
DNA Methylation and Chromatin Accessibility Impact Subgenome Expression Dominance in the Common Carp (Cyprinus carpio)
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
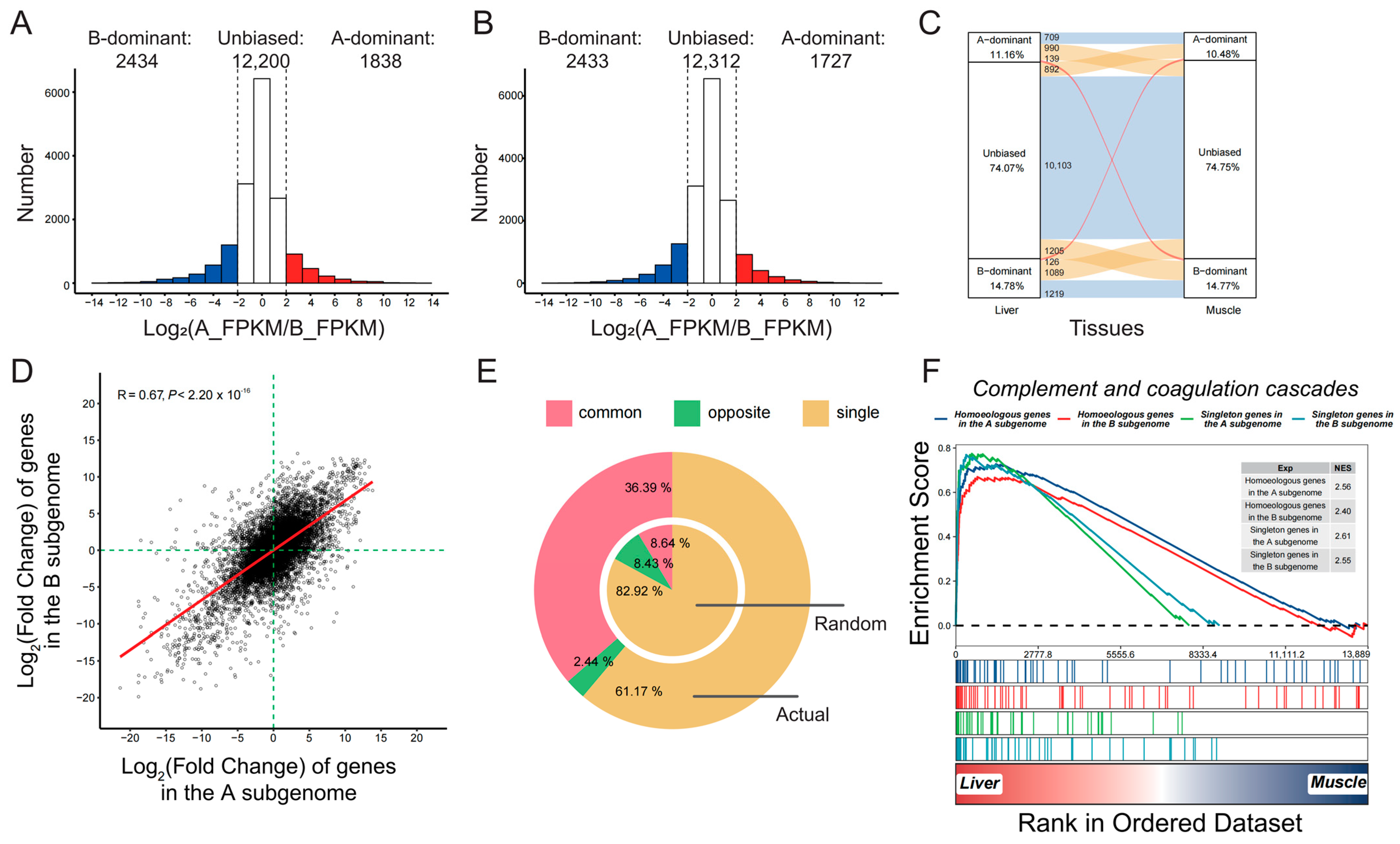
2.1. Biased Differential Expression and Function between the Two Subgenomes
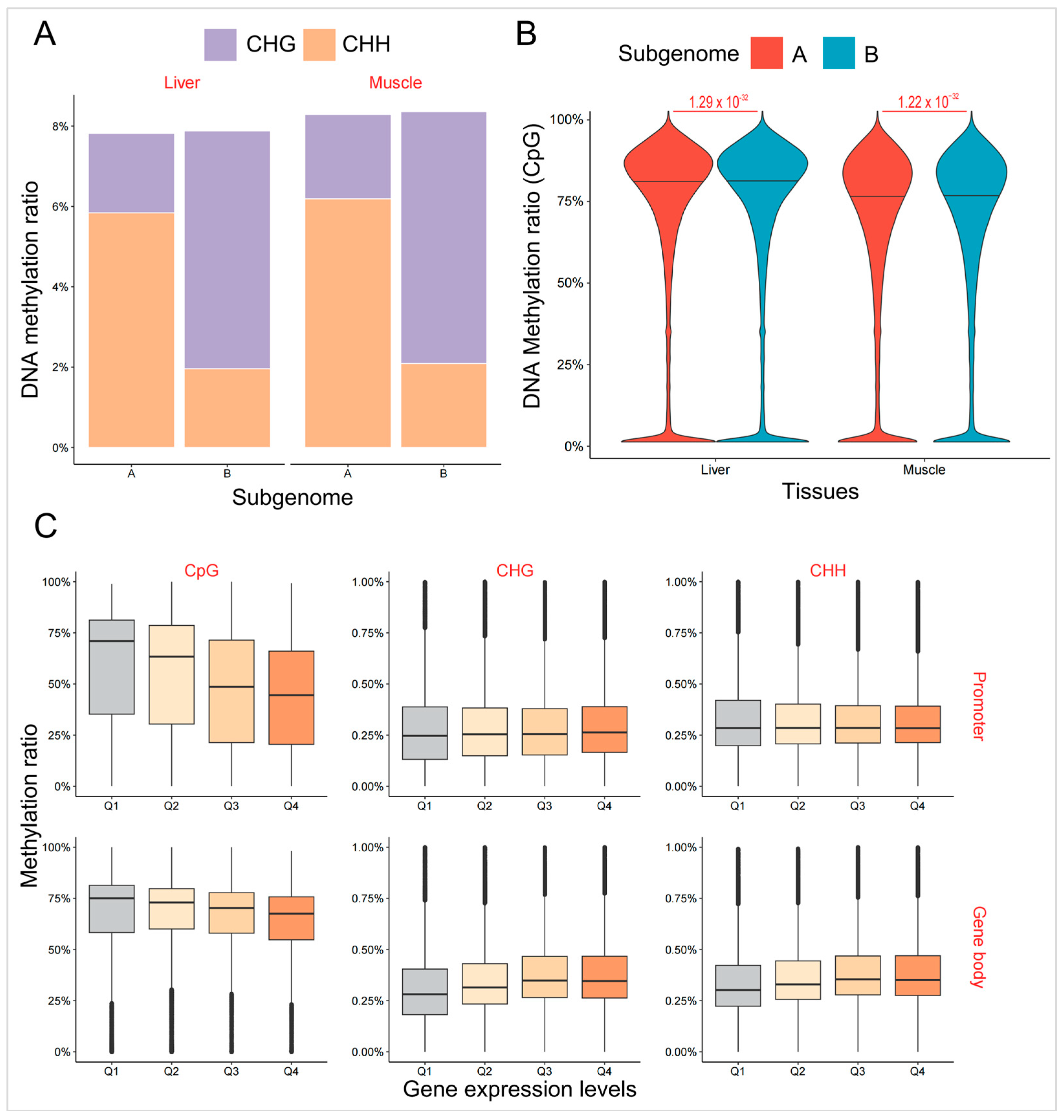
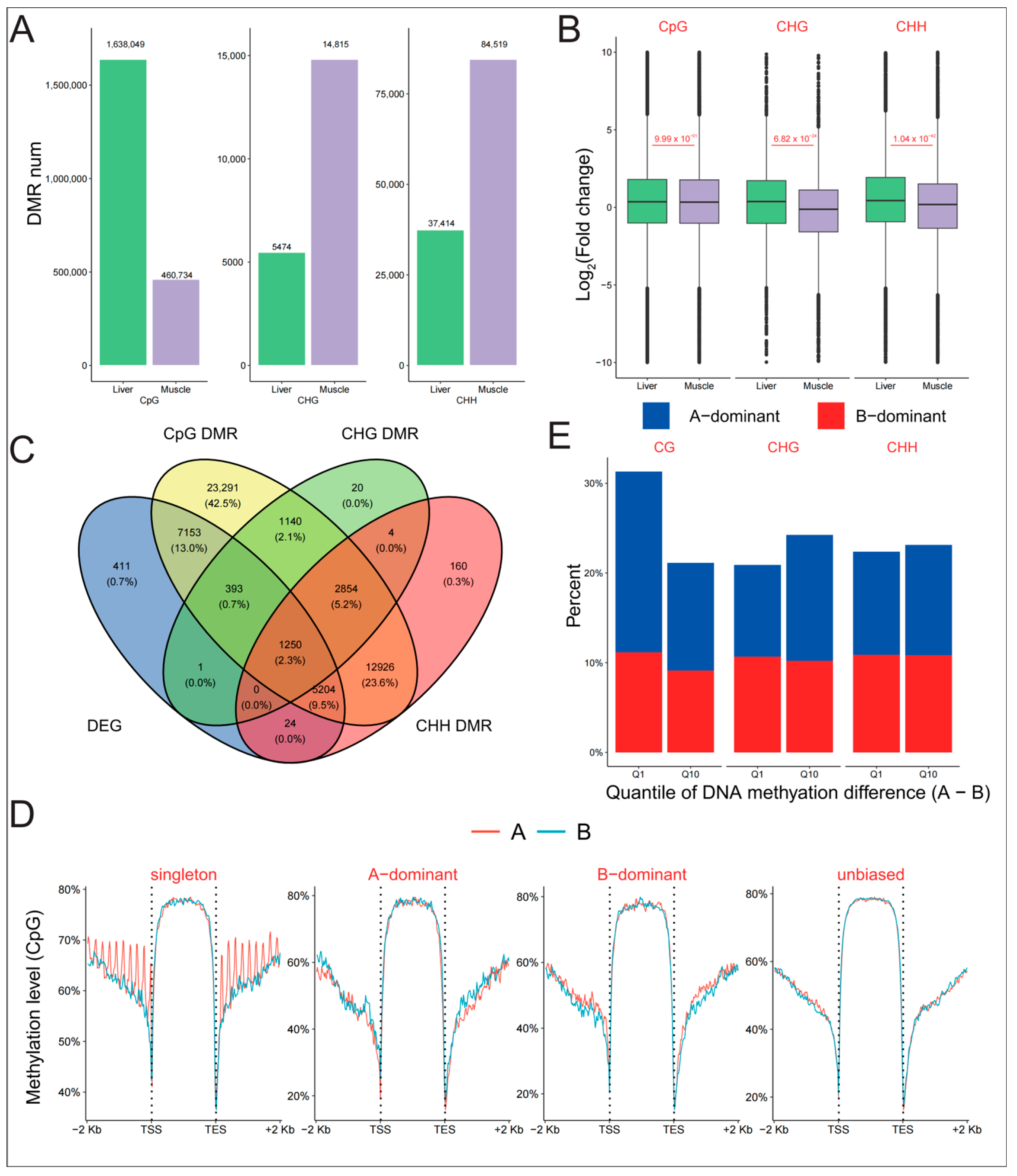
2.2. Asymmetric DNA Methylation Did Not Contribute to Biased Expression of Homoeologous Genes
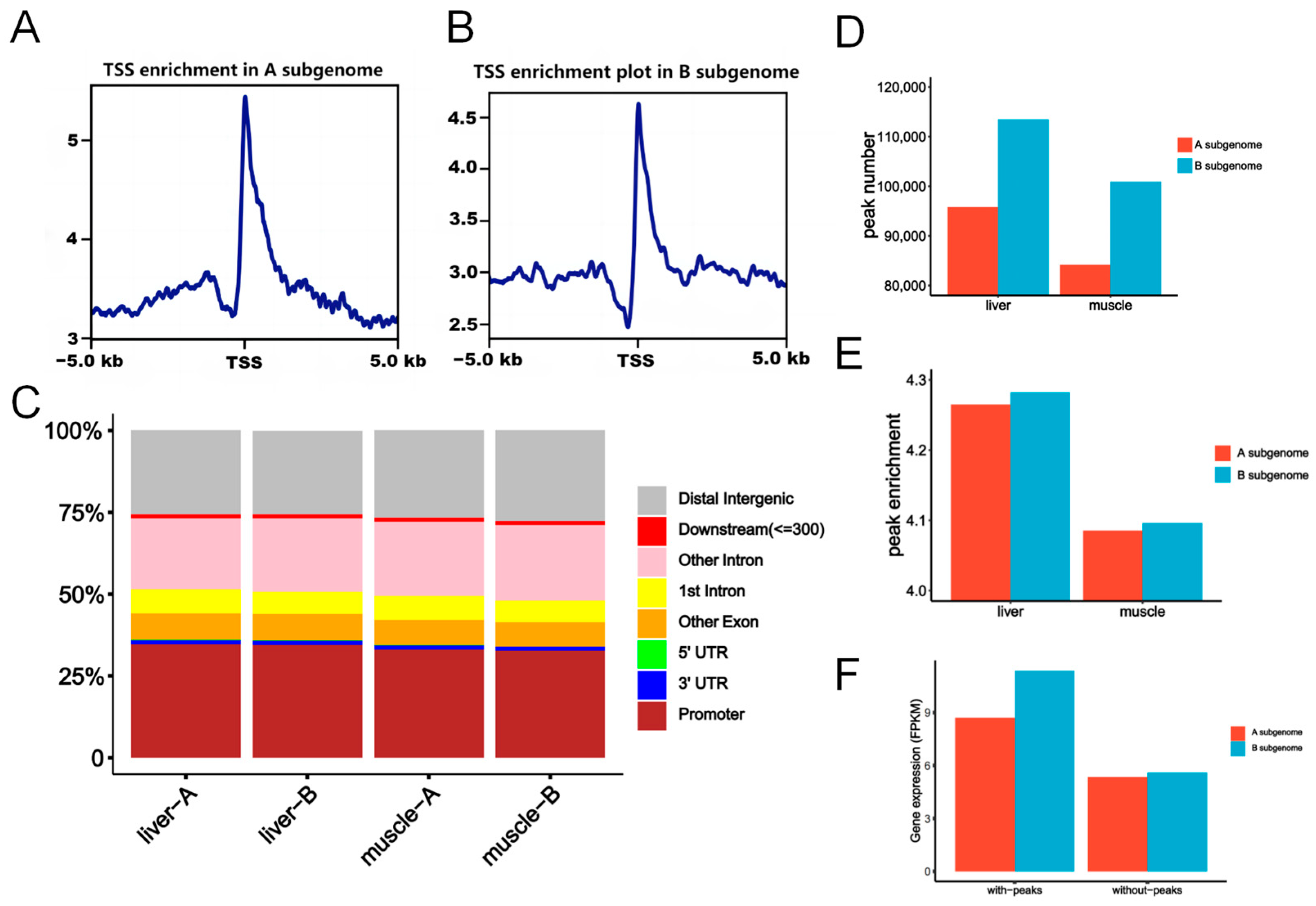
2.3. Chromatin Accessibility Was Correlated with Subgenome Expression Dominance
3. Discussion
4. Materials and Methods
4.1. Experimental Fish and Tissue Collection
4.2. RNA Extraction and Sequencing
4.3. Quantification of Gene Expression and Identification of Differentially Expressed Genes
4.4. Whole-Genome Bisulfite Sequencing (WGBS)
4.5. DNA Methylation Sequencing Data Analysis
4.6. Chromatin Accessibility Profiling by ATAC-Seq
4.7. Analysis of Open Chromatin Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ren, L.; Gao, X.; Cui, J.; Zhang, C.; Dai, H.; Luo, M.; He, S.; Qin, Q.; Luo, K.; Tao, M.; et al. Symmetric subgenomes and balanced homoeolog expression stabilize the establishment of allopolyploidy in cyprinid fish. BMC Biol. 2022, 20, 200. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Cheng, F.; Li, P.; Xin, X.; Wang, W.; Yu, Y.; Zhang, D.; Zhao, X.; Yu, S.; et al. Subgenome dominance and its evolutionary implications in crop domestication and breeding. Hortic. Res. 2022, 9, uhac090. [Google Scholar] [CrossRef]
