
Charcot–Marie–Tooth Disease with Myelin Protein Zero Mutation Presenting as Painful, Predominant Small-Fiber Neuropathy


 ,
,  , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Case Study
2.2. Review of the Literature
3. Results
3.1. Case Report
3.2. Cases in the Literature
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laurá, M.; Pipis, M.; Rossor, A.M.; Reilly, M.M. Charcot-Marie-Tooth disease and related disorders: An evolving landscape. Curr. Opin. Neurol. 2019, 32, 641–650. [Google Scholar] [CrossRef]
- Pareyson, D.; Saveri, P.; Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017, 30, 471–480. [Google Scholar] [CrossRef]
- Padua, L.; Cavallaro, T.; Pareyson, D.; Quattrone, A.; Vita, G.; Schenone, A.; Italian CMT QoL Study Group. Charcot-Marie-Tooth and pain: Correlations with neurophysiological, clinical, and disability findings. Neurol. Sci. 2008, 29, 193–194. [Google Scholar] [CrossRef] [PubMed]
- Ribiere, C.; Bernardin, M.; Sacconi, S.; Delmont, E.; Fournier-Mehouas, M.; Rauscent, H.; Benchortane, M.; Staccini, P.; Lantéri-Minet, M.; Desnuelle, C. Pain assessment in Charcot-Marie-Tooth (CMT) disease. Ann. Phys. Rehabil. Med. 2012, 55, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.D.; Barnes, P.R.J.; Mills, K.R.; Bennett, D.L.H. Intermediate Charcot-Marie-Tooth disease due to a novel Trp101Stop myelin protein zero mutation associated with debilitating neuropathic pain. Pain 2012, 153, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.M.; Herrmann, D.N.; McDermott, M.P.; Scherer, S.S.; Shy, M.E.; Reilly, M.M.; Pareyson, D. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Bouhassira, D.; Attal, N.; Alchaar, H.; Boureau, F.; Brochet, B.; Bruxelle, J.; Cunin, G.; Fermanian, J.; Ginies, P.; Grun-Overdyking, A.; et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain 2005, 114, 29–36. [Google Scholar] [CrossRef]
- Bouhassira, D.; Attal, N.; Fermanian, J.; Alchaar, H.; Gautron, M.; Masquelier, E.; Rostaing, S.; Lanteri-Minet, M.; Collin, E.; Grisart, J.; et al. Development and validation of the Neuropathic Pain Symptom Inventory. Pain 2004, 108, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Padua, L.; Briani, C.; Jann, S.; Nobile-Orazio, E.; Pazzaglia, C.; Morini, A.; Mondelli, M.; Ciaramitaro, P.; Cavaletti, G.; Cocito, D.; et al. Validation of the Italian version of the Neuropathic Pain Symptom Inventory in peripheral nervous system diseases. Neurol. Sci. 2009, 30, 99–106. [Google Scholar] [CrossRef]
- Brouwer, B.A.; Bakkers, M.; Hoeijmakers, J.G.; Faber, C.G.; Merkies, I.S. Improving assessment in small fiber neuropathy. J. Peripher. Nerv. Syst. 2015, 20, 333–340. [Google Scholar] [CrossRef]
