
Kynurenines, Neuronal Excitotoxicity, and Mitochondrial Oxidative Stress: Role of the Intestinal Flora




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
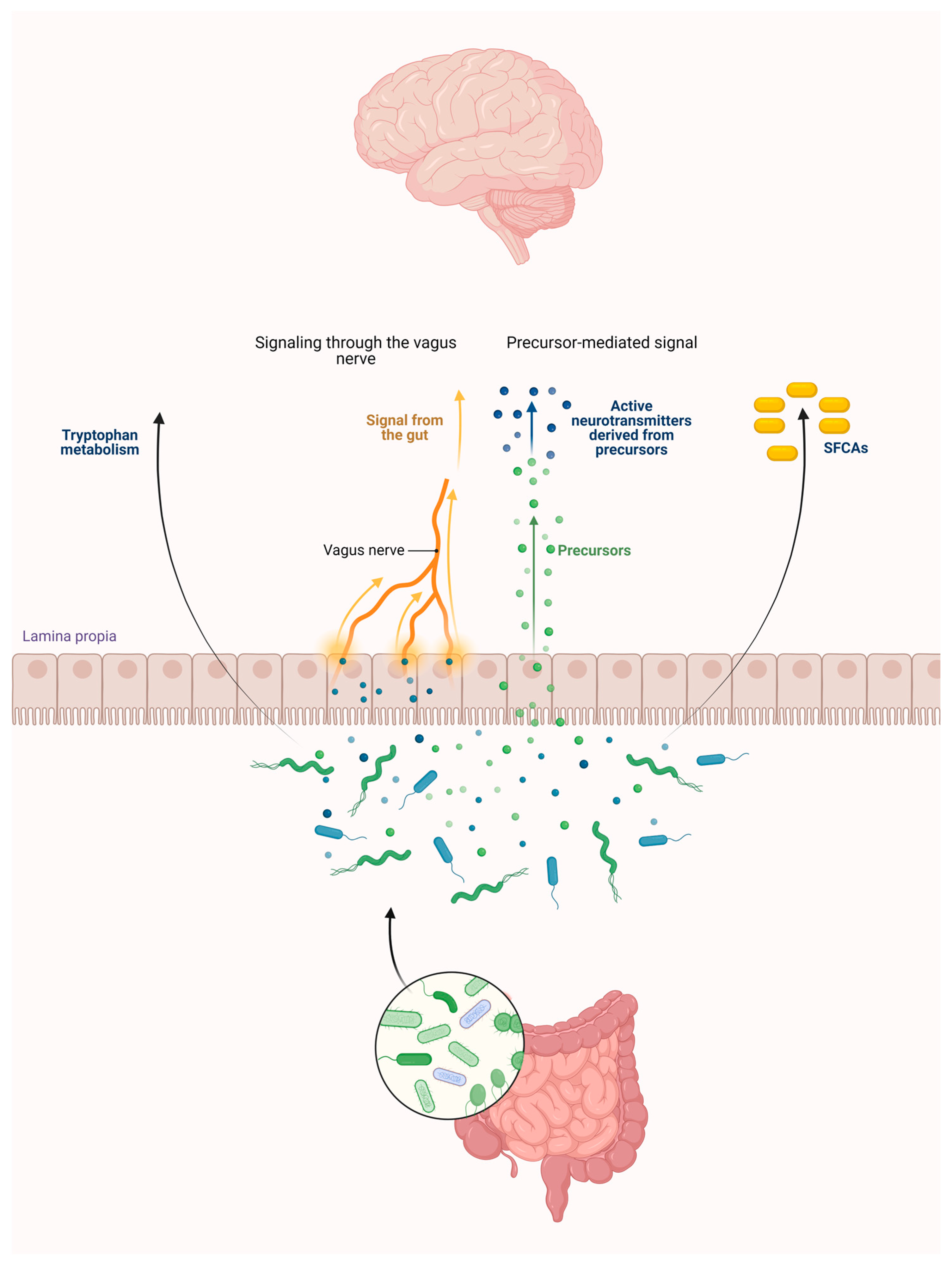
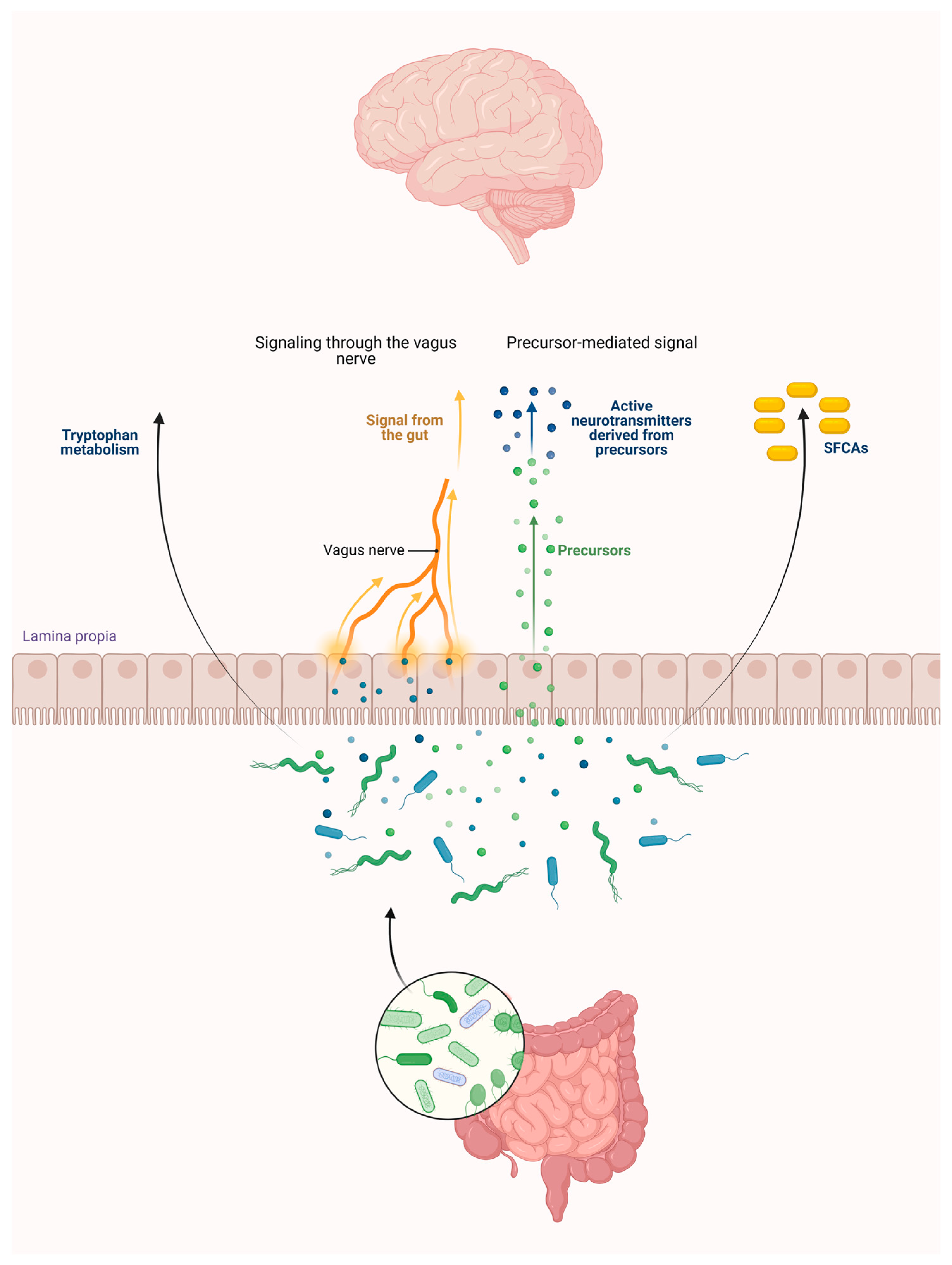
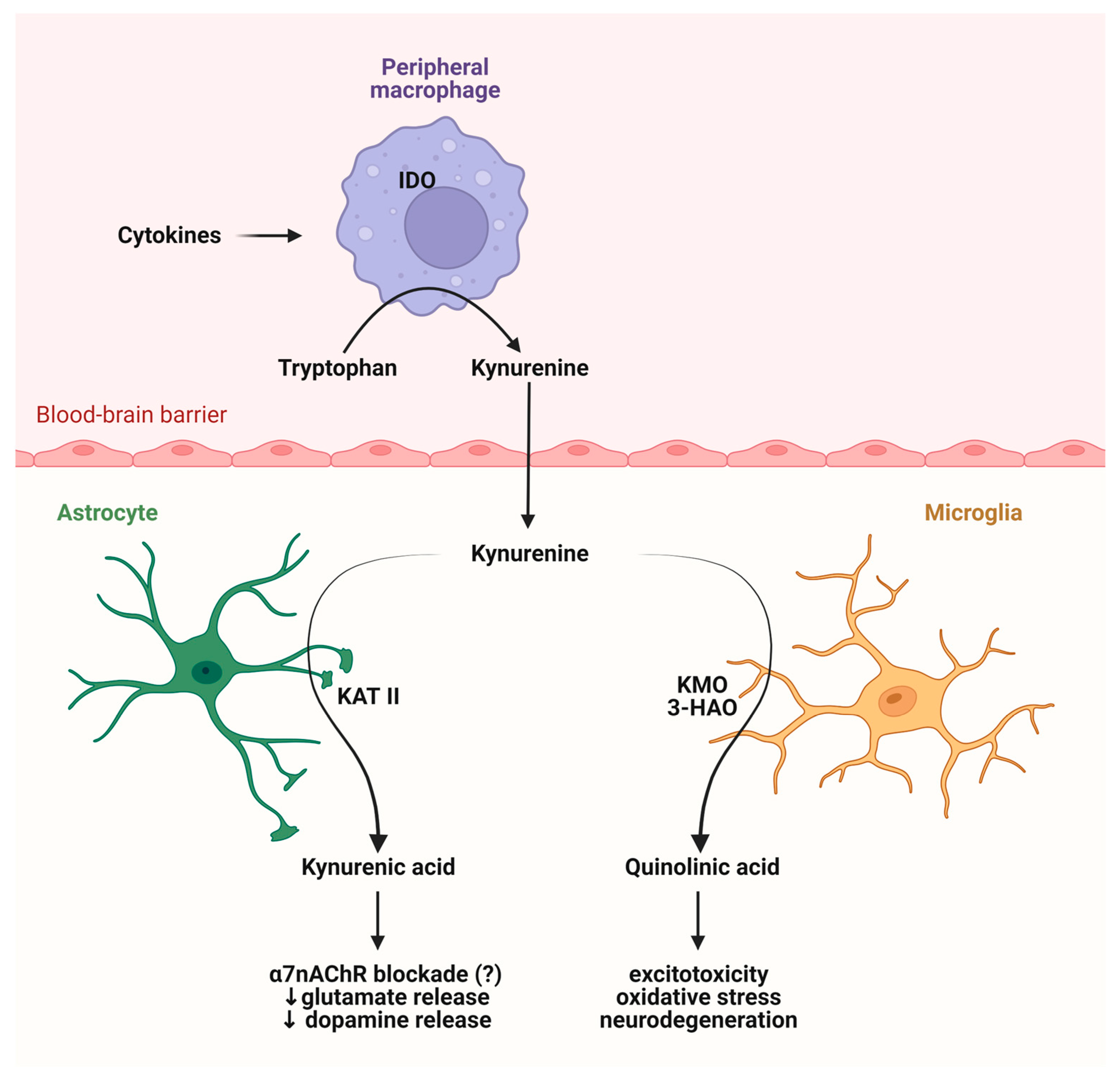
2. Connections between the Intestinal Flora and the KP
2.1. Intestinal Flora
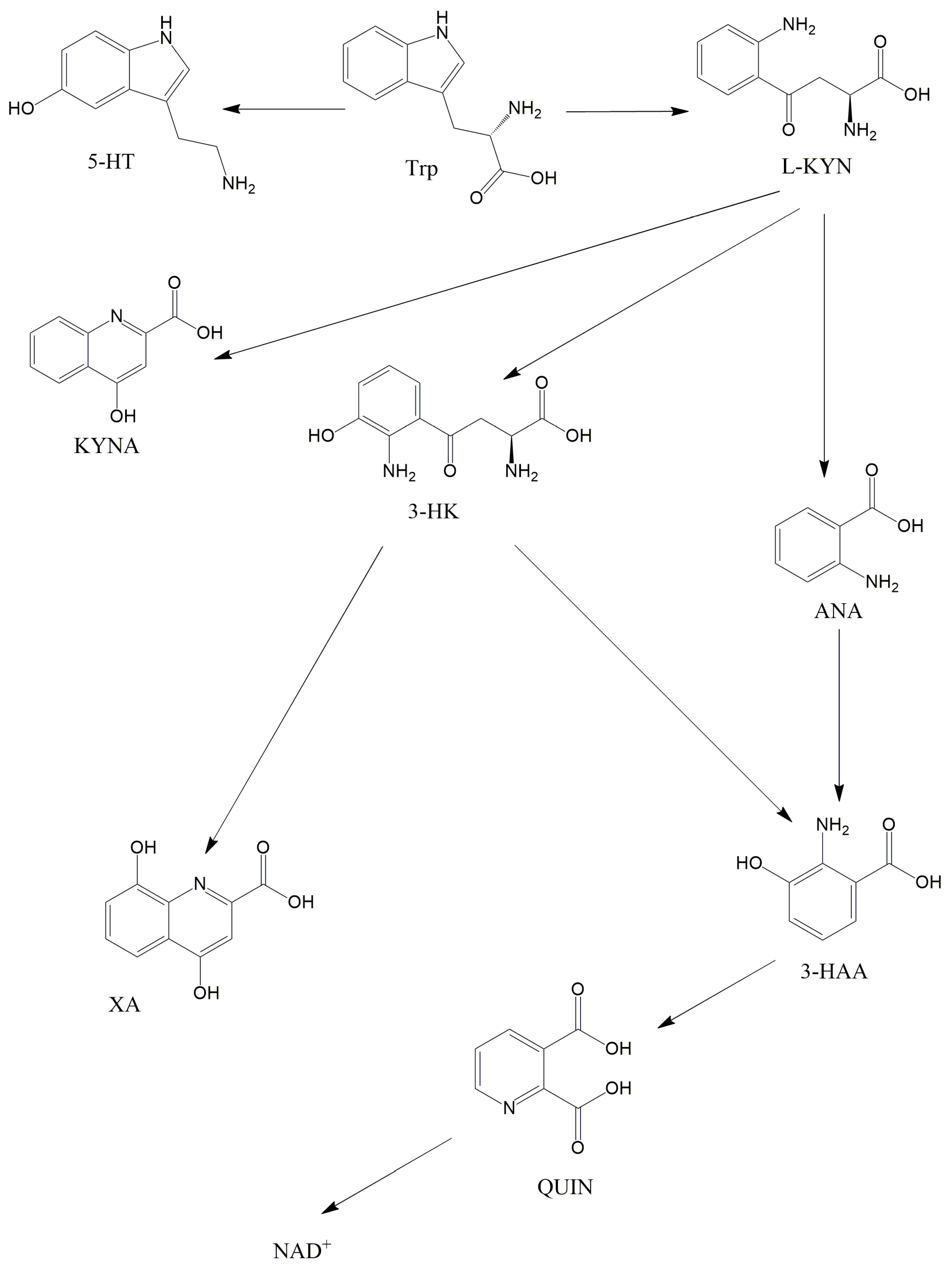
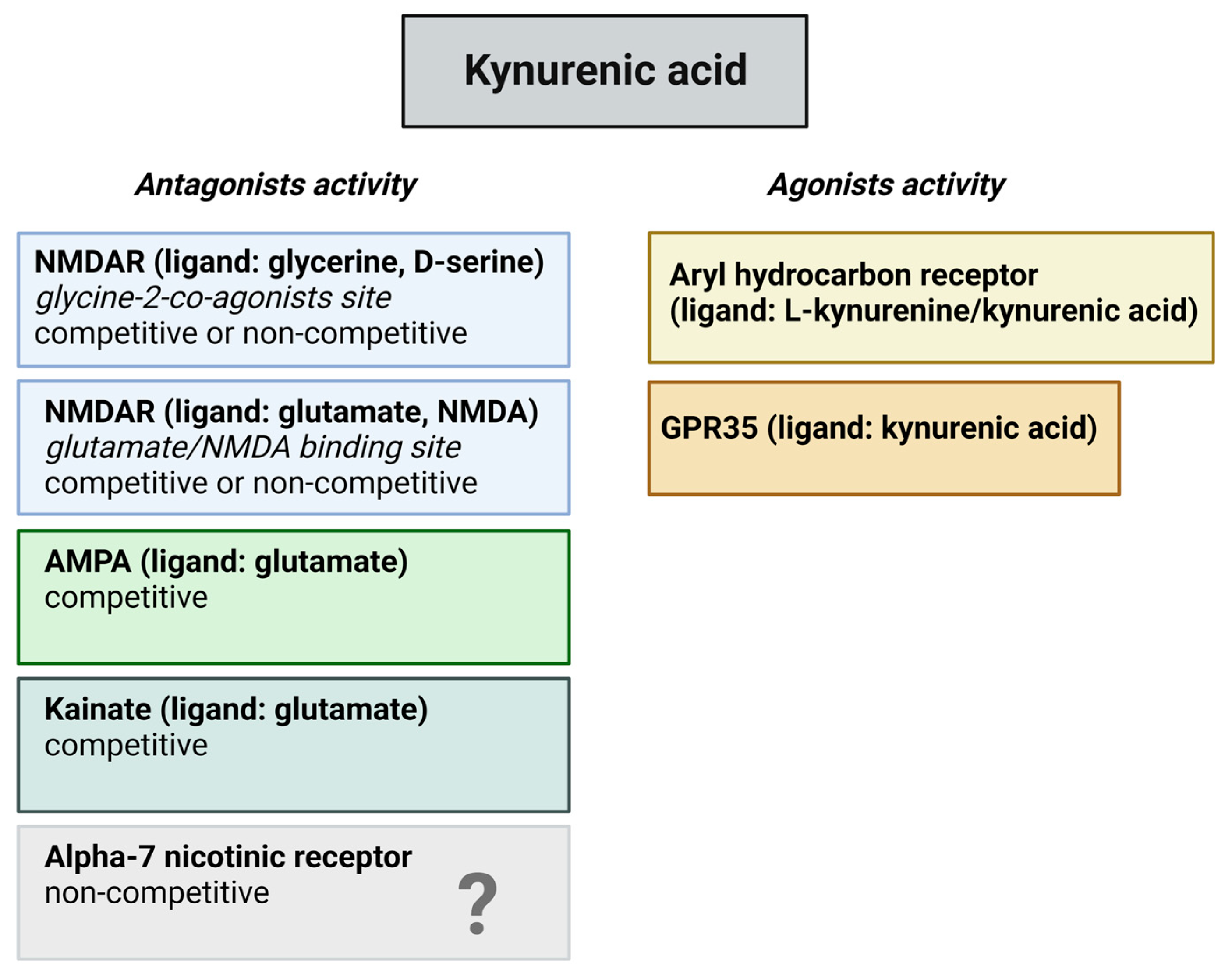
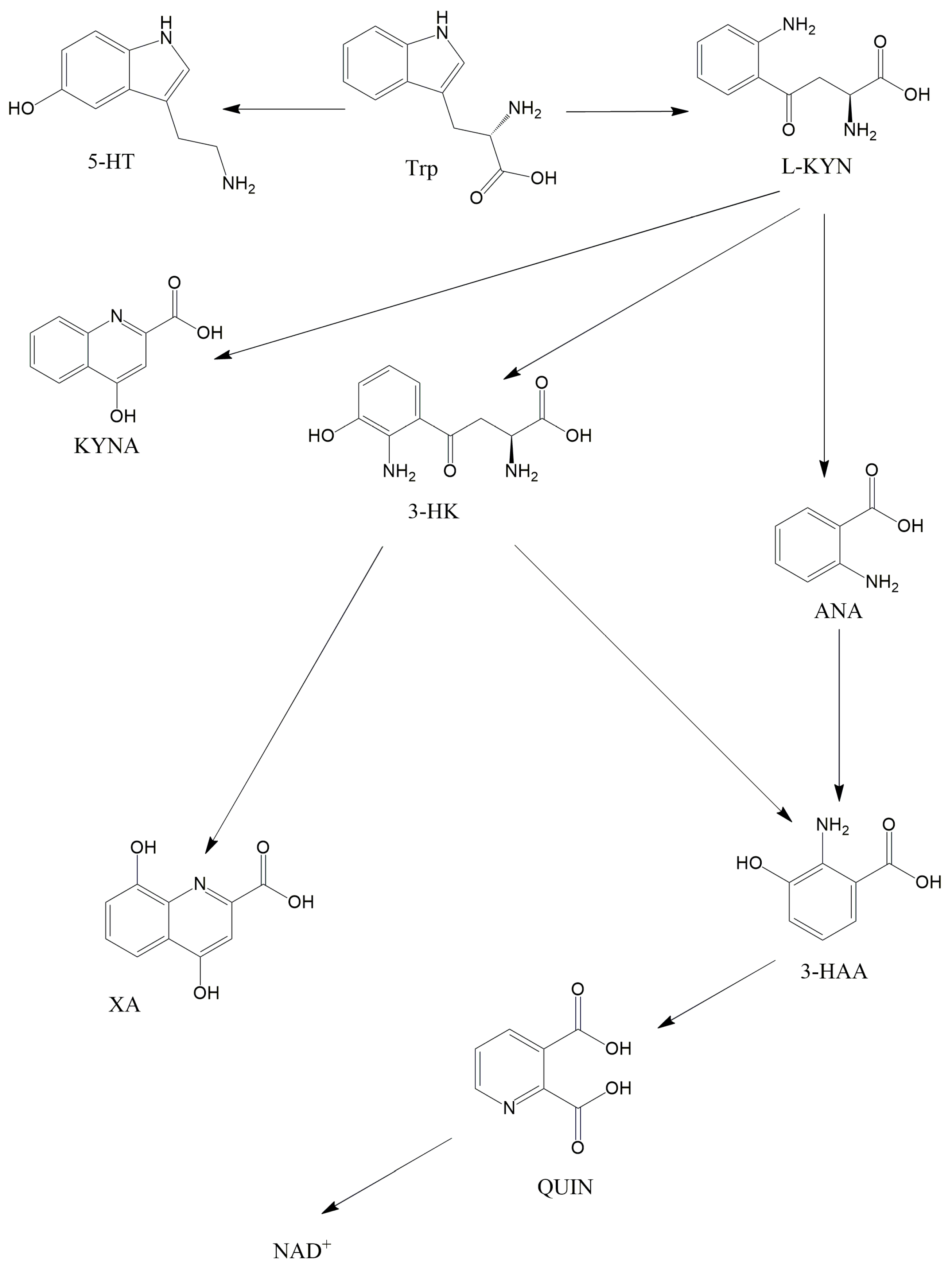
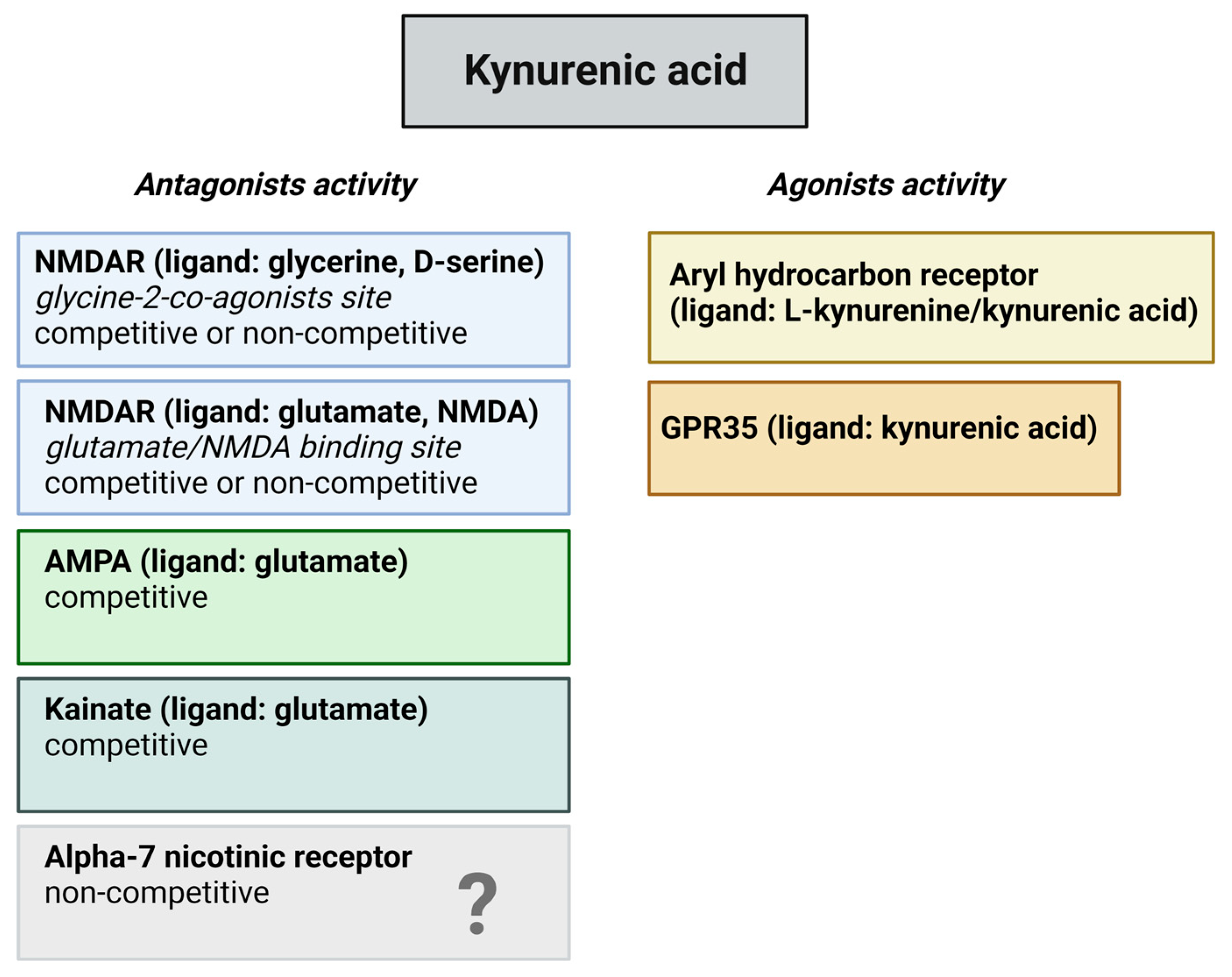
2.2. The KP and Its Receptors
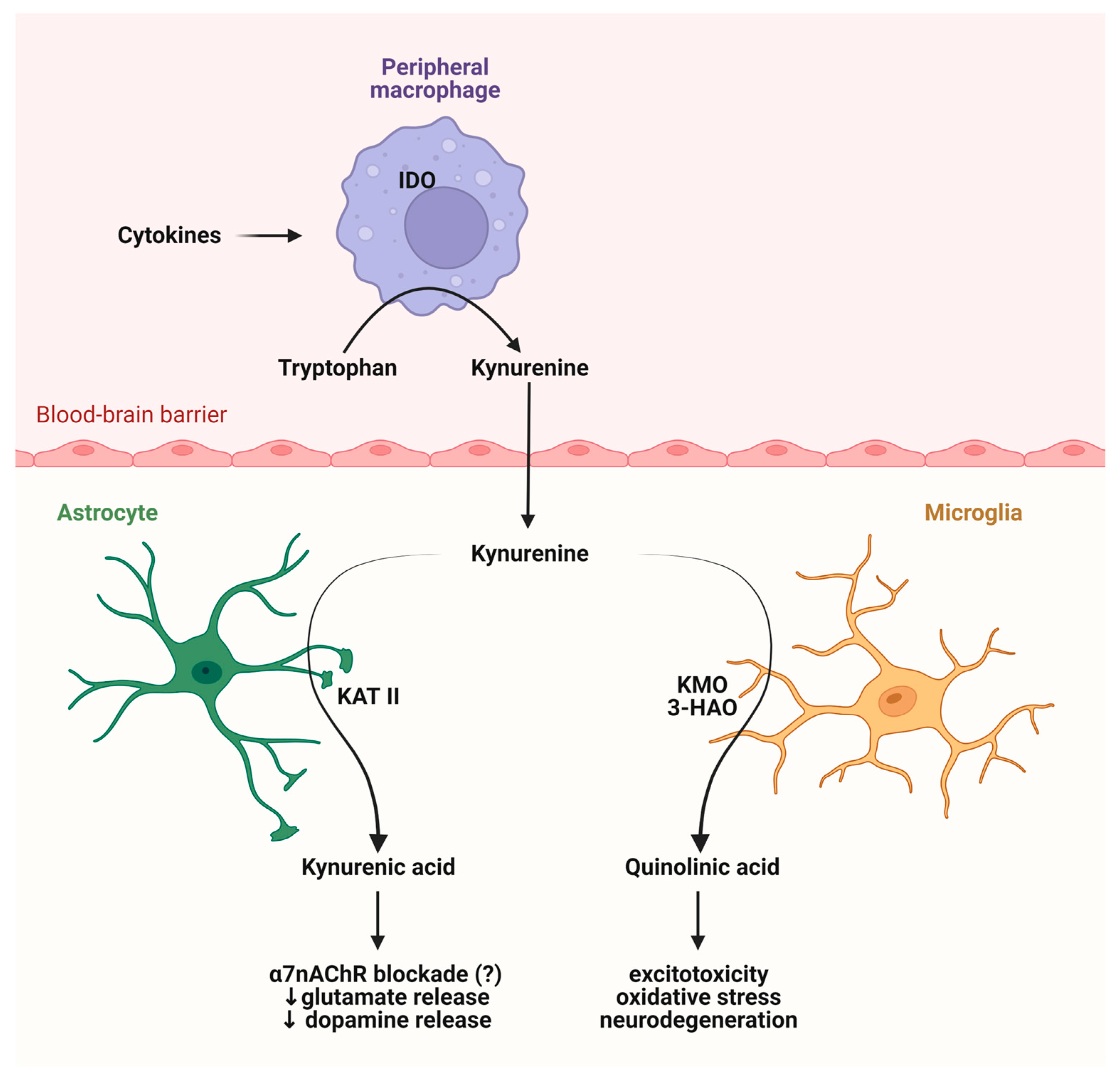
2.3. Role of the Intestinal Flora in the KP Metabolism
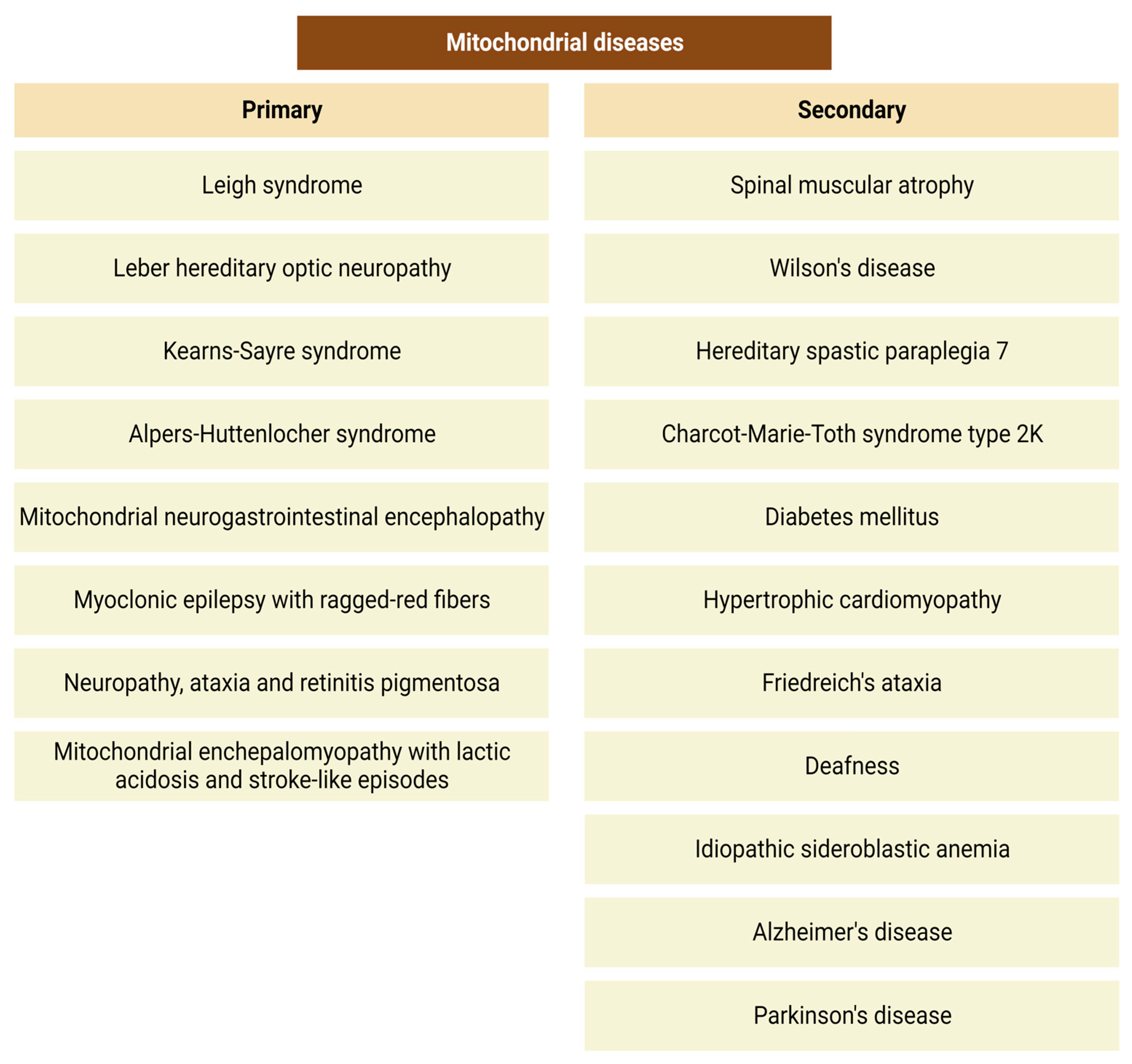
3. Mitochondrial Disorders, and Oxidative Stress: Possible Role of the KP
3.1. Mitochondria
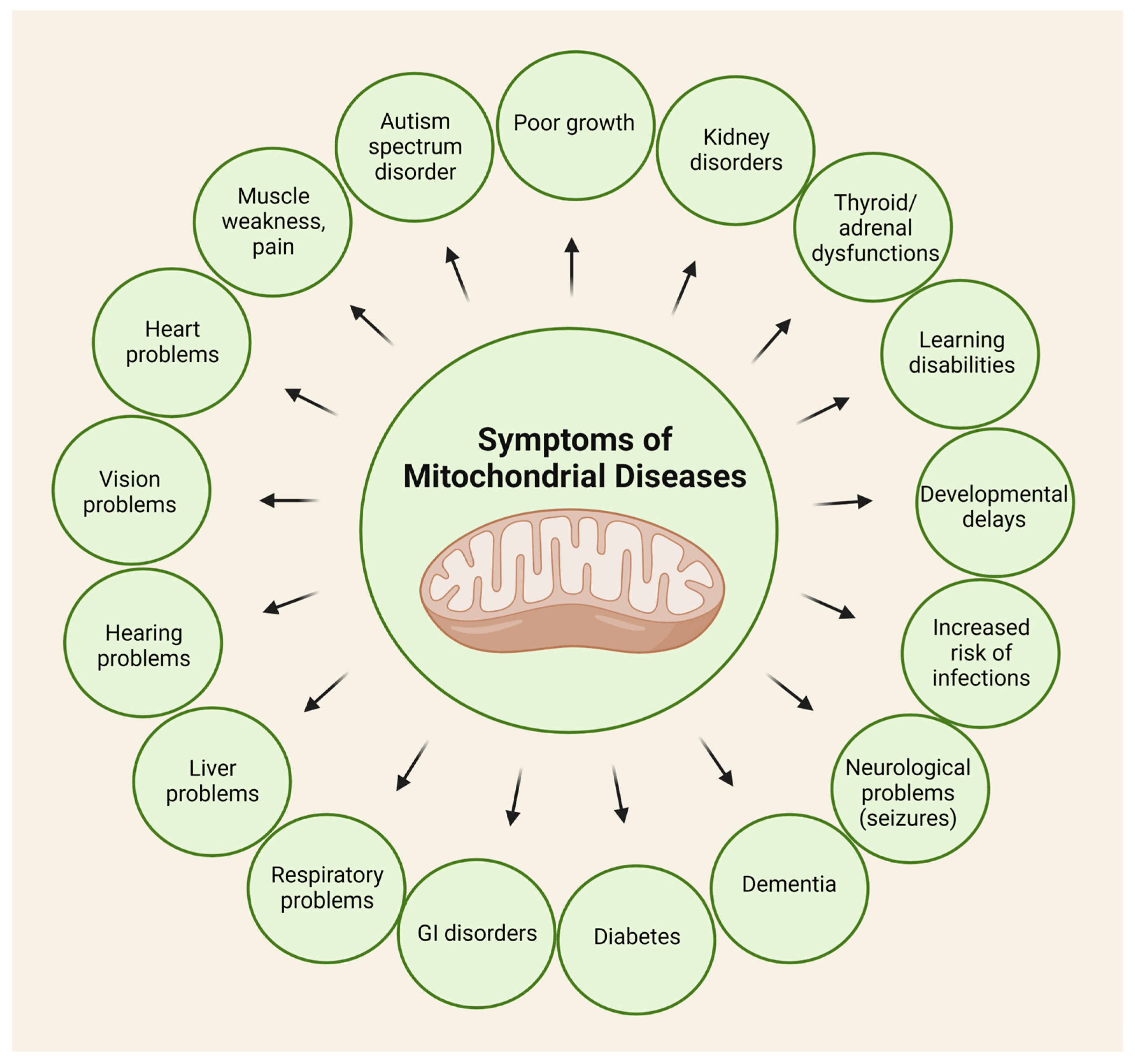
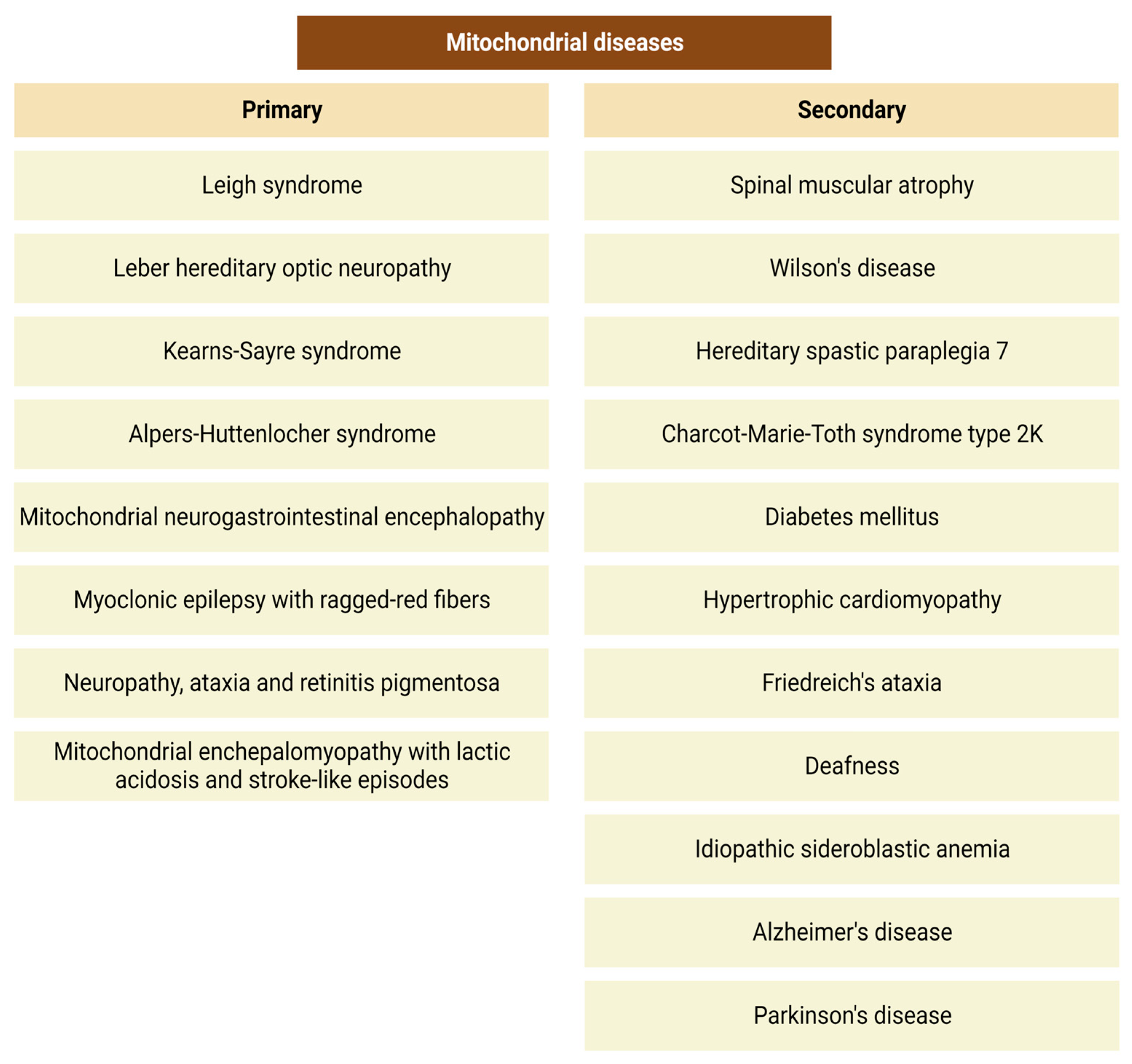
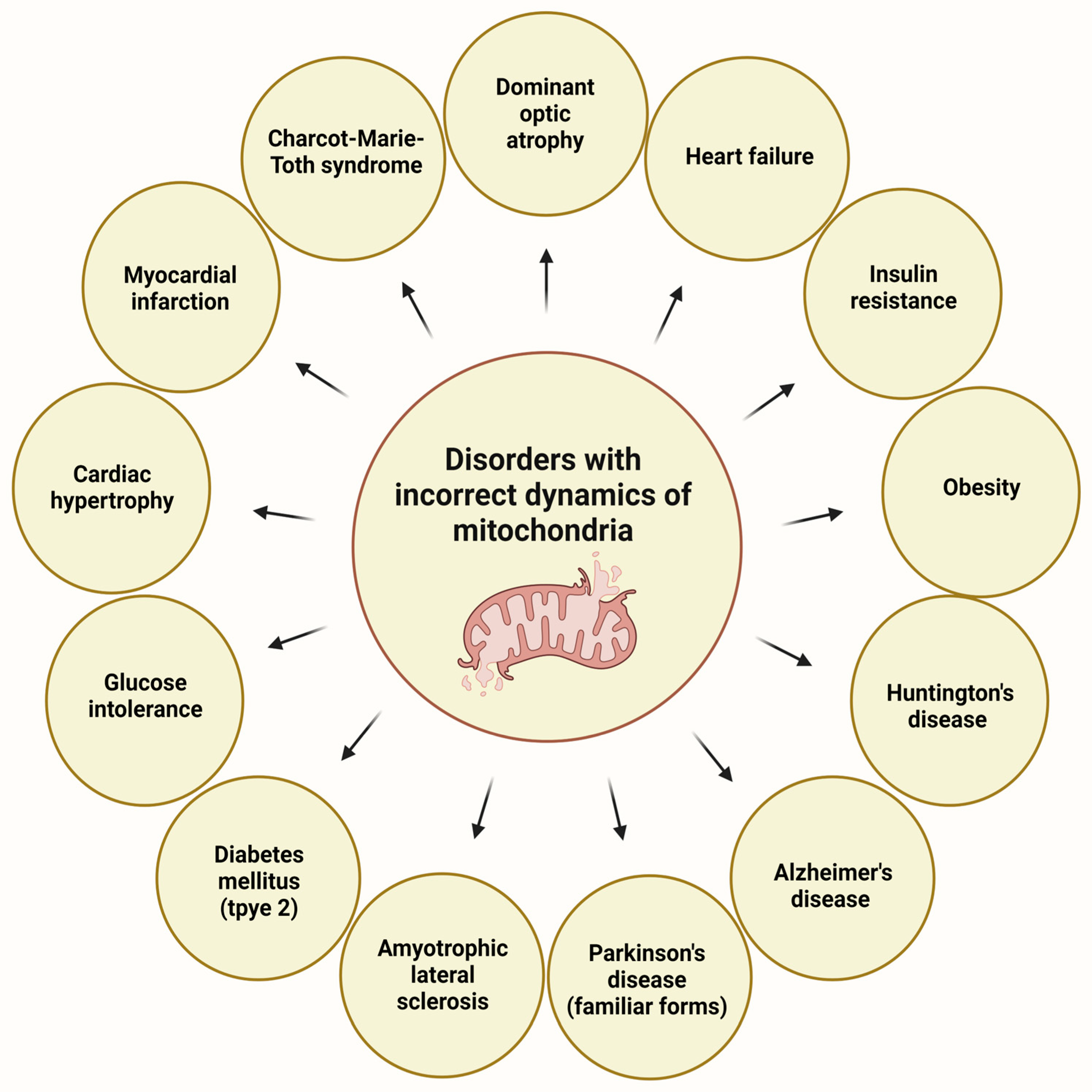
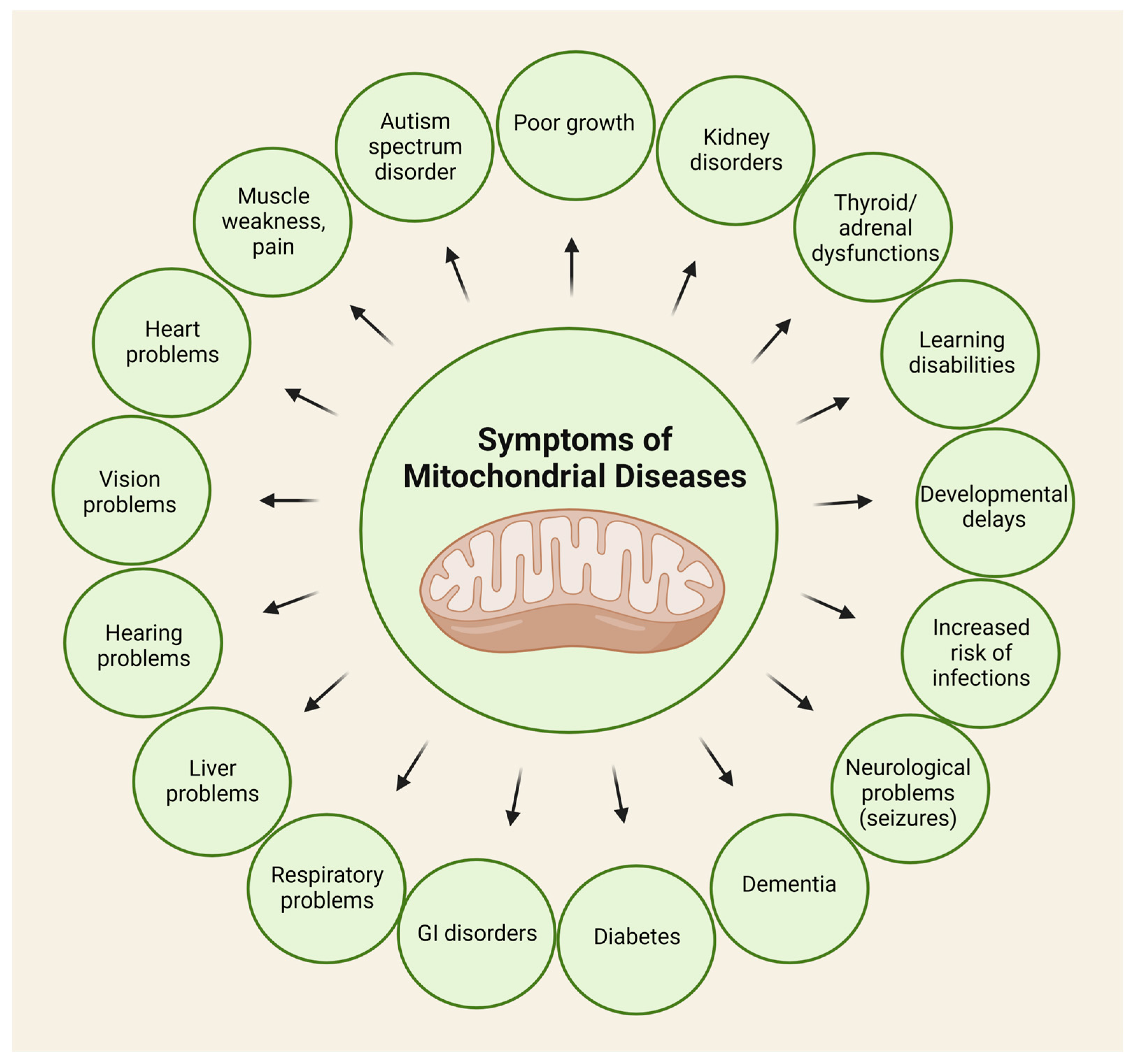
3.2. Mitochondrial Disorders and Oxidative Stress
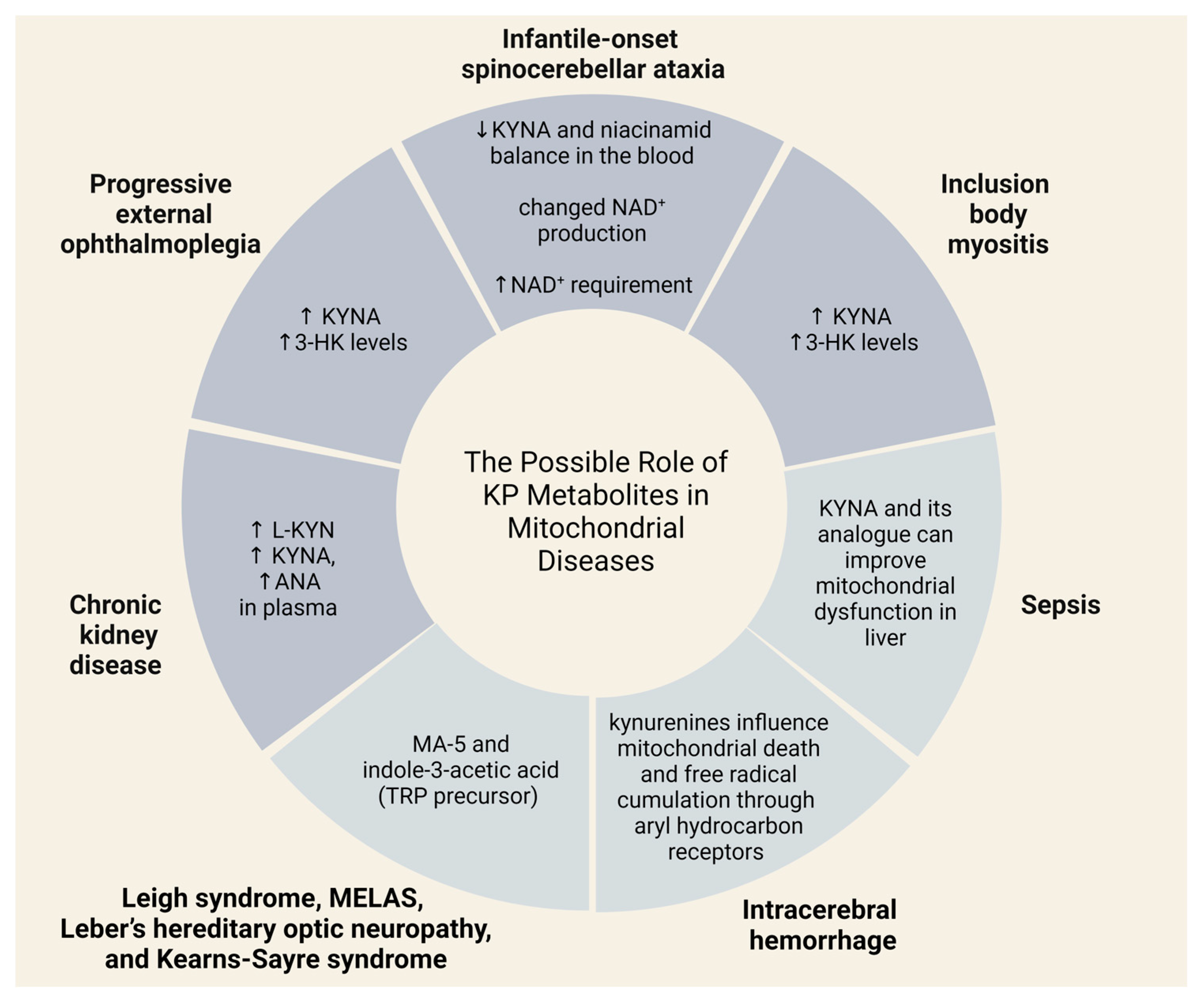
3.3. The Role of the Kynurenine Pathway in the Pathophysiology of Mitochondrial Diseases
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pires, A.S.; Sundaram, G.; Heng, B.; Krishnamurthy, S.; Brew, B.J.; Guillemin, G.J. Recent advances in clinical trials targeting the kynurenine pathway. Pharmacol. Ther. 2022, 236, 108055. [Google Scholar] [CrossRef]
- Huang, Y.S.; Ogbechi, J.; Clanchy, F.I.; Williams, R.O.; Stone, T.W. IDO and Kynurenine Metabolites in Peripheral and CNS Disorders. Front. Immunol. 2020, 11, 388. [Google Scholar] [CrossRef]
- Sas, K.; Szabó, E.; Vécsei, L. Mitochondria, Oxidative Stress and the Kynurenine System, with a Focus on Ageing and Neuroprotection. Molecules 2018, 23, 191. [Google Scholar] [CrossRef]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 2015, 26, 26050. [Google Scholar] [CrossRef] [PubMed]
- Adlerberth, I.; Wold, A.E. Establishment of the gut microbiota in Western infants. Acta Paediatr. 2009, 98, 229–238. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacology 2017, 112 Pt B, 399–412. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef]
- Knights, D.; Ward, T.L.; McKinlay, C.E.; Miller, H.; Gonzalez, A.; McDonald, D.; Knight, R. Rethinking “enterotypes”. Cell Host Microbe 2014, 16, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Sharon, G.; Sampson, T.R.; Geschwind, D.H.; Mazmanian, S.K. The Central Nervous System and the Gut Microbiome. Cell 2016, 167, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Awasthi, A.; Singh, S. Altered gut microbiota and intestinal permeability in Parkinson’s disease: Pathological highlight to management. Neurosci. Lett. 2019, 712, 134516. [Google Scholar] [CrossRef]
- Wang, Y.; Yuan, X.; Kang, Y.; Song, X. Tryptophan-kynurenine pathway as a novel link between gut microbiota and schizophrenia: A review. Trop. J. Pharm. Res. 2019, 18, 897–905. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef]