- Edger, P.P.; Smith, R.; McKain, M.R.; Cooley, A.M.; Vallejo-Marin, M.; Yuan, Y.; Bewick, A.J.; Ji, L.; Platts, A.E.; Bowman, M.J.; et al. Subgenome Dominance in an Interspecific Hybrid, Synthetic Allopolyploid, and a 140-Year-Old Naturally Established Neo-Allopolyploid Monkeyflower. Plant Cell 2017, 29, 2150–2167. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Li, W.; Qin, Q.; Dai, H.; Han, F.; Xiao, J.; Gao, X.; Cui, J.; Wu, C.; Yan, X.; et al. The subgenomes show asymmetric expression of alleles in hybrid lineages of Megalobrama amblycephala × Culter alburnus. Genome Res. 2019, 29, 1805–1815. [Google Scholar] [CrossRef]
- Alger, E.I.; Edger, P.P. One subgenome to rule them all: Underlying mechanisms of subgenome dominance. Curr. Opin. Plant Biol. 2020, 54, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Bird, K.A.; Niederhuth, C.E.; Ou, S.; Gehan, M.; Pires, J.C.; Xiong, Z.; VanBuren, R.; Edger, P.P. Replaying the evolutionary tape to investigate subgenome dominance in allopolyploid Brassica napus. New Phytol. 2021, 230, 354–371. [Google Scholar] [CrossRef]
