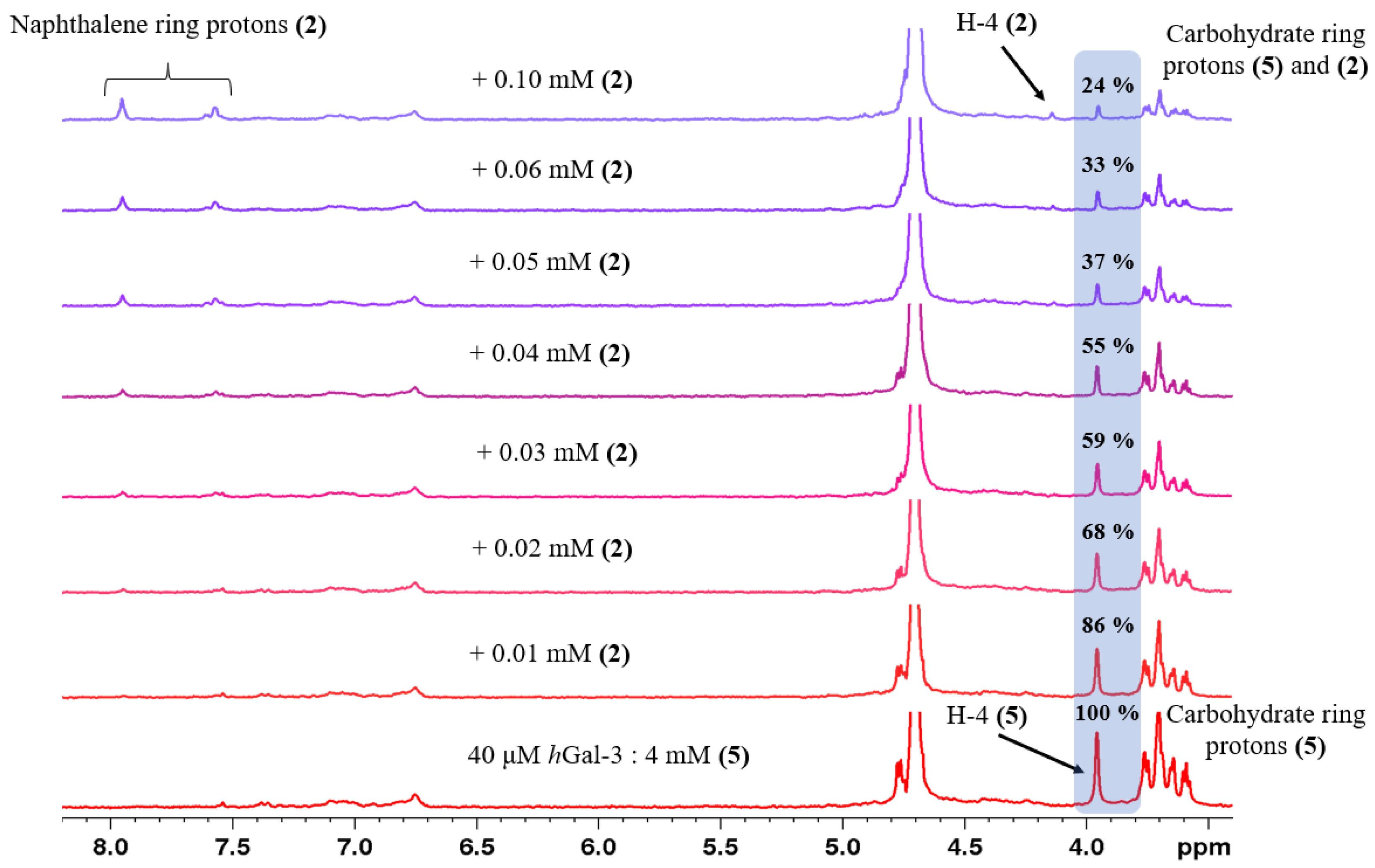
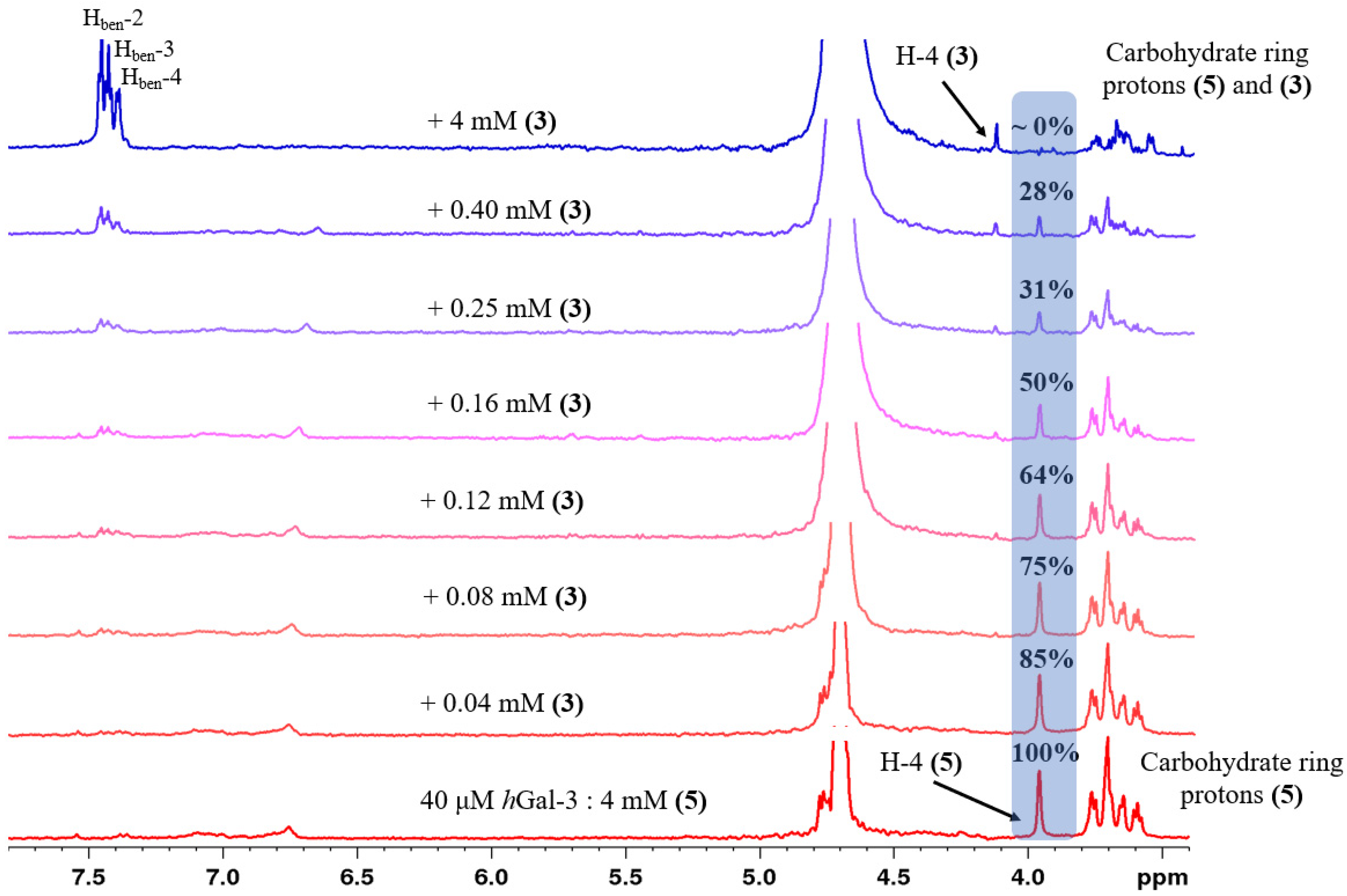
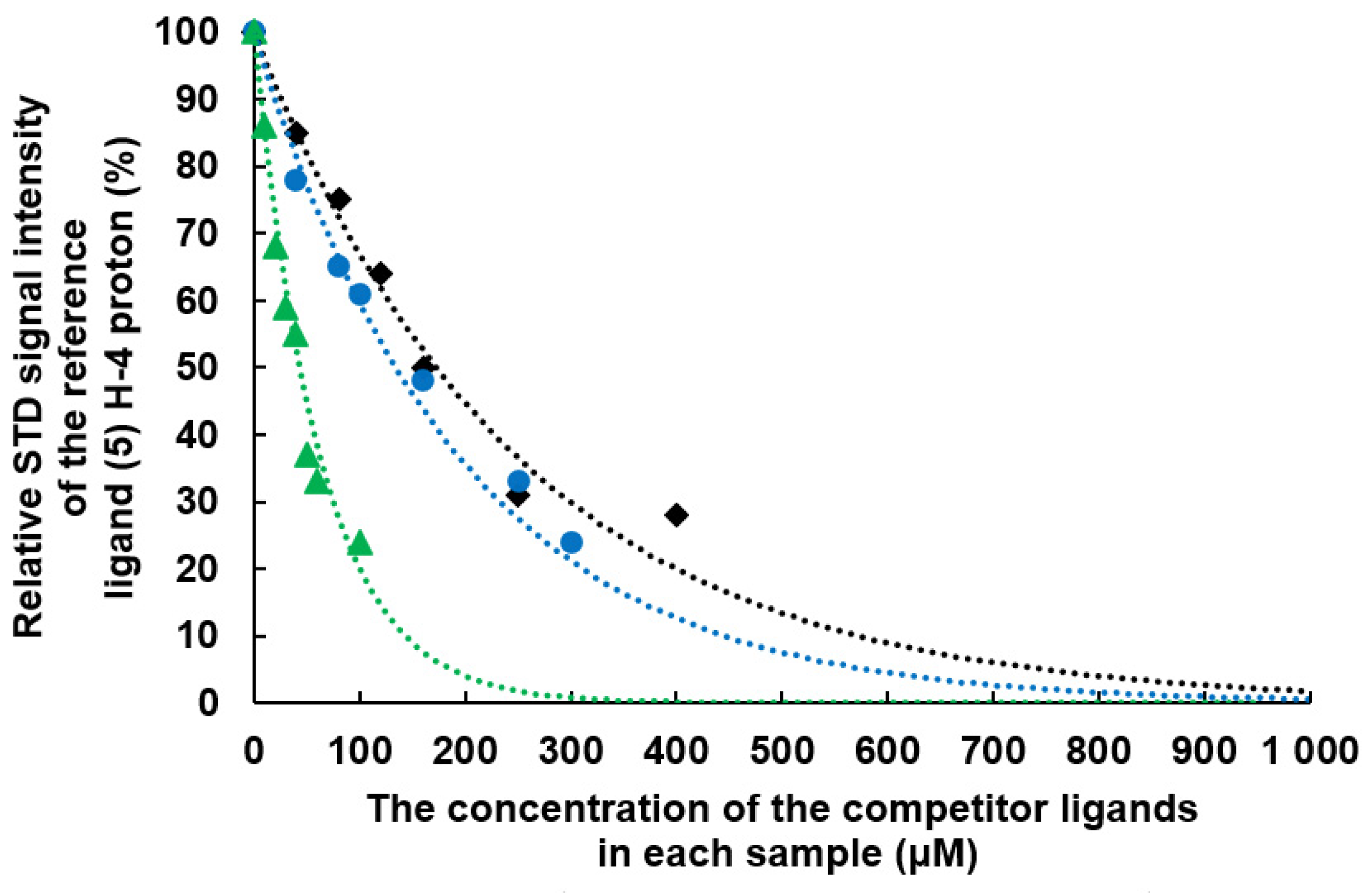
2.2. Determination of the Equilibrium Dissociation Constants (KD) for Aromatic Thiodigalactoside Derivatives Bound to hGal-3 by Competition STD 1H NMR Method
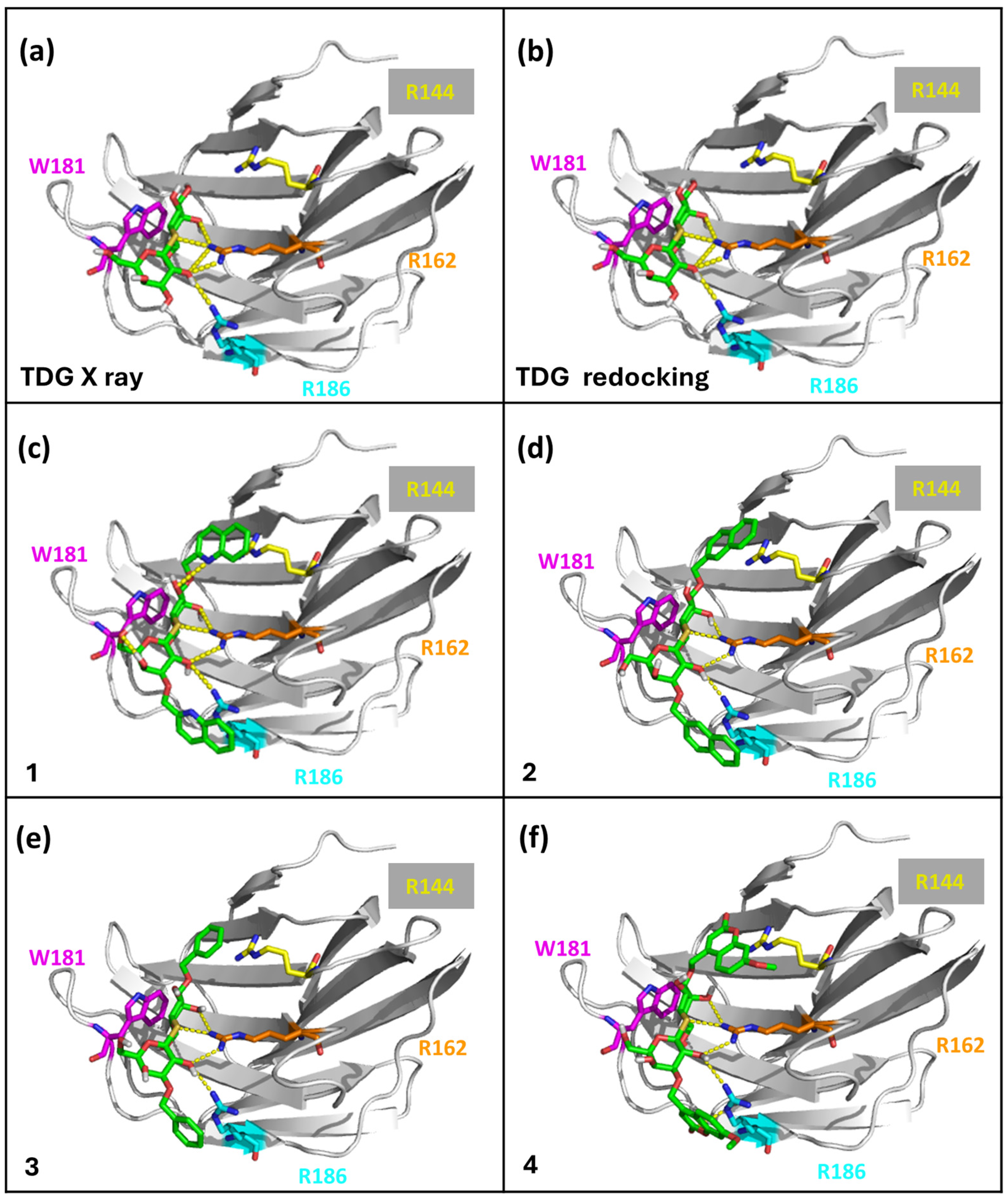
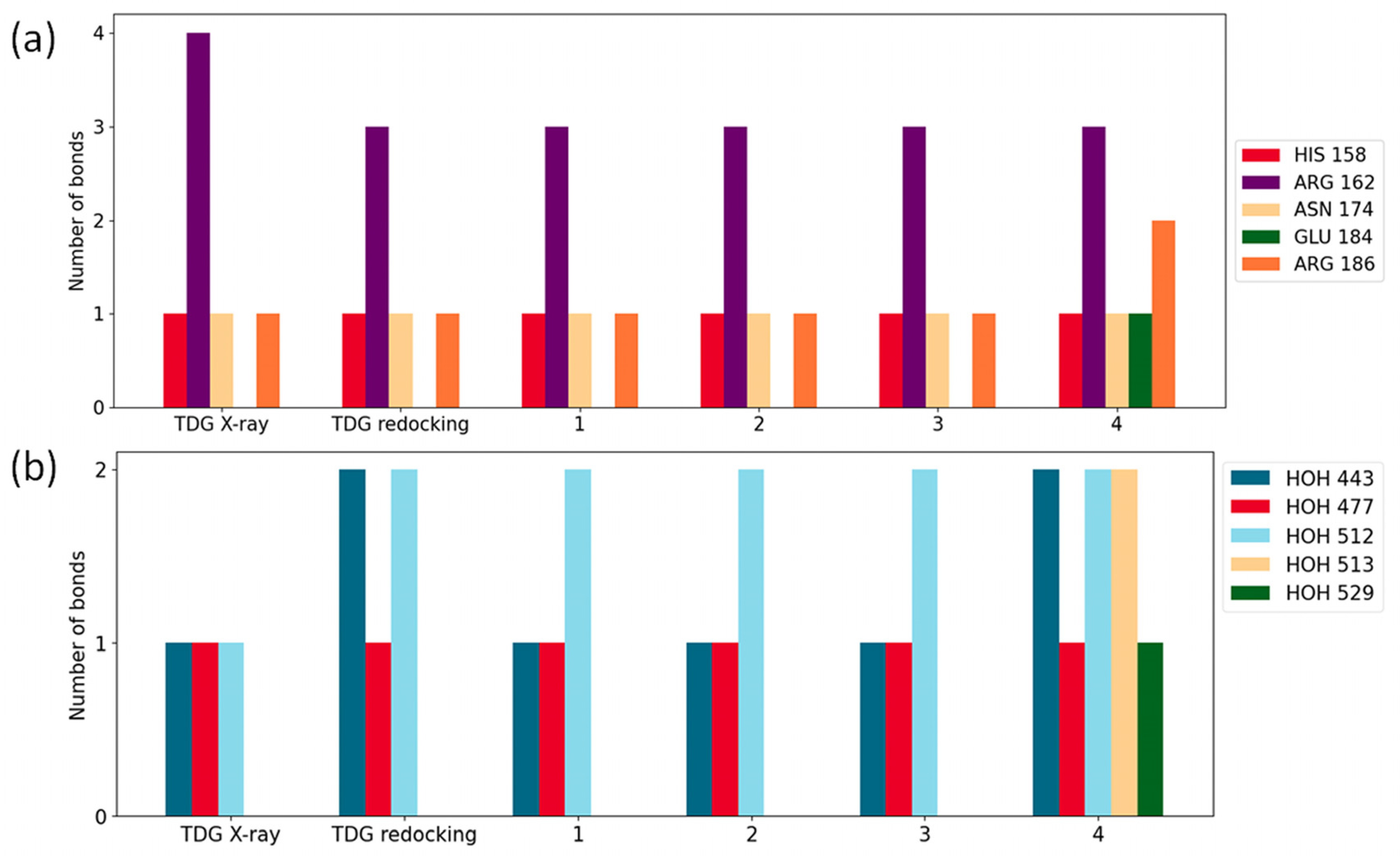
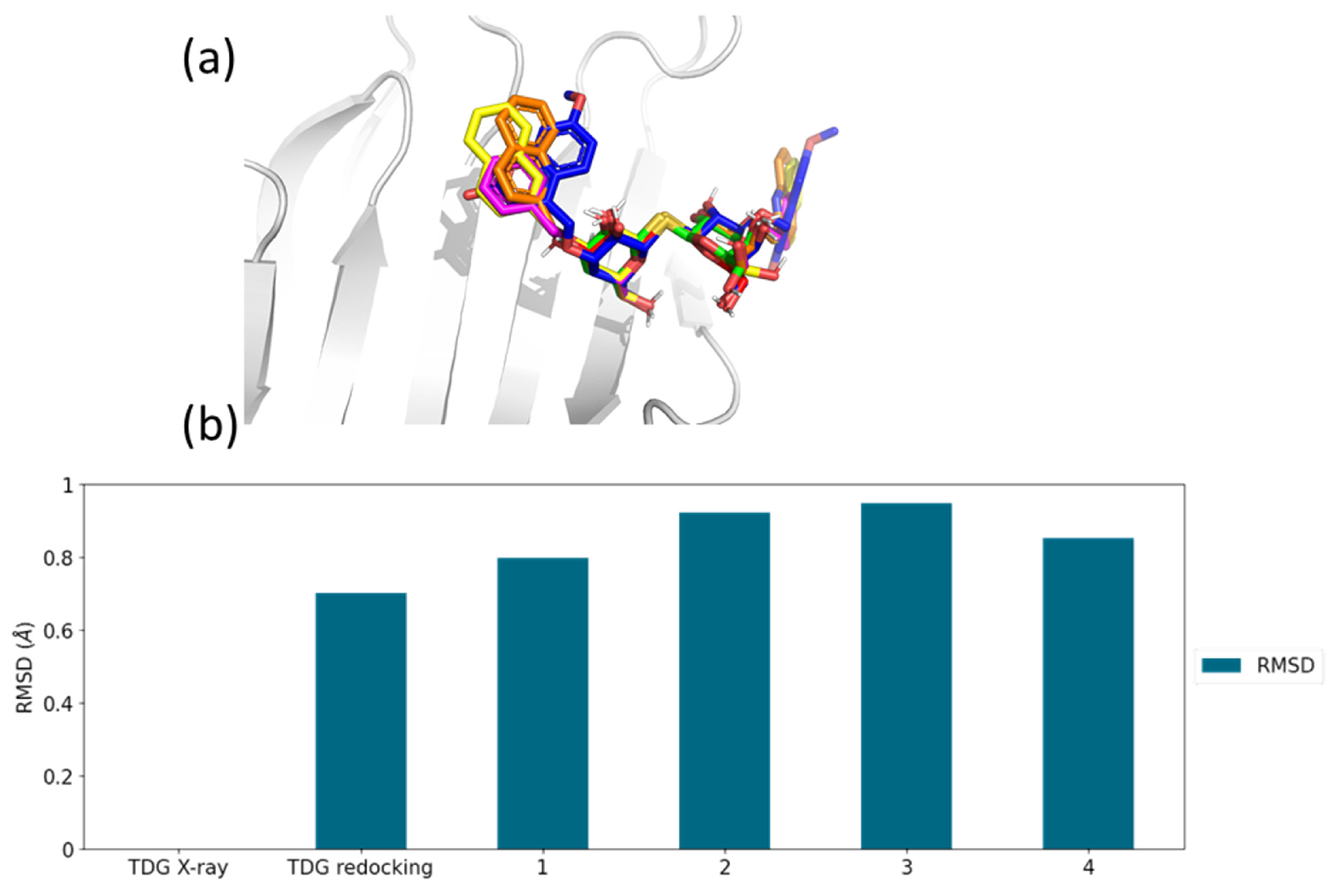
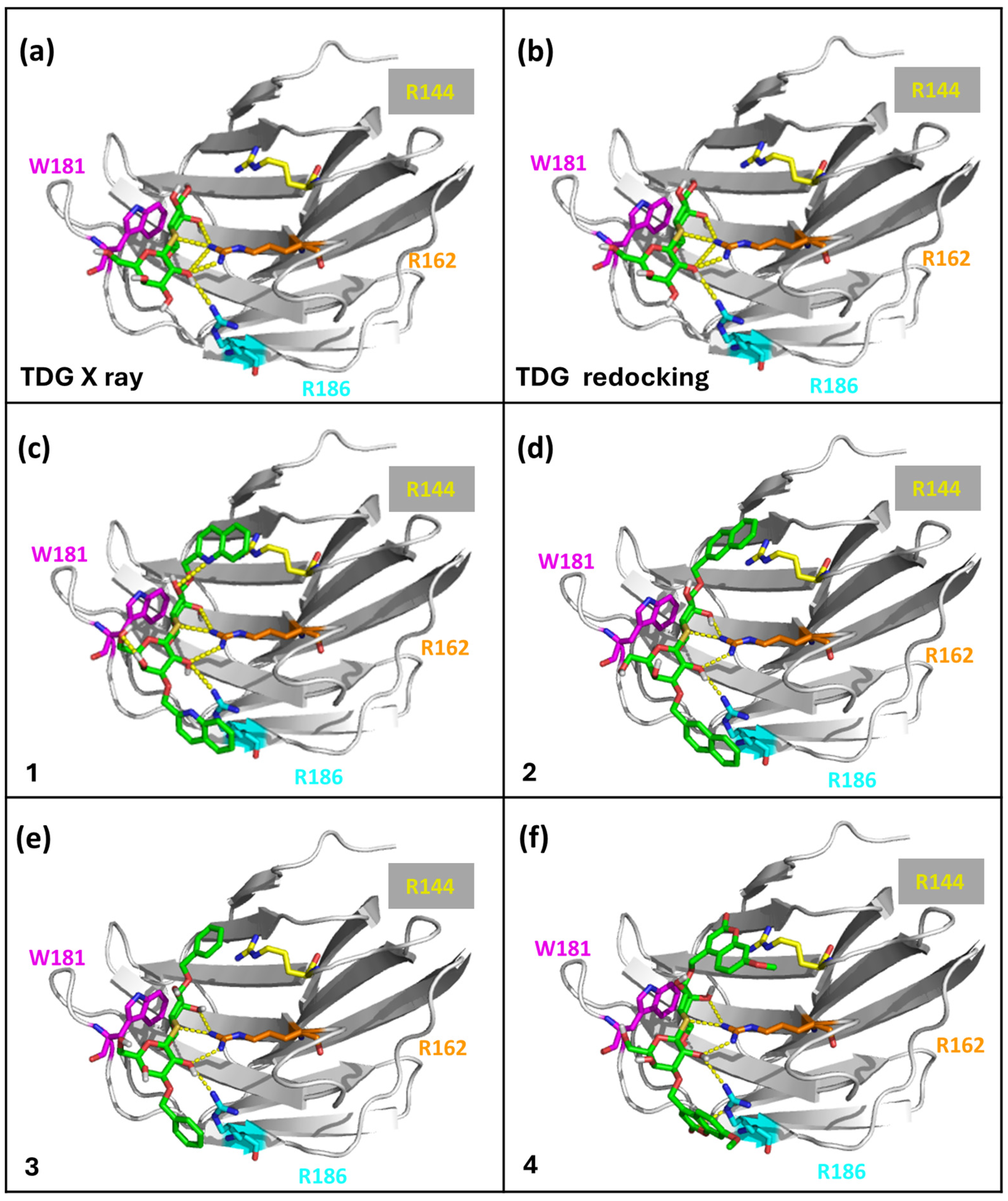
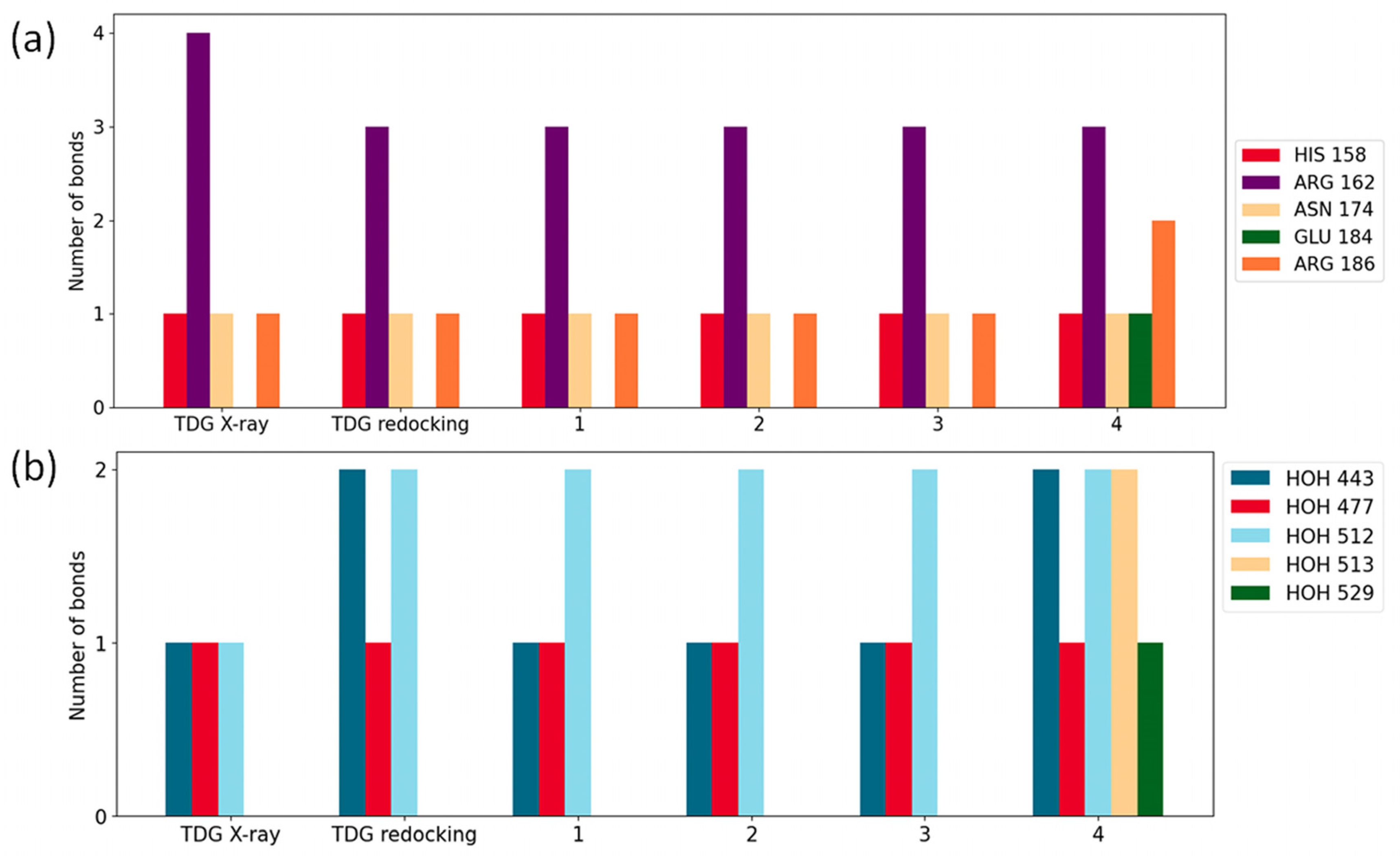
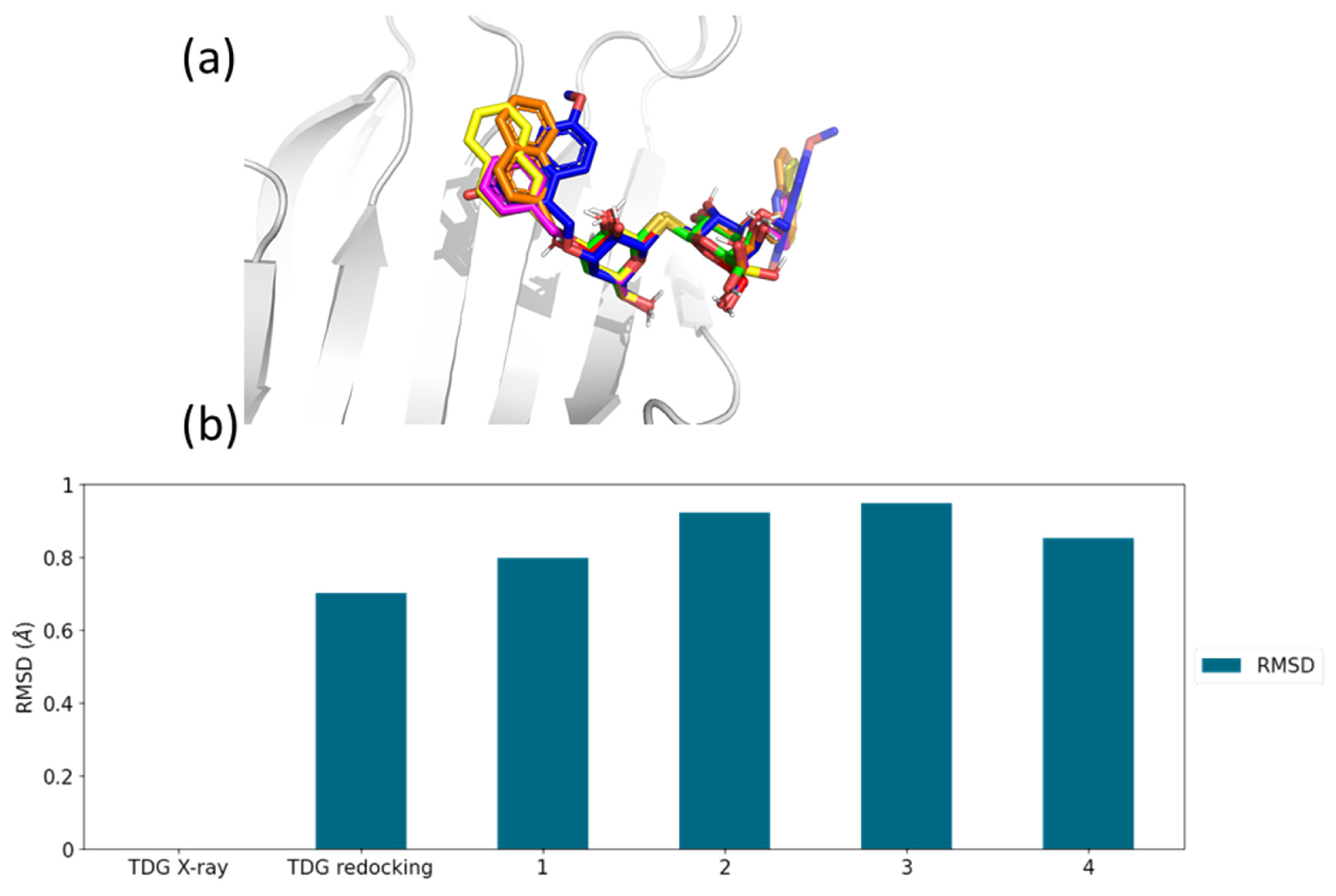
2.3. Molecular Docking Simulations
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. Bis-(2,3,4,6-Tetra-O-Acetyl-β-D-Galactopyranosyl)-Disulfide (8)
3.1.2. Bis-(2,3,4,6-Tetra-O-Acetyl-β-D-Galactopyranosyl)-Sulfane (9)
3.1.3. Bis-(β-D-Galactopyranosyl)-Sulfane (5, Thiodigalactoside, TDG)
3.1.4. General Procedure for the Synthesis of 3,3′-di-O-Aralkyl-Thiodigalactosides 1–4 [11,14]
3.1.5. Bis-{3-O-[(Quinoline-2-yl)Methyl]-β-D-Galactopyranosyl}-Sulfane (1)
3.1.6. Bis-{3-O-[(Naphtalene-2-yl)Methyl]-β-D-Galactopyranosyl}-Sulfane (2)
3.1.7. Bis-(3-O-Benzyl-β-D-Galactopyranosyl)-Sulfane (3)
3.1.8. Bis-{3-O-[(7-Methoxy-2H-1-Benzopyran-2-on-4-yl)Methyl]-β-D-Galactopyranosyl}-Sulfane (4) [37]
3.2. General Methods
3.3. 1H STD STD NMR Experiments
3.4. Docking Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, F.T.; Rabinovich, G.A. Galectins: Regulators of acute and chronic inflammation. Ann. N. Y. Acad. Sci. 2010, 1183, 158–182. [Google Scholar] [CrossRef]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217, e20182041. [Google Scholar] [CrossRef] [PubMed]
- Salatino, M.; Girotti, M.R.; Rabinovich, G.A. Glycans Pave the Way for Immunotherapy in Triple-Negative Breast Cancer. Cancer Cell 2018, 33, 155–157. [Google Scholar] [CrossRef]
