
Emerging Roles of YES1 in Cancer: The Putative Target in Drug Resistance



{kind=link}
{kind=link}
Abstract
1. Introduction
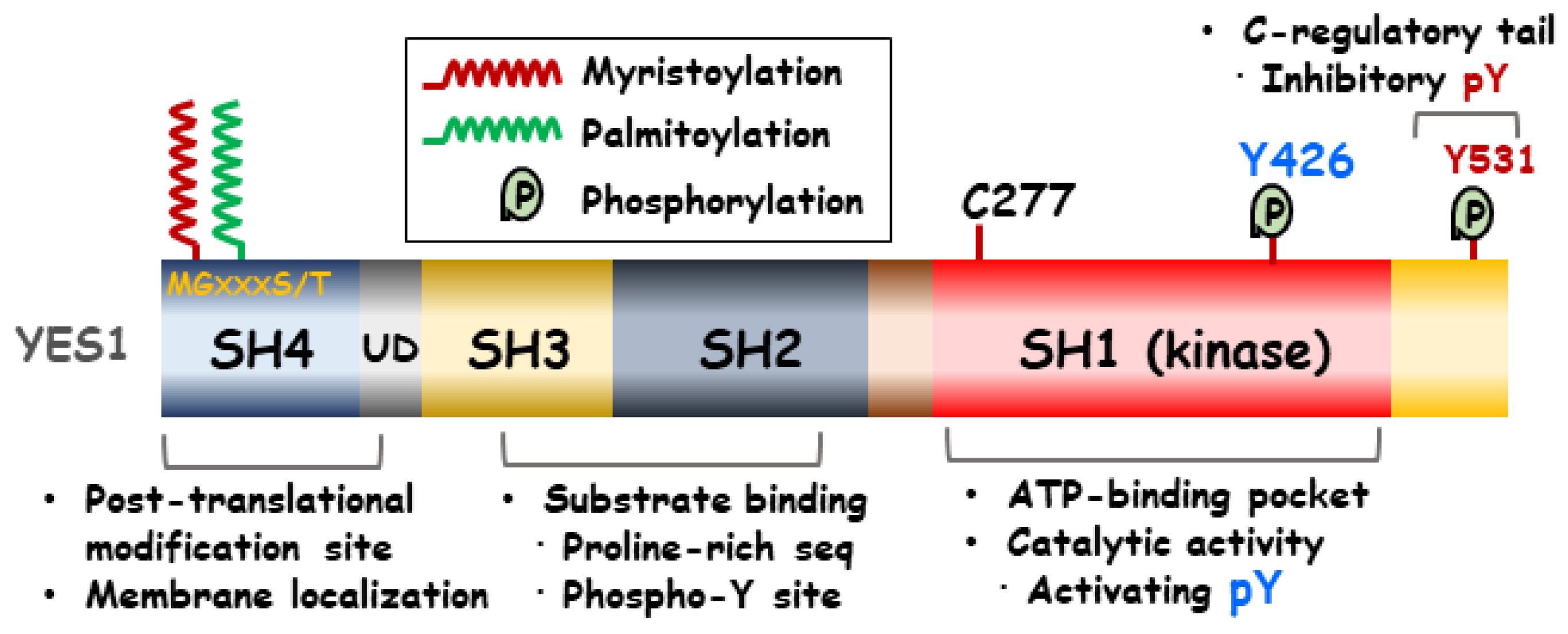
2. Structure of YES1
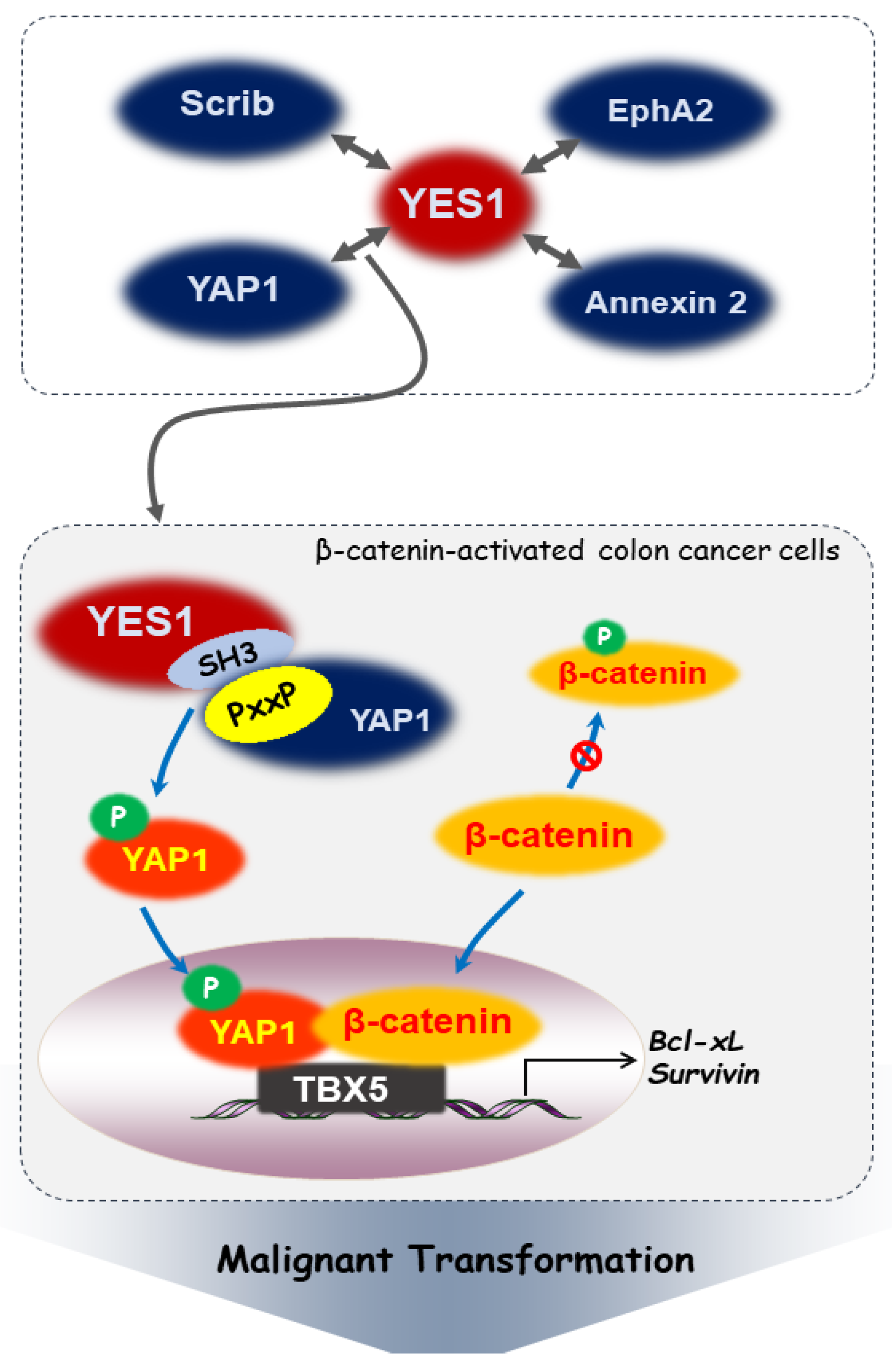
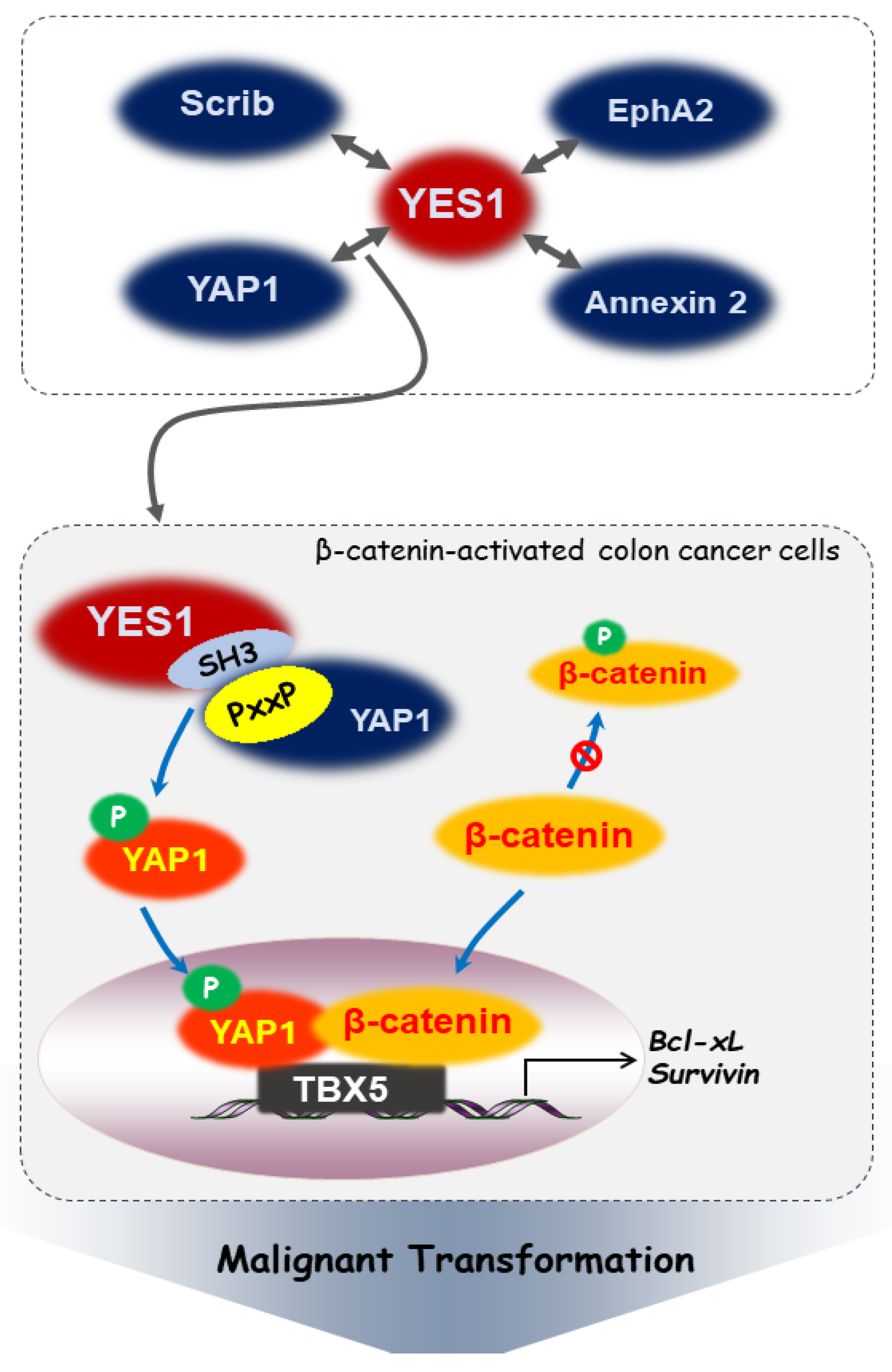
3. Interactions between YES1 and Intracellular Signaling Molecules in Cancer
3.1. YES-Associated Protein 1 (YAP1)
3.2. Scribble (Scrib)
3.3. Erythropoietin-Producing Hepatoma Receptor Tyrosine Kinase A2 (EphA2)
4. Post-Translational Modifications of YES1
5. Role of YES1 Regulation in Tumor Development and Resistance to Cancer Drugs
5.1. Skin Cancer
5.2. Breast Cancer
5.3. Lung Cancer
5.4. Leukemia
5.5. Liver Cancer
5.6. Glioma
5.7. Sarcoma
5.8. Ovarian Cancer
5.9. Prostate Cancer
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lei, Z.N.; Tian, Q.; Teng, Q.X.; Wurpel, J.N.D.; Zeng, L.; Pan, Y.; Chen, Z.S. Understanding and targeting resistance mechanisms in cancer. MedComm 2023, 4, e265. [Google Scholar] [CrossRef] [PubMed]
- Raji, L.; Tetteh, A.; Amin, A. Role of c-Src in carcinogenesis and drug resistance. Cancers 2023, 16, 32. [Google Scholar] [CrossRef]
- Parsons, S.J.; Parsons, J.T. Src family kinases, key regulators of signal transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef]
- Sen, B.; Johnson, F.M. Regulation of SRC family kinases in human cancers. J. Signal Transduct. 2011, 2011, 865819. [Google Scholar] [CrossRef] [PubMed]
- Lapouge, M.; Meloche, S. A renaissance for YES in cancer. Oncogene 2023, 42, 3385–3393. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef]
- Barraclough, J.; Hodgkinson, C.; Hogg, A.; Dive, C.; Welman, A. Increases in c-Yes expression level and activity promote motility but not proliferation of human colorectal carcinoma cells. Neoplasia 2007, 9, 745–754. [Google Scholar] [CrossRef]