- Ren, L.; Yan, X.; Cao, L.; Li, J.; Zhang, X.; Gao, X.; Liu, J.; Cui, J.; Liu, S. Combined effects of dosage compensation and incomplete dominance on gene expression in triploid cyprinids. DNA Res. 2019, 26, 485–494. [Google Scholar] [CrossRef]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noël, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef]
- Deniz, Ö.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Louise Smith, E.; Mok, G.F.; Münsterberg, A. Investigating chromatin accessibility during development and differentiation by ATAC-sequencing to guide the identification of cis-regulatory elements. Biochem. Soc. Trans. 2022, 50, 1167–1177. [Google Scholar] [CrossRef]
- Wu, X.; Yang, Y.; Zhong, C.; Guo, Y.; Wei, T.; Li, S.; Lin, H.; Liu, X. Integration of ATAC-seq and RNA-seq Unravels Chromatin Accessibility during Sex Reversal in Orange-Spotted Grouper (Epinephelus coioides). Int. J. Mol. Sci. 2020, 21, 2800. [Google Scholar] [CrossRef]
- Han, J.; Lopez-Arredondo, D.; Yu, G.; Wang, Y.; Wang, B.; Wall, S.B.; Zhang, X.; Fang, H.; Barragán-Rosillo, A.C.; Pan, X.; et al. Genome-wide chromatin accessibility analysis unveils open chromatin convergent evolution during polyploidization in cotton. Proc. Natl. Acad. Sci. USA 2022, 119, e2209743119. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.-H.; McKenzie, N.; Gardiner, L.-J.; Luo, M.-C.; Hall, A.; Bevan, M.W. Reduced chromatin accessibility underlies gene expression differences in homologous chromosome arms of diploid Aegilops tauschii and hexaploid wheat. GigaScience 2020, 9, giaa070. [Google Scholar] [CrossRef]