- Hisrich, B.V.; Young, R.B.; Sansone, A.M.; Bowens, Z.; Green, L.J.; Lessey, B.A.; Blenda, A.V. Role of Human Galectins in Inflammation and Cancers Associated with Endometriosis. Biomolecules 2020, 10, 230. [Google Scholar] [CrossRef]
- Blanda, V.; Bracale, U.M.; Di Taranto, M.D.; Fortunato, G. Galectin-3 in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 9232. [Google Scholar] [CrossRef] [PubMed]
- Du, X.-J.; Zhao, W.-B.; Nguyen, M.-N.; Lu, Q.; Kiriazis, H. β-Adrenoceptor activation affects galectin-3 as a biomarker and therapeutic target in heart disease. Br. J. Pharmacol. 2019, 176, 2449–2464. [Google Scholar] [CrossRef]
- Mariño, K.V.; Cagnoni, A.J.; Croci, D.O.; Rabinovich, G.A. Targeting galectin-driven regulatory circuits in cancer and fibrosis. Nat. Rev. Drug Discov. 2023, 22, 295–316. [Google Scholar] [CrossRef]
- van Hattum, H.; Branderhorst, H.M.; Moret, E.E.; Nilsson, U.J.; Leffler, H.; Pieters, R.J. Tuning the Preference of Thiodigalactoside- and Lactosamine-Based Ligands to Galectin-3 over Galectin-1. J. Med. Chem. 2013, 56, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Salameh, B.A.; Cumpstey, I.; Sundin, A.; Leffler, H.; Nilsson, U.J. 1H-1,2,3-Triazol-1-yl thiodigalactoside derivatives as high affinity galectin-3 inhibitors. Bioorg. Med. Chem. 2010, 18, 5367–5378. [Google Scholar] [CrossRef]
- Delaine, T.; Collins, P.; MacKinnon, A.; Sharma, G.; Stegmayr, J.; Rajput, V.K.; Mandal, S.; Cumpstey, I.; Larumbe, A.; Salameh, B.A.; et al. Galectin-3-Binding Glycomimetics that Strongly Reduce Bleomycin-Induced Lung Fibrosis and Modulate Intracellular Glycan Recognition. ChemBioChem 2016, 17, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Vašíček, T.; Spiwok, V.; Červený, J.; Petrásková, L.; Bumba, L.; Vrbata, D.; Pelantová, H.; Křen, V.; Bojarová, P. Regioselective 3-O-Substitution of Unprotected Thiodigalactosides: Direct Route to Galectin Inhibitors. Chem. Eur. J. 2020, 26, 9620–9631. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, V.; Borbás, A. 5—Glycomimetics with unnatural glycosidic linkages. In Recent Trends in Carbohydrate Chemistry; Rauter, A.P., Christensen, B.E., Somsák, L., Kosma, P., Adamo, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 161–215. [Google Scholar]