- Anand, N.; Gorantla, V.R.; Chidambaram, S.B. The Role of Gut Dysbiosis in the Pathophysiology of Neuropsychiatric Disorders. Cells 2022, 12, 54. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef] [PubMed]
- de Lau, L.M.; Bornebroek, M.; Witteman, J.C.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Dietary fatty acids and the risk of Parkinson disease: The Rotterdam study. Neurology 2005, 64, 2040–2045. [Google Scholar] [CrossRef]
- Ieraci, A.; Beggiato, S.; Ferraro, L.; Barbieri, S.S.; Popoli, M. Kynurenine pathway is altered in BDNF Val66Met knock-in mice: Effect of physical exercise. Brain Behav. Immun. 2020, 89, 440–450. [Google Scholar] [CrossRef]
- Maqsood, R.; Stone, T.W. The Gut-Brain Axis, BDNF, NMDA and CNS Disorders. Neurochem. Res. 2016, 41, 2819–2835. [Google Scholar] [CrossRef]
- Legan, T.B.; Lavoie, B.; Norberg, E.; Ley, I.C.; Tack, S.; Tompkins, T.A.; Wargo, M.J.; Mawe, G.M. Tryptophan-synthesizing bacteria enhance colonic motility. Neurogastroenterol. Motil. 2023, 35, e14629. [Google Scholar] [CrossRef] [PubMed]
- Liebig, J. Uber Kynurensäure. Ann. Chem. 1853, 86, 125–126. [Google Scholar] [CrossRef]
- Hirai, K.; Kuroyanagi, H.; Tatebayashi, Y.; Hayashi, Y.; Hirabayashi-Takahashi, K.; Saito, K.; Haga, S.; Uemura, T.; Izumi, S. Dual role of the carboxyl-terminal region of pig liver l-kynurenine 3-monooxygenase: Mitochondrial-targeting signal and enzymatic activity. J. Biochem. 2010, 148, 639–650. [Google Scholar] [CrossRef]
- Castro-Portuguez, R.; Sutphin, G.L. Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 2020, 132, 110841. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smythe, G.A.; Veas, L.A.; Takikawa, O.; Brew, B.J. A beta 1-42 induces production of quinolinic acid by human macrophages and microglia. Neuroreport 2003, 14, 2311–2315. [Google Scholar] [CrossRef] [PubMed]
- Behan, W.M.H.; McDonald, M.; Darlington, L.G.; Stone, T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: Protection by melatonin and deprenyl. Br. J. Pharmacol. 1999, 128, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Grócz, G.; Zádor, F.; Dvorácskó, S.; Bohár, Z.; Benyhe, S.; Tömböly, C.; Párdutz, Á.; Vécsei, L. Interactions between the kynurenine and the endocannabinoid system with special emphasis on migraine. Int. J. Mol. Sci. 2017, 18, 1617. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Fernández-Gomez, F.J.; Braidy, N.; Estrada, C.; Costa, C.; Costa, S.; Bessede, A.; Fernandez-Villalba, E.; Zinger, A.; Herrero, M.T.; et al. Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2017, 155, 76–95. [Google Scholar] [CrossRef]
- Zádor, F.; Nagy-Grócz, G.; Kekesi, G.; Dvorácskó, S.; Szűcs, E.; Tömböly, C.; Horvath, G.; Benyhe, S.; Vécsei, L. Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions. Molecules 2019, 24, 3709. [Google Scholar] [CrossRef]
- González Esquivel, D.; Ramírez-Ortega, D.; Pineda, B.; Castro, N.; Ríos, C.; Pérez de la Cruz, V. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 2017, 112 Pt B, 331–345. [Google Scholar] [CrossRef]