- Lauria, G.; Hsieh, S.T.; Johansson, O.; Kennedy, W.R.; Leger, J.M.; Mellgren, S.I.; Nolano, M.; Merkies, I.S.J.; Polydefkis, M.; European Federation of Neurological Societies; et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur. J. Neurol. 2010, 17, 903-e49. [Google Scholar]
- Saporta, A.S.; Sottile, S.L.; Miller, L.J.; Feely, S.M.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 2011, 69, 22–33. [Google Scholar] [CrossRef]
- Galosi, E.; Falco, P.; Di Pietro, G.; Leone, C.; Esposito, N.; De Stefano, G.; Di Stefano, G.; Truini, A. The diagnostic accuracy of the small fiber neuropathy symptoms inventory questionnaire (SFN-SIQ) for identifying pure small fiber neuropathy. J. Peripher. Nerv. Syst. 2022, 27, 283–290. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; ACMG Laboratory Quality Assurance Committee; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- De Jonghe, P.; Timmerman, V.; Ceuterick, C.; Nelis, E.; De Vriendt, E.; Löfgren, A.; Vercruyssen, A.; Verellen, C.; Van Maldergem, L.; Martin, J.-J.; et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain 1999, 122, 281–290. [Google Scholar] [CrossRef]
- Lagueny, A.; Latour, P.; Vital, A.; Le Masson, G.; Rouanet, M.; Ferrer, X.; Vital, C.; Vandenberghe, A. Mild recurrent neuropathy in CMT1B with a novel nonsense mutation in the extracellular domain of the MPZ gene. J. Neurol. Neurosurg. Psychiatry 2001, 70, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Hanemann, C.O.; Gabreëls-Festen, A.A.; De Jonghe, P. Axon damage in CMT due to mutation in myelin protein P0. Neuromuscul. Disord. 2001, 11, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Züchner, S.; Siska, E.; Mechler, F.; Molnar, M.J. Marked phenotypic variation in a family with a new myelin protein zero mutation. Neuromuscul. Disord. 2005, 15, 760–763. [Google Scholar] [CrossRef]
- Burns, T.M.; Phillips, L.H., 2nd; Dimberg, E.L.; Vaught, B.K.; Klein, C.J. Novel myelin protein zero mutation (Arg36Trp) in a patient with acute onset painful neuropathy. Neuromuscul. Disord. 2006, 16, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Kilfoyle, D.H.; Dyck, P.J.; Wu, Y.; Litchy, W.J.; Klein, D.M.; Dyck, P.J.B.; Kumar, N.; Cunningham, J.M.; Klein, C.J. Myelin protein zero mutation His39Pro: Hereditary motor and sensory neuropathy with variable onset, hearing loss, restless legs and multiple sclerosis. J. Neurol. Neurosurg. Psychiatry. 2006, 77, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Mazzeo, A.; Muglia, M.; Rodolico, C.; Toscano, A.; Patitucci, A.; Quattrone, A.; Messina, C.; Vita, G. Charcot-Marie-Tooth disease type 1B: Marked phenotypic variation of the Ser78Leu mutation in five Italian families. Acta Neurol. Scand. 2008, 118, 328–332. [Google Scholar] [CrossRef]