- Mandal, S.; Nilsson, U.J. Tri-isopropylsilyl thioglycosides as masked glycosyl thiol nucleophiles for the synthesis of S-linked glycosides and glyco-conjugates. Org. Biomol. Chem. 2014, 12, 4816–4819. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; Joosten, J.A.F.; el Maate, F.A.; Liskamp, R.M.J.; Pieters, R.J. Microwave-assisted, tin-mediated, regioselective 3-O-alkylation of galactosides. Tetrahedron Lett. 2004, 45, 6685–6687. [Google Scholar] [CrossRef]
- André, S.; Kövér, K.E.; Gabius, H.-J.; Szilágyi, L. Thio- and selenoglycosides as ligands for biomedically relevant lectins: Valency–activity correlations for benzene-based dithiogalactoside clusters and first assessment for (di)selenodigalactosides. Bioorg. Med. Chem. Lett. 2015, 25, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Kaltner, H.; Szabó, T.; Fehér, K.; André, S.; Balla, S.; Manning, J.C.; Szilágyi, L.; Gabius, H.J. Bivalent O-glycoside mimetics with S/disulfide/Se substitutions and aromatic core: Synthesis, molecular modeling and inhibitory activity on biomedically relevant lectins in assays of increasing physiological relevance. Bioorg. Med. Chem. 2017, 25, 3158–3170. [Google Scholar] [CrossRef]
- Raics, M.; Balogh, Á.K.; Kishor, C.; Timári, I.; Medrano, F.J.; Romero, A.; Go, R.M.; Blanchard, H.; Szilágyi, L.; Kövér, K.E.; et al. Investigation of the Molecular Details of the Interactions of Selenoglycosides and Human Galectin-3. Int. J. Mol. Sci. 2022, 23, 2494. [Google Scholar] [CrossRef]