- Baran, H.; Staniek, K.; Bertignol-Spörr, M.; Attam, M.; Kronsteiner, C.; Kepplinger, B. Effects of Various Kynurenine Metabolites on Respiratory Parameters of Rat Brain, Liver and Heart Mitochondria. Int. J. Tryptophan Res. 2016, 9, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Maddison, D.C.; Alfonso-Núñez, M.; Swaih, A.M.; Breda, C.; Campesan, S.; Allcock, N.; Straatman-Iwanowska, A.; Kyriacou, C.P.; Giorgini, F. A novel role for kynurenine 3-monooxygenase in mitochondrial dynamics. PLoS Genet. 2020, 16, e1009129. [Google Scholar] [CrossRef]
- Birch, P.J.; Grossman, C.J.; Hayes, A.G. Kynurenic acid antagonises responses to NMDA via an action at the strychnine-insensitive glycine receptor. Eur. J. Pharmacol. 1988, 154, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A Glycine Site Associated with N-Methyl-d-Aspartic Acid Receptors: Characterization and Identification of a New Class of Antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rózsa, E.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef]
- Bessede, A.; Gargaro, M.; Pallotta, M.T.; Matino, D.; Servillo, G.; Brunacci, C.; Bicciato, S.; Mazza, E.M.; Macchiarulo, A.; Vacca, C.; et al. Aryl hydrocarbon receptor control of a disease tolerance defence pathway. Nature 2014, 511, 184–190. [Google Scholar] [CrossRef]
- Jaronen, M.; Quintana, F.J. Immunological Relevance of the Coevolution of IDO1 and AHR. Front. Immunol. 2014, 5, 521. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Appelman, B.; Mooij-Kalverda, K.; Houtkooper, R.H.; van Weeghel, M.; Vaz, F.M.; Dijkhuis, A.; Dekker, T.; Smids, B.S.; Duitman, J.W.; et al. COVID-19 Biobank study Group. Prolonged indoleamine 2,3-dioxygenase-2 activity and associated cellular stress in post-acute sequelae of SARS-CoV-2 infection. EBioMedicine 2023, 94, 104729. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [PubMed]
- Spekker, E.; Nagy-Grócz, G. All Roads Lead to the Gut: The Importance of the Microbiota and Diet in Migraine. Neurol. Int. 2023, 15, 1174–1190. [Google Scholar] [CrossRef]
- Cronin, P.; Joyce, S.A.; O’Toole, P.W.; O’Connor, E.M. Dietary Fibre Modulates the Gut Microbiota. Nutrients 2021, 13, 1655. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms’ Footprint in Neurodegenerative Diseases. Front. Cell Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673. [Google Scholar] [CrossRef]
- Desbonnet, L.; Clarke, G.; Traplin, A.; O’Sullivan, O.; Crispie, F.; Moloney, R.D.; Cotter, P.D.; Dinan, T.G.; Cryan, J.F. Gut microbiota depletion from early adolescence in mice: Implications for brain and behaviour. Brain Behav. Immun. 2015, 48, 165–173. [Google Scholar] [CrossRef]
- Sun, P.; Wang, M.; Liu, Y.X.; Li, L.; Chai, X.; Zheng, W.; Chen, S.; Zhu, X.; Zhao, S. High-fat diet-disturbed gut microbiota-colonocyte interactions contribute to dysregulating peripheral tryptophan-kynurenine metabolism. Microbiome 2023, 11, 154. [Google Scholar] [CrossRef]