- Li, J.-T.; Wang, Q.; Huang Yang, M.-D.; Li, Q.-S.; Cui, M.-S.; Dong, Z.-J.; Wang, H.-W.; Yu, J.-H.; Zhao, Y.-J.; Yang, C.-R.; et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021, 53, 1493–1503. [Google Scholar] [CrossRef]
- Xu, P.; Xu, J.; Liu, G.; Chen, L.; Zhou, Z.; Peng, W.; Jiang, Y.; Zhao, Z.; Jia, Z.; Sun, Y.; et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 2019, 10, 4625. [Google Scholar] [CrossRef]
- Ni, J.; Zhu, P.; Mo, Q.; Luo, W.; Du, Z.; Jiang, J.; Yang, S.; Zhao, L.; Gong, Q.; Wang, Y. Dynamic Transcriptomic Profiling during Liver Development in Schizothorax prenanti. Front. Physiol. 2022, 13, 928858. [Google Scholar] [CrossRef]
- Gerry, S.P.; Ellerby, D.J. Resolving Shifting Patterns of Muscle Energy Use in Swimming Fish. PLoS ONE 2014, 9, e106030. [Google Scholar] [CrossRef] [PubMed]
- Amara, U.; Rittirsch, D.; Flierl, M.; Bruckner, U.; Klos, A.; Gebhard, F.; Lambris, J.D.; Huber-Lang, M. Interaction between the Coagulation and Complement System. In Current Topics in Complement II; Lambris, J.D., Ed.; Springer: New York, NY, USA, 2008; pp. 68–76. [Google Scholar]
- Kon, T.; Fukuta, K.; Chen, Z.; Kon-Nanjo, K.; Suzuki, K.; Ishikawa, M.; Tanaka, H.; Burgess, S.M.; Noguchi, H.; Toyoda, A.; et al. Single-cell transcriptomics of the goldfish retina reveals genetic divergence in the asymmetrically evolved subgenomes after allotetraploidization. Commun. Biol. 2022, 5, 1404. [Google Scholar] [CrossRef]