- Timári, I.; Balla, S.; Fehér, K.; Kövér, K.E.; Szilágyi, L. 77Se-Enriched Selenoglycoside Enables Significant Enhancement in NMR Spectroscopic Monitoring of Glycan–Protein Interactions. Pharmaceutics 2022, 14, 201. [Google Scholar] [CrossRef]
- Raics, M.; Timári, I.; Diercks, T.; Szilágyi, L.; Gabius, H.-J.; Kövér, K.E. Selenoglycosides as Lectin Ligands: 77Se-Edited CPMG-HSQMBC NMR Spectroscopy To Monitor Biomedically Relevant Interactions. ChemBioChem 2019, 20, 1688–1692. [Google Scholar] [CrossRef]
- Di Carluccio, C.; Forgione, M.C.; Martini, S.; Berti, F.; Molinaro, A.; Marchetti, R.; Silipo, A. Investigation of protein-ligand complexes by ligand-based NMR methods. Carbohydr. Res. 2021, 503, 108313. [Google Scholar] [CrossRef]
- Valverde, P.; Quintana, J.I.; Santos, J.I.; Ardá, A.; Jiménez-Barbero, J. Novel NMR Avenues to Explore the Conformation and Interactions of Glycans. ACS Omega 2019, 4, 13618–13630. [Google Scholar] [CrossRef]
- Walter, B.; Chaitanya, B.K.; Nina, G.; Klaus, Z. Investigating Protein–Ligand Interactions by Solution Nuclear Magnetic Resonance Spectroscopy. ChemPhysChem 2018, 19, 895–906. [Google Scholar] [CrossRef]
- Arda, A.; Jimenez-Barbero, J. The recognition of glycans by protein receptors. Insights from NMR spectroscopy. Chem. Commun. 2018, 54, 4761–4769. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Konuma, T.; Yanaka, S.; Sugase, K. Quantitative analysis of protein-ligand interactions by NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 96, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Meyer, B.; Klein, J.; Mayer, M.; Meinecke, R.; Möller, H.; Neffe, A.; Schuster, O.; Wülfken, J.; Ding, Y.; Knaie, O.; et al. Saturation Transfer Difference NMR Spectroscopy for Identifying Ligand Epitopes and Binding Specificities; Ernst Schering Research Foundation Workshop; Springer: Berlin/Heidelberg, Germany, 2004; pp. 149–167. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef]