- Desbonnet, L.; Garrett, L.; Clarke, G.; Bienenstock, J.; Dinan, T.G. The probiotic Bifidobacteria infantis: An assessment of potential antidepressant properties in the rat. J. Psychiatr. Res. 2008, 43, 164–174. [Google Scholar] [CrossRef]
- Kennedy, P.J.; Cryan, J.F.; Dinan, T.G.; Clarke, G. Irritable bowel syndrome: A microbiome-gut-brain axis disorder? World J. Gastroenterol. 2014, 20, 14105–14125. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; McKernan, D.P.; Gaszner, G.; Quigley, E.M.; Cryan, J.F.; Dinan, T.G. A Distinct Profile of Tryptophan Metabolism along the Kynurenine Pathway Downstream of Toll-Like Receptor Activation in Irritable Bowel Syndrome. Front. Pharmacol. 2012, 3, 90. [Google Scholar] [CrossRef]
- Fitzgerald, P.; Cassidy, E.M.; Clarke, G.; Scully, P.; Barry, S.; Quigley, E.M.M.; Shanahan, F.; Cryan, J.; Dinan, T.G. Tryptophan catabolism in females with irritable bowel syndrome: Relationship to interferon-gamma, severity of symptoms and psychiatric co-morbidity. Neurogastroenterol. Motil. 2008, 20, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Bernstein, C.N. Treatment of IBD: Where we are and where we are going. Am. J. Gastroenterol. 2015, 110, 114–126. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Zhang, S.; Qi, C.; Zhang, Z.; Ma, R.; Xiang, L.; Chen, L.; Zhu, Y.; Tang, C.; et al. Oxidative stress gene expression, DNA methylation, and gut microbiota interaction trigger Crohn’s disease: A multi-omics Mendelian randomization study. BMC Med. 2023, 21, 179. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Du, Y.; Li, C.; Zhang, H.; Lai, W.; Li, S.; Ye, Z.; Fu, W.; Li, S.; Li, X.G.; et al. Association between metabolites in tryptophan-kynurenine pathway and inflammatory bowel disease: A two-sample Mendelian randomization. Sci. Rep. 2024, 14, 201. [Google Scholar] [CrossRef]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update: Recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef]
- Kerrison, J.B.; Miller, N.R.; Hsu, F.; Beaty, T.H.; Maumenee, I.H.; Smith, K.H.; Savino, P.J.; Stone, E.M.; Newman, N.J. A case-control study of tobacco and alcohol consumption in Leber hereditary optic neuropathy. Am. J. Ophthalmol. 2000, 130, 803–812. [Google Scholar] [CrossRef]
- Giordano, L.; Deceglie, S.; d’Adamo, P.; Valentino, M.L.; La Morgia, C.; Fracasso, F.; Roberti, M.; Cappellari, M.; Petrosillo, G.; Ciaravolo, S.; et al. Cigarette toxicity triggers Leber’s hereditary optic neuropathy by affecting mtDNA copy number, oxidative phosphorylation and ROS detoxification pathways. Cell Death Dis. 2015, 6, e2021. [Google Scholar] [CrossRef]