- Han, N.M.; Curley, S.A.; Gallick, G.E. Differential activation of pp60(c-src) and pp62(c-yes) in human colorectal carcinoma liver metastases. Clin. Cancer Res. 1996, 2, 1397–1404. [Google Scholar]
- Garmendia, I.; Redin, E.; Montuenga, L.M.; Calvo, A. YES1: A Novel Therapeutic Target and Biomarker in Cancer. Mol. Cancer Ther. 2022, 21, 1371–1380. [Google Scholar] [CrossRef]
- Resh, M.D. Myristylation and palmitylation of Src family members: The fats of the matter. Cell 1994, 76, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein-protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Arbesu, M.; Maffei, M.; Cordeiro, T.N.; Teixeira, J.M.; Perez, Y.; Bernado, P.; Roche, S.; Pons, M. The unique domain forms a fuzzy intramolecular complex in Src family kinases. Structure 2017, 25, 630–640.e4. [Google Scholar] [CrossRef] [PubMed]
- Barkho, S.; Pierce, L.C.; McGlone, M.L.; Li, S.; Woods, V.L., Jr.; Walker, R.C.; Adams, J.A.; Jennings, P.A. Distal loop flexibility of a regulatory domain modulates dynamics and activity of C-terminal SRC kinase (csk). PLoS Comput. Biol. 2013, 9, e1003188. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.E. Structural insights into redox-active cysteine residues of the Src family kinases. Redox Biol. 2021, 41, 101934. [Google Scholar] [CrossRef]
- Sudol, M.; Chen, H.I.; Bougeret, C.; Einbond, A.; Bork, P. Characterization of a novel protein-binding module—The WW domain. FEBS Lett. 1995, 369, 67–71. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef]
- Santoni, M.J.; Kashyap, R.; Camoin, L.; Borg, J.P. The Scribble family in cancer: Twentieth anniversary. Oncogene 2020, 39, 7019–7033. [Google Scholar] [CrossRef]