- Ren, L.; Li, W.; Tao, M.; Qin, Q.; Luo, J.; Chai, J.; Tang, C.; Xiao, J.; Tang, X.; Lin, G.; et al. Homoeologue expression insights into the basis of growth heterosis at the intersection of ploidy and hybridity in Cyprinidae. Sci. Rep. 2016, 6, 27040. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, J.; Sun, X.; Xu, P. Research advances and future perspectives of genomics and genetic improvement in allotetraploid common carp. Rev. Aquac. 2022, 14, 957–978. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, B.; Lisch, D.; Ma, J. Patterns and Consequences of Subgenome Differentiation Provide Insights into the Nature of Paleopolyploidy in Plants. Plant Cell 2017, 29, 2974–2994. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Elhamamsy, A.R. Role of DNA methylation in imprinting disorders: An updated review. J. Assist. Reprod. Genet. 2017, 34, 549–562. [Google Scholar] [CrossRef]
- Sun, H.; Wu, S.; Zhang, G.; Jiao, C.; Guo, S.; Ren, Y.; Zhang, J.; Zhang, H.; Gong, G.; Jia, Z.; et al. Karyotype Stability and Unbiased Fractionation in the Paleo-Allotetraploid Cucurbita Genomes. Mol. Plant 2017, 10, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, L.; Cui, Y.; Yang, Y.; Wu, J.; Liang, J.; Li, X.; Zhang, X.; Zhang, Y.; Guo, Z.; et