- Hwang, J.M.; Park, H.W. Carbon monoxide poisoning as an epigenetic factor for Leber’s hereditary optic neuropathy. Korean J. Ophthalmol. 1996, 10, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Ta, C.; Basham, A.A.; Mansour, S. Leber hereditary optic neuropathy associated with antiretroviral therapy for human immunodeficiency virus infection. Am. J. Ophthalmol. 2001, 131, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Jeyakumar, A. Genetic susceptibility to aminoglycoside ototoxicity. Int. J. Pediatr. Otorhinolaryngol. 2019, 120, 15–19. [Google Scholar] [CrossRef]
- Klopstock, T.; Priglinger, C.; Yilmaz, A.; Kornblum, C.; Distelmaier, F.; Prokisch, H. Mitochondrial Disorders. Dtsch. Arztebl. Int. 2021, 118, 741–748. [Google Scholar] [CrossRef]
- Luft, R. The development of mitochondrial medicine. Proc. Natl. Acad. Sci. USA 1994, 91, 8731–8738. [Google Scholar] [CrossRef]
- Koenig, M.K. Presentation and diagnosis of mitochondrial disorders in children. Pediatr. Neurol. 2008, 38, 305–313. [Google Scholar] [CrossRef]
- Parikh, S.; Karaa, A.; Goldstein, A.; Bertini, E.S.; Chinnery, P.F.; Christodoulou, J.; Cohen, B.H.; Davis, R.L.; Falk, M.J.; Fratter, C.; et al. Diagnosis of ‘possible’ mitochondrial disease: An existential crisis. J. Med. Genet. 2019, 56, 123–130. [Google Scholar] [CrossRef]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Anupam, K.; Kaushal, J.; Prabhakar, N.; Bhatnagar, A. Effect of redox status of peripheral blood on immune signature of circulating regulatory and cytotoxic T cells in streptozotocin induced rodent model of type I diabetes. Immunobiology 2018, 223, 586–597. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Bayrami, G.; Alihemmati, A.; Karimi, P.; Javadi, A.; Keyhanmanesh, R.; Mohammadi, M.; Zadi-Heydarabad, M.; Badalzadeh, R. Combination of Vildagliptin and Ischemic Postconditioning in Diabetic Hearts as a Working Strategy to Reduce Myocardial Reperfusion Injury by Restoring Mitochondrial Function and Autophagic Activity. Adv. Pharm. Bull. 2018, 8, 319–329. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Wang, J.; Chen, P.; Cao, Q.; Wang, W.; Chang, X. Traditional Chinese Medicine Ginseng Dingzhi Decoction Ameliorates Myocardial Fibrosis and High Glucose-Induced Cardiomyocyte Injury by Regulating Intestinal Flora and Mitochondrial Dysfunction. Oxid. Med. Cell Longev. 2022, 2022, 9205908. [Google Scholar] [CrossRef] [PubMed]
- Novak, E.A.; Mollen, K.P. Mitochondrial dysfunction in inflammatory bowel disease. Front. Cell Dev. Biol. 2015, 3, 62. [Google Scholar] [CrossRef]
- Kłos, P.; Dabravolski, S.A. The Role of Mitochondria Dysfunction in Inflammatory Bowel Diseases and Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 11673. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Parrott, J.M.; O’Connor, J.C. Kynurenine 3-Monooxygenase: An Influential Mediator of Neuropathology. Front. Psychiatry 2015, 6, 116. [Google Scholar] [CrossRef]