- Zhan, L.; Rosenberg, A.; Bergami, K.C.; Yu, M.; Xuan, Z.; Jaffe, A.B.; Allred, C.; Muthuswamy, S.K. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 2008, 135, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yin, Z.; Soellner, M.B.; Martin, B.R. Scribble sub-cellular localization modulates recruitment of YES1 to regulate YAP1 phosphorylation. Cell Chem. Biol. 2021, 28, 1235–1241.e5. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Sakurai, H. Emerging and diverse functions of the EphA2 noncanonical pathway in cancer progression. Biol. Pharm. Bull. 2017, 40, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Xiao, D.; Li, G.; Ma, J.; Chen, P.; Yuan, W.; Hou, F.; Ge, J.; Zhong, M.; Tang, Y.; et al. EphA2 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer cells. Oncogene 2014, 33, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Chen, L.; Wu, W.; Wang, J.; Zheng, X.; Chen, Z.; Jiang, Q.; Han, J.; Wei, L.; Wang, L.; et al. EPH receptor A2 governs a feedback loop that activates Wnt/beta-catenin signaling in gastric cancer. Cell Death Dis. 2018, 9, 1146. [Google Scholar] [CrossRef]
- Mao, L.; Yuan, W.; Cai, K.; Lai, C.; Huang, C.; Xu, Y.; Zhong, S.; Yang, C.; Wang, R.; Zeng, P.; et al. EphA2-YES1-ANXA2 pathway promotes gastric cancer progression and metastasis. Oncogene 2021, 40, 3610–3623. [Google Scholar] [CrossRef]
- Kpetemey, M.; Dasgupta, S.; Rajendiran, S.; Das, S.; Gibbs, L.D.; Shetty, P.; Gryczynski, Z.; Vishwanatha, J.K. MIEN1, a novel interactor of Annexin A2, promotes tumor cell migration by enhancing AnxA2 cell surface expression. Mol. Cancer 2015, 14, 156. [Google Scholar] [CrossRef]