- Schneider-Gold, C.; Kötting, J.; Epplen, J.T.; Gold, R.; Gerding, W.M. Unusual Charcot-Marie-Tooth phenotype due to a mutation within the intracellular domain of myelin protein zero. Muscle Nerve 2010, 41, 550–554. [Google Scholar] [CrossRef]
- Corrado, L.; Magri, S.; Bagarotti, A.; Carecchio, M.; Piscosquito, G.; Pareyson, D.; Varrasi, C.; Vecchio, D.; Zonta, A.; Cantello, R.; et al. A novel synonymous mutation in the MPZ gene causing an aberrant splicing pattern and Charcot-Marie-Tooth disease type 1b. Neuromuscul. Disord. 2016, 26, 516–520. [Google Scholar] [CrossRef]
- Subréville, M.; Bonello-Palot, N.; Yahiaoui, D.; Beloribi-Djefaflia, S.; Fernandes, S.; Stojkovic, T.; Cassereau, J.; Péréon, Y.; Echaniz-Laguna, A.; Violleau, M.; et al. Genotype-phenotype correlation in French patients with myelin protein zero gene-related inherited neuropathy. Eur. J. Neurol. 2021, 28, 2913–2921. [Google Scholar] [CrossRef]
- D’Urso, D.; Brophy, P.J.; Staugaitis, S.M.; Gillespie, C.S.; Frey, A.B.; Stempak, J.G.; Colman, D.R. Protein zero of peripheral nerve myelin: Biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron 1990, 4, 449–460. [Google Scholar] [CrossRef]
- Grandis, M.; Vigo, T.; Passalacqua, M.; Jain, M.; Scazzola, S.; La Padula, V.; Brucal, M.; Benvenuto, F.; Nobbio, L.; Cadoni, A.; et al. Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations. Hum. Mol. Genet. 2008, 17, 1877–1889. [Google Scholar] [CrossRef]
- Sanmaneechai, O.; Feely, S.; Scherer, S.S.; Herrmann, D.N.; Burns, J.; Muntoni, F.; Li, J.; Siskind, C.E.; Day, J.W.; Inherited Neuropathies Consortium—Rare Disease Clinical Research Consortium (INC-RDCRC); et al. Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain 2015, 138, 3180–3192. [Google Scholar] [CrossRef]
- Callegari, I.; Gemelli, C.; Geroldi, A.; Veneri, F.; Mandich, P.; D’antonio, M.; Pareyson, D.; Shy, M.E.; Schenone, A.; Prada, V.; et al. Mutation update for myelin protein zero-related neuropathies and the increasing role of variants causing a late-onset phenotype. J. Neurol. 2019, 266, 2629–2645. [Google Scholar] [CrossRef] [PubMed]