- Seetharaman, J.; Kanigsberg, A.; Slaaby, R.; Leffler, H.; Barondes, S.H.; Rini, J.M. X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-A resolution. J. Biol. Chem. 1998, 273, 13047–13052. [Google Scholar] [CrossRef]
- Dnyandev, K.M.; Babasaheb, G.V.; Chandrashekhar, K.V.; Chandrakant, M.A.; Vasant, O.K. A Review on Molecular Docking. Int. Res. J. Pure Appl. Chem. 2021, 22, 60–68. [Google Scholar] [CrossRef]
- Moor, L.F.E.; Vasconcelos, T.R.A.; da R Reis, R.; Pinto, L.S.S.; da Costa, T.M. Quinoline: An Attractive Scaffold in Drug Design. Mini-Rev. Med. Chem. 2021, 21, 2209–2226. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Martorana, A.; La Monica, G.; Lauria, A. Quinoline-Based Molecules Targeting c-Met, EGF, and VEGF Receptors and the Proteins Involved in Related Carcinogenic Pathways. Molecules 2020, 25, 4279. [Google Scholar] [CrossRef]
- Marella, A.; Tanwar, O.P.; Saha, R.; Ali, M.R.; Srivastava, S.; Akhter, M.; Shaquiquzzaman, M.; Alam, M.M. Quinoline: A versatile heterocyclic. Saudi Pharm. J. 2013, 21, 1–12. [Google Scholar] [CrossRef]
- Hopkins, K.L.; Davies, R.H.; Threlfall, E.J. Mechanisms of quinolone resistance in Escherichia coli and Salmonella: Recent developments. Int. J. Antimicrob. Agents 2005, 25, 358–373. [Google Scholar] [CrossRef]
- Di Gaetano, S.; Bedini, E.; Landolfi, A.; Pedone, E.; Pirone, L.; Saviano, M.; Traboni, S.; Capasso, D.; Iadonisi, A. Synthesis of diglycosylated (di)sulfides and comparative evaluation of their antiproliferative effect against tumor cell lines: A focus on the nature of sugar-recognizing mediators involved. Carbohydr. Res. 2019, 482, 107740. [Google Scholar] [CrossRef] [PubMed]
- Kirihara, M.; Asai, Y.; Ogawa, S.; Noguchi, T.; Hatano, A.; Hirai, Y. A Mild and Environmentally Benign Oxidation of Thiols to Disulfides. Synthesis 2007, 2007, 3286–3289. [Google Scholar] [CrossRef]