al. The lack of negative association between TE load and subgenome dominance in synthesized Brassica allotetraploids. Proc. Natl. Acad. Sci. USA 2023, 120, e2305208120. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.W.; He, F.; de Soto, M.F.; Akhunova, A.; Akhunov, E. Differential chromatin accessibility landscape reveals structural and functional features of the allopolyploid wheat chromosomes. Genome Biol. 2020, 21, 176. [Google Scholar] [CrossRef]
- Bottani, S.; Zabet, N.R.; Wendel, J.F.; Veitia, R.A. Gene Expression Dominance in Allopolyploids: Hypotheses and Models. Trends Plant Sci. 2018, 23, 393–402. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Lim, J.-Q.; Sung, W.-K.; Li, G. An integrated package for bisulfite DNA methylation data analysis with Indel-sensitive mapping. BMC Bioinform. 2019, 20, 47. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, M.; Wu, T.; Zhan, L.; Li, L.; Chen, M.; Xie, W.; Xie, Z.; Hu, E.; Xu, S.; et al. Exploring Epigenomic Datasets by ChIPseeker. Curr. Protoc. 2022, 2, e585. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Stark, R.; Teschendorff, A.E.; Holmes, K.A.; Ali, H.R.; Dunning, M.J.; Brown, G.D.; Gojis, O.; Ellis, I.O.; Green, A.R.; et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012, 481, 389–393. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.