- Shy, M.E.; Jáni, A.; Krajewski, K.; Grandis, M.; Lewis, R.A.; Li, J.; Shy, R.R.; Balsamo, J.; Lilien, J.; Garbern, J.Y.; et al. Phenotypic clustering in MPZ mutations. Brain 2004, 127, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Magot, A.; Latour, P.; Mussini, J.M.; Mourtada, R.; Guiheneuc, P.; Pereon, Y. A new MPZ mutation associated with a mild CMT1 phenotype presenting with recurrent nerve compression. Muscle Nerve 2008, 38, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, N.F.; Kumar, K.R.; Ng, K. Myelin protein zero-related autosomal dominant peripheral neuropathy presenting as hereditary neuropathy with liability to pressure palsies. Muscle Nerve 2023, 68, E34–E36. [Google Scholar] [CrossRef]
- Howard, P.; Feely, S.M.E.; Grider, T.; Bacha, A.; Scarlato, M.; Fazio, R.; Quattrini, A.; Shy, M.E.; Previtali, S.C. Loss of function MPZ mutation causes milder CMT1B neuropathy. J. Peripher. Nerv. Syst. 2021, 26, 177–183. [Google Scholar] [CrossRef]
- Karam, C.; Scelsa, S.N. Clinical Reasoning: A 34-year-old woman with recurrent bouts of acral paresthesias. Neurology 2010, 74, 775–778. [Google Scholar] [CrossRef]
- Taniguchi, T.; Ando, M.; Okamoto, Y.; Yoshimura, A.; Higuchi, Y.; Hashiguchi, A.; Shiga, K.; Hayashida, A.; Hatano, T.; Ishiura, H.; et al. Genetic spectrum of Charcot-Marie-Tooth disease associated with myelin protein zero gene variants in Japan. Clin. Genet. 2021, 99, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Fridman, V.; Sillau, S.; Bockhorst, J.; Smith, K.; Moroni, I.; Pagliano, E.; Pisciotta, C.; Piscosquito, G.; Laurá, M.; Inherited Neuropathies Consortium-Rare Diseases Clinical Research Network; et al. Disease Progression in Charcot-Marie-Tooth Disease Related to MPZ Mutations: A Longitudinal Study. Ann. Neurol. 2023, 93, 563–576. [Google Scholar] [CrossRef]
- Lindeboom, R.G.H.; Vermeulen, M.; Lehner, B.; Supek, F. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 2019, 51, 1645–1651. [Google Scholar] [CrossRef]
- Bremer, J.; Meinhardt, A.; Katona, I.; Senderek, J.; Kämmerer-Gassler, E.K.; Roos, A.; Ferbert, A.; Schröder, J.M.; Nikolin, S.; Nolte, K.; et al. Myelin protein zero mutation-related hereditary neuropathies: Neuropathological insight from a new nerve biopsy cohort. Brain Pathol. 2024, 34, e13200. [Google Scholar] [CrossRef] [PubMed]
- Senderek, J.; Ramaekers, V.T.; Zerres, K.; Rudnik-Schöneborn, S.; Schröder, J.M.; Bergmann, C. Phenotypic variation of a novel nonsense mutation in the P0 intracellular domain. J. Neurol. Sci. 2001, 192, 49–51. [Google Scholar] [CrossRef]
- Marques, W.; Hanna, M.G., Jr.; Marques, S.R.; Sweeney, M.G.; Thomas, P.K.; Wood, N.W. Phenotypic variation of a new P0 mutation in genetically identical twins. J. Neurol. 1999, 246, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Gemignani, F. Restless legs syndrome from the spinal cord perspective: A flexor reflex circuitopathy? J. Sleep. Res. 2018, 27, e12704. [Google Scholar] [CrossRef]