- Rajput, V.K.; MacKinnon, A.; Mandal, S.; Collins, P.; Blanchard, H.; Leffler, H.; Sethi, T.; Schambye, H.; Mukhopadhyay, B.; Nilsson, U.J. A Selective Galactose–Coumarin-Derived Galectin-3 Inhibitor Demonstrates Involvement of Galectin-3-glycan Interactions in a Pulmonary Fibrosis Model. J. Med. Chem. 2016, 59, 8141–8147. [Google Scholar] [CrossRef]
- Černý, M.; Staněk, J.; Pacák, J. 2,3,4,6-Tetra-O-acetyl-β-d-galaktopyranosylmercaptan und dessen Anwendung zur Synthese von β-d-Thiogalaktosiden. Monatshefte Für Chem. Verwandte Teile Anderer Wiss. 1963, 94, 290–294. [Google Scholar] [CrossRef]
- Staněk, J.; Šindlerová, M.; Černý, M. Derivatives of D-thioxylopyranose and of some reducing 1-deoxy-1-thiodisaccharides. Collect. Czech. Chem. Commun. 1965, 30, 297–303. [Google Scholar] [CrossRef]
- Fehér, K.; Groves, P.; Batta, G.; Jiménez-Barbero, J.; Muhle-Goll, C.; Kövér, K.E. Competition Saturation Transfer Difference Experiments Improved with Isotope Editing and Filtering Schemes in NMR-Based Screening. J. Am. Chem. Soc. 2008, 130, 17148–17153. [Google Scholar] [CrossRef]
- Wang, Y.S.; Liu, D.; Wyss, D.F. Competition STD NMR for the detection of high-affinity ligands and NMR-based screening. Magn. Reson. Chem. 2004, 42, 485–489. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Diercks, T.; Medrano, F.J.; FitzGerald, F.G.; Beckwith, D.; Pedersen, M.J.; Reihill, M.; Ludwig, A.K.; Romero, A.; Oscarson, S.; Cudic, M.; et al. Galectin-Glycan Interactions: Guidelines for Monitoring by 77Se NMR Spectroscopy, and Solvent (H2O/D2O) Impact on Binding. Chem. Eur. J. 2021, 27, 316–325. [Google Scholar] [CrossRef]
- Ninković, D.B.; Vojislavljević-Vasilev, D.Z.; Medaković, V.B.; Hall, M.B.; Brothers, E.N.; Zarić, S.D. Aliphatic–aromatic stacking interactions in cyclohexane–benzene are stronger than aromatic–aromatic interaction in the benzene dimer. Phys. Chem. Chem. Phys. 2016, 18, 25791–25795. [Google Scholar] [CrossRef]