- Skorobogatov, K.; De Picker, L.; Verkerk, R.; Coppens, V.; Leboyer, M.; Müller, N.; Morrens, M. Brain Versus Blood: A Systematic Review on the Concordance Between Peripheral and Central Kynurenine Pathway Measures in Psychiatric Disorders. Front. Immunol. 2021, 12, 716980. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Maldonado, P.D.; Santamaría, A. 3-Hydroxykynurenine: An intriguing molecule exerting dual actions in the central nervous system. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef]
- García-Lara, L.; Pérez-Severiano, F.; González-Esquivel, D.; Elizondo, G.; Segovia, J. Absence of aryl hydrocarbon receptors increases endogenous kynurenic acid levels and protects mouse brain against excitotoxic insult and oxidative stress. J. Neurosci. Res. 2015, 93, 1423–1433. [Google Scholar] [CrossRef]
- Bryleva, E.Y.; Brundin, L. Kynurenine pathway metabolites and suicidality. Neuropharmacology 2017, 112 Pt B, 324–330. [Google Scholar] [CrossRef]
- Mishra, J.; Kumar, A. Improvement of mitochondrial function by paliperidone attenuates quinolinic acid-induced behavioural and neurochemical alterations in rats: Implications in Huntington’s disease. Neurotox. Res. 2014, 26, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, Q.; Gao, W.; Sehgal, S.A.; Wu, H. The multifaceted regulation of mitophagy by endogenous metabolites. Autophagy 2022, 18, 1216–1239. [Google Scholar] [CrossRef]
- Marzetti, E.; Csiszar, A.; Dutta, D.; Balagopal, G.; Calvani, R.; Leeuwenburgh, C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: From mechanisms to therapeutics. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H459–H476. [Google Scholar] [CrossRef]
- Ivatt, R.M.; Sanchez-Martinez, A.; Godena, V.K.; Brown, S.; Ziviani, E.; Whitworth, A.J. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Thompson Legault, J.; Strittmatter, L.; Tardif, J.; Sharma, R.; Tremblay-Vaillancourt, V.; Aubut, C.; Boucher, G.; Clish, C.B.; Cyr, D.; Daneault, C.; et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep. 2015, 13, 981–989. [Google Scholar] [CrossRef]
- Rozen, T.D. Can the effects of the mitochondrial DNA mutations found in Leber’s hereditary optic neuropathy be protective against the development of cluster headache in smokers? Cephalalgia Rep. 2020, 3. [Google Scholar] [CrossRef]
- Arbatova, J.; D’Amato, E.; Vaarmann, A.; Zharkovsky, A.; Reeben, M. Reduced serotonin and 3-hydroxyanthranilic acid levels in serum of cystatin B-deficient mice, a model system for progressive myoclonus epilepsy. Epilepsia 2005, 46 (Suppl. S5), 49–51. [Google Scholar] [CrossRef]
- Buzkova, J.; Nikkanen, J.; Ahola, S.; Hakonen, A.H.; Sevastianova, K.; Hovinen, T.; Yki-Järvinen, H.; Pietiläinen, K.H.; Lönnqvist, T.; Velagapudi, V.; et al. Metabolomes of mitochondrial diseases and inclusion body myositis patients: Treatment targets and biomarkers. EMBO Mol. Med. 2018, 10, e9091. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, K.; Tankiewicz, J.; Mysliwiec, M.; Pawlak, D. Tissue factor/its pathway inhibitor system and kynurenines in chronic kidney disease patients on conservative treatment. Blood Coagul. Fibrinolysis 2009, 20, 590–594. [Google Scholar] [CrossRef]