- Carenini, S.; Mäurer, M.; Werner, A.; Blazyca, H.; Toyka, K.V.; Schmid, C.D.; Raivich, G.; Martini, R. The role of macrophages in demyelinating peripheral nervous system of mice heterozygously deficient in p0. J. Cell Biol. 2001, 152, 301–308. [Google Scholar] [CrossRef]
- Shy, M.E.; Arroyo, E.; Sladky, J.; Menichella, D.; Jiang, H.; Xu, W.; Kamholz, J.; Scherer, S.S. Heterozygous P0 knockout mice develop a peripheral neuropathy that resembles chronic inflammatory demyelinating polyneuropathy (CIDP). J. Neuropathol. Exp. Neurol. 1997, 56, 811–821. [Google Scholar] [CrossRef]
- Dacci, P.; Taroni, F.; Bella, E.D.; Milani, M.; Pareyson, D.; Morbin, M.; Lauria, G. Myelin protein zero Arg36Gly mutation with very late onset and rapidly progressive painful neuropathy. J. Peripher. Nerv. Syst. 2012, 17, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, M.; Sisodiya, S.M.; Kennett, R.; McDonald, B.; Haites, N.; Bell, C. Steroid responsive polyneuropathy in a family with a novel myelin protein zero mutation. J. Neurol. Neurosurg. Psychiatry 2000, 69, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Yamamoto, N.; Ohkoshi, N.; Nagata, H.; Kohno, Y.; Hayashi, A.; Tamaoka, A.; Shoji, S. Corticosteroid- responsive asymmetric neuropathy with a myelin protein zero gene mutation. Neurology 2002, 59, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Kong, X.; Lu, Z.; Wang, L.; Zheng, C. A Rare Phenotype of Uncommon Charcot-Marie-Tooth Genotypes Complicated with Inflammation Evaluated by Genetics and Magnetic Resonance Neurography. Front. Genet. 2022, 13, 873641. [Google Scholar] [CrossRef] [PubMed]
- Abbott, M.G.; Allawi, Z.; Hofer, M.; Ansorge, O.; Brady, S.; Fadic, R.; Torres, G.; Knight, R.; Calvo, M.; Bennett, D.L.; et al. Acute small fiber neuropathy after Oxford-AstraZeneca ChAdOx1-S vaccination: A report of three cases and review of the literature. J. Peripher. Nerv. Syst. 2022, 27, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Small fiber neuropathy as a complication of SARS-CoV-2 vaccinations. J. Family Med. Prim. Care 2022, 11, 4071–4073. [Google Scholar] [CrossRef] [PubMed]
- Watad, A.; De Marco, G.; Mahajna, H.; Druyan, A.; Eltity, M.; Hijazi, N.; Haddad, A.; Elias, M.; Zisman, D.; Naffaa, M.E.; et al. Immune-Mediated Disease Flares or New-Onset Disease in 27 Subjects Following mRNA/DNA SARS-CoV-2 Vaccination. Vaccines 2021, 9, 435. [Google Scholar] [CrossRef]