- Dougherty, D.A. The cation-π interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef]
- Infield, D.T.; Rasouli, A.; Galles, G.D.; Chipot, C.; Tajkhorshid, E.; Ahern, C.A. Cation-π Interactions and their Functional Roles in Membrane Proteins. J. Mol. Biol. 2021, 433, 167035. [Google Scholar] [CrossRef] [PubMed]
- Gyöngyösi, T.; Timári, I.; Sinnaeve, D.; Luy, B.; Kövér, K.E. Expedited Nuclear Magnetic Resonance Assignment of Small- to Medium-Sized Molecules with Improved HSQC−CLIP−COSY Experiments. Anal. Chem. 2021, 93, 3096–3102. [Google Scholar] [CrossRef] [PubMed]
- Bum-Erdene, K.; Gagarinov, I.A.; Collins, P.M.; Winger, M.; Pearson, A.G.; Wilson, J.C.; Leffler, H.; Nilsson, U.J.; Grice, I.D.; Blanchard, H. Investigation into the feasibility of thioditaloside as a novel scaffold for galectin-3-specific inhibitors. ChemBioChem 2013, 14, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.K.; Todd, A.; Millam, J. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of 1 in the Sample [μM] | Relative STD Intensity of H-4 of TDG Titrated by 1 [%] | Concentration of 2 in the Sample [μM] | Relative STD Intensity of H-4 of TDG Titrated by 2 [%] | Concentration of 3 in the Sample [μM] | Relative STD Intensity of H-4 of TDG Titrated by 3 [%] |
|---|---|---|---|---|---|
| 0 | 100 | 0 | 100 | 0 | 100 |
| 40 | 78 | 10 | 86 | 40 | 85 |
| 80 | 65 | 20 | 68 | 80 | 75 |
| 100 | 61 | 30 | 59 | 120 | 64 |
| 160 | 48 | 40 | 55 | 160 | 50 |
| 250 | 33 | 50 | 37 | 250 | 31 |
| 300 | 24 | 60 | 33 | 400 | 28 |
| 4000 | 0 | 100 | 24 | 4000 | 0 |
| Inhibitor Ligand | IC50 [μM] | Kd [μM] | Relative Affinity 2 | Valency 3 |
|---|---|---|---|---|
| Compound (5) 1 | - | 51.4 [43] | 1 | 2 |
| Compound (1) | 134 | 1.70 | 30 | 2 |
| Compound (2) | 43 | 0.55 | 94 | 2 |
| Compound (3) | 172 | 2.19 | 24 | 2 |
| Inhibitor Ligand | Score [Kcal/mol] | Kd-score [μM] | Kd [μM] |