- De Paepe, B. Sporadic Inclusion Body Myositis: An Acquired Mitochondrial Disease with Extras. Biomolecules 2019, 9, 15. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Trushina, E.; McMurray, C.T. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Solvang, S.H.; Nordrehaug, J.E.; Aarsland, D.; Lange, J.; Ueland, P.M.; McCann, A.; Midttun, Ø.; Tell, G.S.; Giil, L.M. Kynurenines, Neuropsychiatric Symptoms, and Cognitive Prognosis in Patients with Mild Dementia. Int. J. Tryptophan Res. 2019, 12, 1178646919877883. [Google Scholar] [CrossRef]
- Boros, F.A.; Vécsei, L. Immunomodulatory Effects of Genetic Alterations Affecting the Kynurenine Pathway. Front. Immunol. 2019, 10, 2570. [Google Scholar] [CrossRef]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef]
- Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells 2021, 10, 1603. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Saito, K.; Lackner, A.; Wiley, C.A.; Achim, C.L.; Markey, S.P. Sources of the neurotoxin quinolinic acid in the brain of HIV-1-infected patients and retrovirus-infected macaques. FASEB J. 1998, 12, 881–896. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Gekker, G.; Lokensgard, J.R.; Heyes, M.P.; Peterson, P.K. U50,488 protection against HIV-1-related neurotoxicity: Involvement of quinolinic acid suppression. Neuropharmacology 2000, 39, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Rangel-López, E.; Colín-González, A.L.; Paz-Loyola, A.L.; Pinzón, E.; Torres, I.; Serratos, I.N.; Castellanos, P.; Wajner, M.; Souza, D.O.; Santamaría, A. Cannabinoid receptor agonists reduce the short-term mitochondrial dysfunction and oxidative stress linked to excitotoxicity in the rat brain. Neuroscience 2015, 285, 97–106. [Google Scholar] [CrossRef]
- Zádor, F.; Nagy-Grócz, G.; Dvorácskó, S.; Bohár, Z.; Cseh, E.K.; Zádori, D.; Párdutz, Á.; Szűcs, E.; Tömböly, C.; Borsodi, A.; et al. Long-term systemic administration of kynurenic acid brain region specifically elevates the abundance of functional CB1 receptors in rats. Neurochem. Int. 2020, 138, 104752. [Google Scholar] [CrossRef] [PubMed]
- Juhász, L.; Rutai, A.; Fejes, R.; Tallósy, S.P.; Poles, M.Z.; Szabó, A.; Szatmári, I.; Fülöp, F.; Vécsei, L.; Boros, M.; et al. Divergent Effects of the N-Methyl-D-Aspartate Receptor Antagonist Kynurenic Acid and the Synthetic Analog SZR-72 on Microcirculatory and Mitochondrial Dysfunction in Experimental Sepsis. Front. Med. 2020, 7, 566582. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, Y.; Izumi, R.; Koide, M.; Hagiwara, Y.; Kanzaki, M.; Suzuki, N.; Kikuchi, K.; Matsuhashi, T.; Akiyama, Y.; Ichijo, M.; et al. Mitochondrial dysfunction underlying sporadic inclusion body myositis is ameliorated by the mitochondrial homing drug MA-5. PLoS ONE 2020, 15, e0231064. [Google Scholar] [CrossRef]