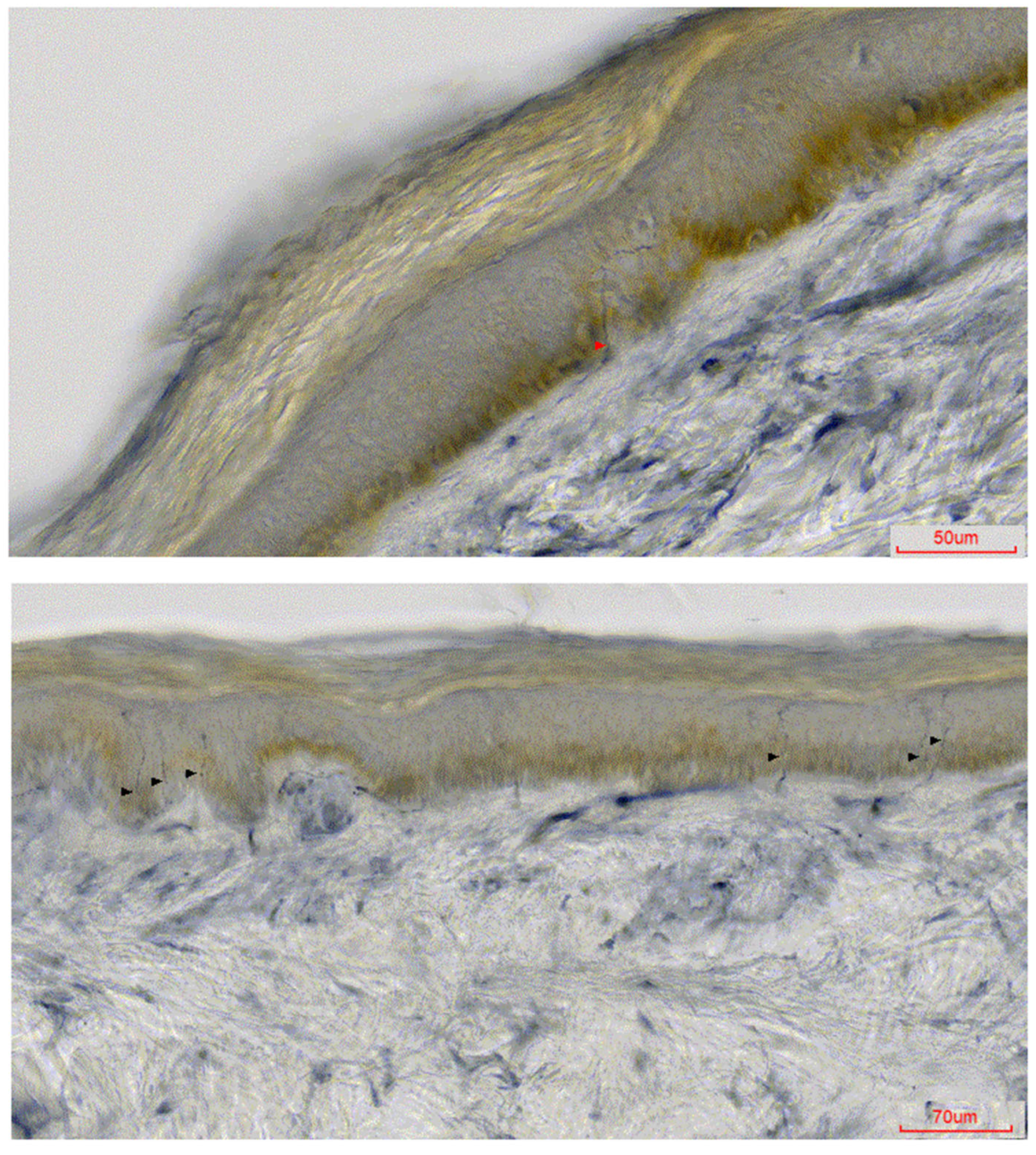{kind=link}
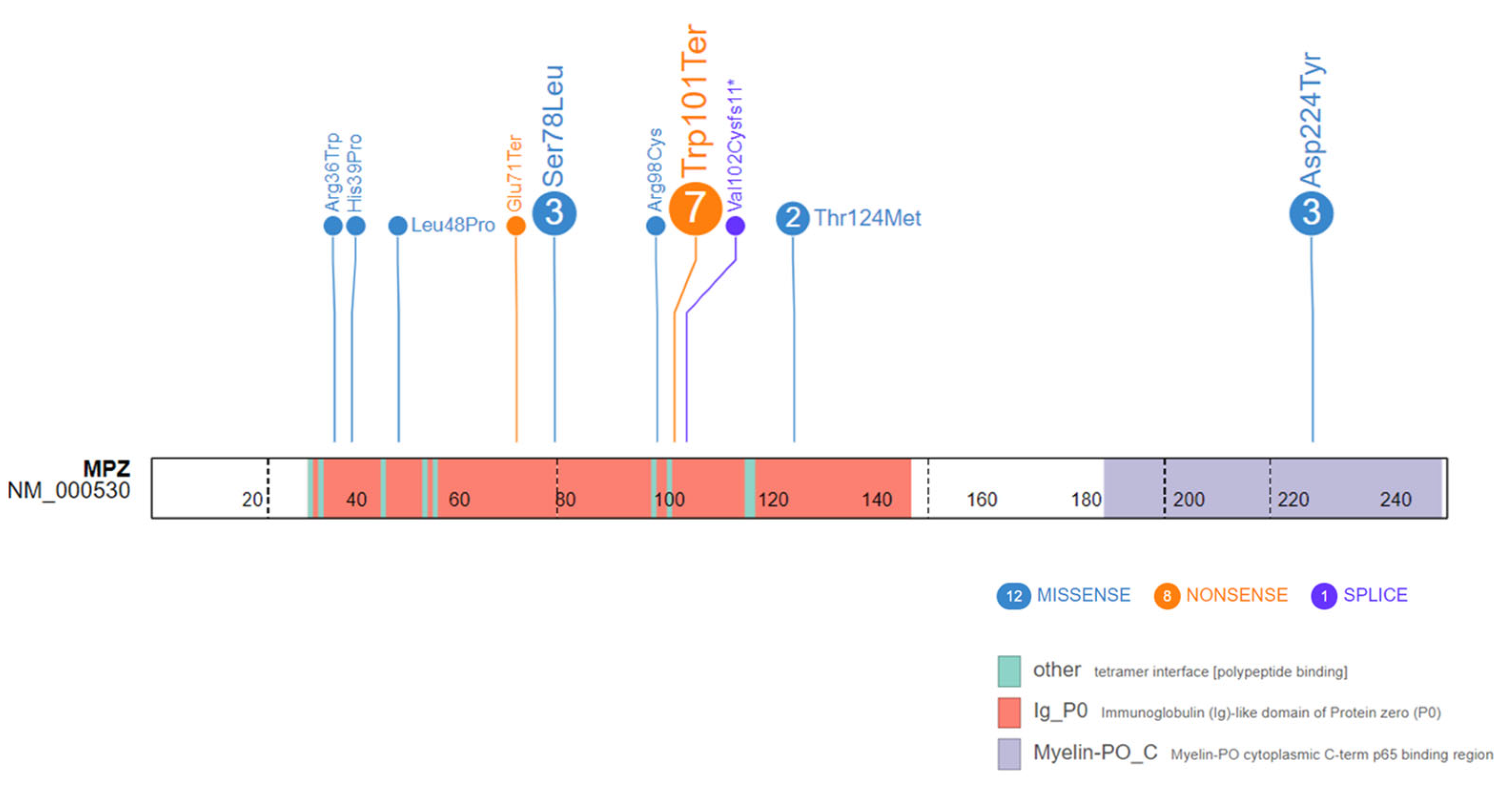{kind=link}
| Ref | Patient | Age at Onset | Pain Features | Distribution | Motor Signs | Associated Features | NCV | Nerve/Skin * Biopsy | Treatment | Family History | MPZ Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| [15] | PN-54 III.1 M | 4th dec | Pains and paraesthesias | Lower limbs | Slight weakness of peroneal muscles | No | Axon | ND | NR | AD variable severity, hearing loss | Thr124Met c.371C>T |
| [16] | W 36 y | 36 y | Burning—recurrent | Soles to lower legs | No weakness | No | Interm | De/remyelination, axonal loss, uncompacted myelin lamellae | NR | AD Father asymptomatic | Glu71ter c.211G>T |
| [17] | Index M 48 y | 39 y | Dysesthesia | Feet | Normal | Unreactive pupils | Axon | Marked fiber loss, clusters | NR | AD Dysesthesia in 3, motor impairment variable | Thr124Met c.371C>T |
| [18] | Pt2 W 26 y | 18 y | Pain | Feet and legs | No weakness | No | Interm | ND | NR | AD marked phenotypic variations in 5 members | Leu48Pro c.143T>C |
| [19] | M 47 y | 47 y | Acute lancinating pain, recurrent | Arms and dorsal forearms, thighs and legs, to buttocks and face. | Normal strength | No | Interm | ND | IVIG ineffective | AD Mother paucisymptomatic | Arg36Trp c.106A>T |
| [20] | IV-103 W 30 y | 30 y | Acute onset, severe burning and shock-like pain | More in the distal feet than in the hands | No | No | Interm | Normal myelinated fibre density | Pain management NS | AD- RLS in 8 of 10, hearing loss 7; asymptomatic 4 | His39Pro c.116A>C |
| [21] | II-1 M 50 y | 28 y | Severe cramps and painful paresthesia | NR | Mild weakness | No | Dem | ND | NR | AD family 2, all patients with cramps and painful paresthesia | Ser78Leu c.233C>T |
| II-4 M 46 y | 26 y | Severe cramps and painful paresthesia | NR | Moderate weakness | No | Dem | ND | NR | Ser78Leu c.233C>T | ||
| III-3 M 11 y | 8 y | Severe cramps and painful paresthesia | NR | Mild weakness | No | Dem | ND | NR | Ser78Leu c.233C>T | ||
| [22] | III-1 W 62 y | 45 y | Painful sensations | Legs | Mild distal weakness | RLS | Interm | Mild large fiber loss, regeneration clusters | IVIg, gabapentin, pregabalin-improvement | AD RLS without pain in 1 | Asp224Tyr c.670G>T |
| IV-1 W 41 y | 30 y | Cramps and pain | Legs | Mild distal weakness | No | Interm | ND | NR | Asp195Tyr c.670G>T | ||
| IV-2 W 35 y | 35 y | Cramps and burning pain | Legs and arms | Mild distal weakness | RLS | Interm | ND | NR | Asp195Tyr c.670G>T | ||