|---|---|---|---|
| 1 | −7.548 | 3.60 | 1.70 |
| 2 | −7.303 | 5.40 | 0.55 |
| 3 | −6.556 | 18.68 | 2.19 |
| 4 | −7.700 | 2.79 | n.a. |
| TDG-redocking | −6.116 | 38.79 | 51.40 |
| TDG X-ray | −5.788 | 66.88 | 51.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hőgye, F.; Farkas, L.B.; Balogh, Á.K.; Szilágyi, L.; Alnukari, S.; Bajza, I.; Borbás, A.; Fehér, K.; Illyés, T.Z.; Timári, I. Saturation Transfer Difference NMR and Molecular Docking Interaction Study of Aralkyl-Thiodigalactosides as Potential Inhibitors of the Human-Galectin-3 Protein. Int. J. Mol. Sci. 2024, 25, 1742. https://doi.org/10.3390/ijms25031742
Hőgye F, Farkas LB, Balogh ÁK, Szilágyi L, Alnukari S, Bajza I, Borbás A, Fehér K, Illyés TZ, Timári I. Saturation Transfer Difference NMR and Molecular Docking Interaction Study of Aralkyl-Thiodigalactosides as Potential Inhibitors of the Human-Galectin-3 Protein. International Journal of Molecular Sciences. 2024; 25(3):1742. https://doi.org/10.3390/ijms25031742
Chicago/Turabian StyleHőgye, Fanni, László Bence Farkas, Álex Kálmán Balogh, László Szilágyi, Samar Alnukari, István Bajza, Anikó Borbás, Krisztina Fehér, Tünde Zita Illyés, and István Timári. 2024. "Saturation Transfer Difference NMR and Molecular Docking Interaction Study of Aralkyl-Thiodigalactosides as Potential Inhibitors of the Human-Galectin-3 Protein" International Journal of Molecular Sciences 25, no. 3: 1742. https://doi.org/10.3390/ijms25031742
APA StyleHőgye, F., Farkas, L. B., Balogh, Á. K., Szilágyi, L., Alnukari, S., Bajza, I., Borbás, A., Fehér, K., Illyés, T. Z., & Timári, I. (2024). Saturation Transfer Difference NMR and Molecular Docking Interaction Study of Aralkyl-Thiodigalactosides as Potential Inhibitors of the Human-Galectin-3 Protein. International Journal of Molecular Sciences, 25(3), 1742. https://doi.org/10.3390/ijms25031742