- Suzuki, T.; Yamaguchi, H.; Kikusato, M.; Matsuhashi, T.; Matsuo, A.; Sato, T.; Oba, Y.; Watanabe, S.; Minaki, D.; Saigusa, D.; et al. Mitochonic acid 5 (MA-5), a derivative of the plant hormone indole-3-acetic acid, improves survival of fibroblasts from patients with mitochondrial diseases. Tohoku J. Exp. Med. 2015, 236, 225–232. [Google Scholar] [CrossRef]
- Ren, R.; Fang, Y.; Sherchan, P.; Lu, Q.; Lenahan, C.; Zhang, J.H.; Zhang, J.; Tang, J. Kynurenine/Aryl Hydrocarbon Receptor Modulates Mitochondria-Mediated Oxidative Stress and Neuronal Apoptosis in Experimental Intracerebral Hemorrhage. Antioxid. Redox Signal. 2022, 37, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, K.; Beal, M.F. Neuroprotective effects of L-kynurenine on hypoxia-ischemia and NMDA lesions in neonatal rats. J. Cereb. Blood Flow Metab. 1992, 12, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Gigler, G.; Szénási, G.; Simó, A.; Lévay, G.; Hársing, L.G.; Sas, K.; Vécsei, L.; Toldi, J. Neuroprotective effect of L-kynurenine sulfate administered before focal cerebral ischemia in mice and global cerebral ischemia in gerbils. Eur. J. Pharmacol. 2007, 564, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Fejes-Szabó, A.; Bohár, Z.; Nagy-Grócz, G.; Vámos, E.; Tar, L.; Pődör, B.; Tajti, J.; Toldi, J.; Vécsei, L.; Párdutz, Á. Effect of probenecid on the pain-related behaviour and morphological markers in orofacial formalin test of the rat. CNS Neurol. Disord. Drug Targets 2015, 14, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Parli, C.J.; Krieter, P.; Schmidt, B. Metabolism of 6-chlorotryptophan to 4-chloro-3-hydroxyanthranilic acid: A potent inhibitor of 3-hydroxyanthranilic acid oxidase. Arch. Biochem. Biophys. 1980, 203, 161–166. [Google Scholar] [CrossRef]
- Luhavaya, H.; Sigrist, R.; Chekan, J.R.; McKinnie, S.M.K.; Moore, B.S. Biosynthesis of l-4-Chlorokynurenine, an Antidepressant Prodrug and a Non-Proteinogenic Amino Acid Found in Lipopeptide Antibiotics. Angew. Chem. Int. Ed. Engl. 2019, 58, 8394–8399. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy-Grócz, G.; Spekker, E.; Vécsei, L. Kynurenines, Neuronal Excitotoxicity, and Mitochondrial Oxidative Stress: Role of the Intestinal Flora. Int. J. Mol. Sci. 2024, 25, 1698. https://doi.org/10.3390/ijms25031698
Nagy-Grócz G, Spekker E, Vécsei L. Kynurenines, Neuronal Excitotoxicity, and Mitochondrial Oxidative Stress: Role of the Intestinal Flora. International Journal of Molecular Sciences. 2024; 25(3):1698. https://doi.org/10.3390/ijms25031698
Chicago/Turabian StyleNagy-Grócz, Gábor, Eleonóra Spekker, and László Vécsei. 2024. "Kynurenines, Neuronal Excitotoxicity, and Mitochondrial Oxidative Stress: Role of the Intestinal Flora" International Journal of Molecular Sciences 25, no. 3: 1698. https://doi.org/10.3390/ijms25031698
APA StyleNagy-Grócz, G., Spekker, E., & Vécsei, L. (2024). Kynurenines, Neuronal Excitotoxicity, and Mitochondrial Oxidative Stress: Role of the Intestinal Flora. International Journal of Molecular Sciences, 25(3), 1698. https://doi.org/10.3390/ijms25031698