- Wang, T.; Wang, Z.; Niu, R.; Wang, L. Crucial role of Anxa2 in cancer progression: Highlights on its novel regulatory mechanism. Cancer Biol. Med. 2019, 16, 671–687. [Google Scholar] [CrossRef]
- Pan, S.; Chen, R. Pathological implication of protein post-translational modifications in cancer. Mol. Asp. Med. 2022, 86, 101097. [Google Scholar] [CrossRef]
- Scott, J.D.; Pawson, T. Cell signaling in space and time: Where proteins come together and when they’re apart. Science 2009, 326, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Bi, B.; Qiu, M.; Liu, P.; Wang, Q.; Wen, Y.; Li, Y.; Li, B.; Li, Y.; He, Y.; Zhao, J. Protein post-translational modifications: A key factor in colorectal cancer resistance mechanisms. Biochim. Biophys. Acta Gene Regul. Mech. 2023, 1866, 194977. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.Y.; Fu, S.H.; Chien, M.W.; Liu, Y.W.; Chen, S.J.; Sytwu, H.K. Post-translational modifications of transcription factors harnessing the etiology and pathophysiology in colonic diseases. Int. J. Mol. Sci. 2020, 21, 3207. [Google Scholar] [CrossRef] [PubMed]
- Sethi, M.K.; Hancock, W.S.; Fanayan, S. Identifying N-glycan biomarkers in colorectal cancer by mass spectrometry. Acc. Chem. Res. 2016, 49, 2099–2106. [Google Scholar] [CrossRef] [PubMed]
- Sethi, M.K.; Kim, H.; Park, C.K.; Baker, M.S.; Paik, Y.K.; Packer, N.H.; Hancock, W.S.; Fanayan, S.; Thaysen-Andersen, M. In-depth N-glycome profiling of paired colorectal cancer and non-tumorigenic tissues reveals cancer-, stage- and EGFR-specific protein N-glycosylation. Glycobiology 2015, 25, 1064–1078. [Google Scholar] [CrossRef]