-T.; Zhao, R.; Sun, X.-Q.; Hou, M.-X.; Cao, Y.-M.; Zhang, J.; Chen, Y.-J.; Wang, K.-K.; Zhang, Y.; Li, J.-T.; et al. DNA Methylation and Chromatin Accessibility Impact Subgenome Expression Dominance in the Common Carp (Cyprinus carpio). Int. J. Mol. Sci. 2024, 25, 1635. https://doi.org/10.3390/ijms25031635
Yu S-T, Zhao R, Sun X-Q, Hou M-X, Cao Y-M, Zhang J, Chen Y-J, Wang K-K, Zhang Y, Li J-T, et al. DNA Methylation and Chromatin Accessibility Impact Subgenome Expression Dominance in the Common Carp (Cyprinus carpio). International Journal of Molecular Sciences. 2024; 25(3):1635. https://doi.org/10.3390/ijms25031635
Chicago/Turabian StyleYu, Shuang-Ting, Ran Zhao, Xiao-Qing Sun, Ming-Xi Hou, Yi-Ming Cao, Jin Zhang, Ying-Jie Chen, Kai-Kuo Wang, Yan Zhang, Jiong-Tang Li, and et al. 2024. "DNA Methylation and Chromatin Accessibility Impact Subgenome Expression Dominance in the Common Carp (Cyprinus carpio)" International Journal of Molecular Sciences 25, no. 3: 1635. https://doi.org/10.3390/ijms25031635
APA StyleYu, S.-T., Zhao, R., Sun, X.-Q., Hou, M.-X., Cao, Y.-M., Zhang, J., Chen, Y.-J., Wang, K.-K., Zhang, Y., Li, J.-T., & Wang, Q. (2024). DNA Methylation and Chromatin Accessibility Impact Subgenome Expression Dominance in the Common Carp (Cyprinus carpio). International Journal of Molecular Sciences, 25(3), 1635. https://doi.org/10.3390/ijms25031635