| [5] | Pt 1 W 18 y | 1st dec | Deep, burning, aching, shooting, throbbing pain NRS 7–10 | Feet, ankles | No | No | ND | ND | tricyclic antidepressants, gabapentinoids, opiates, lidocaine plaster—partially effective | AD Similar features in all | Trp101ter |
| Pt 2 W 23 y | 1st dec | Mild distal weakness | No | Interm | ND | Trp101ter c.302G>A | |||||
| Pt 3 W 25 y | 2nd dec | No | No | Interm | ND | Trp101ter | |||||
| Pt 4 W 40 y | 3rd dec | No | No | ND | ND | Trp101ter | |||||
| Pt 5 W 42y | 3rd dec | No | No | Interm | Normal * | Trp101ter | |||||
| Pt 6 W 44 y | 4th dec | No | No | Interm | ND | Trp101ter | |||||
| [23] | F1-III.1 W 53 y | 15 y | Burning | Lower extremities | Normal strength | No | Dem | ND | NR | AD Mild symptoms and signs in other 2 | Val10Cysfs11 * c.309G >T |
| [24] | NA | Adult | Pain | Distal | No motor symptoms | No | Dem | ND | NR | NR | Arg98Cys c.292C>T |
| CR | W 57 y | 46 y | Burning pain, shock-like sensations, NRS 8. | Mainly distal, occasionally proximal | No motor symptoms and signs | RLS | Interm | Abnormal * | Paracetamol, clonazepam—partial improvement | Sporadic | Trp101ter c.302G>A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gemignani, F.; Percesepe, A.; Gualandi, F.; Allegri, I.; Bellanova, M.F.; Nuredini, A.; Saccani, E.; Ambrosini, E.; Barili, V.; Uliana, V. Charcot–Marie–Tooth Disease with Myelin Protein Zero Mutation Presenting as Painful, Predominant Small-Fiber Neuropathy. Int. J. Mol. Sci. 2024, 25, 1654. https://doi.org/10.3390/ijms25031654
Gemignani F, Percesepe A, Gualandi F, Allegri I, Bellanova MF, Nuredini A, Saccani E, Ambrosini E, Barili V, Uliana V. Charcot–Marie–Tooth Disease with Myelin Protein Zero Mutation Presenting as Painful, Predominant Small-Fiber Neuropathy. International Journal of Molecular Sciences. 2024; 25(3):1654. https://doi.org/10.3390/ijms25031654
Chicago/Turabian StyleGemignani, Franco, Antonio Percesepe, Francesca Gualandi, Isabella Allegri, Maria Federica Bellanova, Andi Nuredini, Elena Saccani, Enrico Ambrosini, Valeria Barili, and Vera Uliana. 2024. "Charcot–Marie–Tooth Disease with Myelin Protein Zero Mutation Presenting as Painful, Predominant Small-Fiber Neuropathy" International Journal of Molecular Sciences 25, no. 3: 1654. https://doi.org/10.3390/ijms25031654
APA StyleGemignani, F., Percesepe, A., Gualandi, F., Allegri, I., Bellanova, M. F., Nuredini, A., Saccani, E., Ambrosini, E., Barili, V., & Uliana, V. (2024). Charcot–Marie–Tooth Disease with Myelin Protein Zero Mutation Presenting as Painful, Predominant Small-Fiber Neuropathy. International Journal of Molecular Sciences, 25(3), 1654. https://doi.org/10.3390/ijms25031654