- Dian, C.; Perez-Dorado, I.; Riviere, F.; Asensio, T.; Legrand, P.; Ritzefeld, M.; Shen, M.; Cota, E.; Meinnel, T.; Tate, E.W.; et al. High-resolution snapshots of human N-myristoyltransferase in action illuminate a mechanism promoting N-terminal Lys and Gly myristoylation. Nat. Commun. 2020, 11, 1132. [Google Scholar] [CrossRef] [PubMed]
- Farazi, T.A.; Waksman, G.; Gordon, J.I. The biology and enzymology of protein N-myristoylation. J. Biol. Chem. 2001, 276, 39501–39504. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, P.; Resh, M.D. Myristoylation and membrane binding regulate c-Src stability and kinase activity. Mol. Cell. Biol. 2010, 30, 4094–4107. [Google Scholar] [CrossRef]
- Rajala, R.V.; Dehm, S.; Bi, X.; Bonham, K.; Sharma, R.K. Expression of N-myristoyltransferase inhibitor protein and its relationship to c-Src levels in human colon cancer cell lines. Biochem. Biophys. Res. Commun. 2000, 273, 1116–1120. [Google Scholar] [CrossRef]
- Shoji, S.; Kurosawa, T.; Inoue, H.; Funakoshi, T.; Kubota, Y. Human cellular src gene product: Identification of the myristoylated pp60c-src and blockage of its myristoyl acylation with N-fatty acyl compounds resulted in the suppression of colony formation. Biochem. Biophys. Res. Commun. 1990, 173, 894–901. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X.; Wang, J.; Qiao, Y. The role of N-myristoyltransferase 1 in tumour development. Ann. Med. 2023, 55, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Goya Grocin, A.; Serwa, R.A.; Morales Sanfrutos, J.; Ritzefeld, M.; Tate, E.W. Whole proteome profiling of N-myristoyltransferase activity and inhibition using sortase A. Mol. Cell. Proteom. 2019, 18, 115–126. [Google Scholar] [CrossRef] [PubMed]
- McCabe, J.B.; Berthiaume, L.G. N-terminal protein acylation confers localization to cholesterol, sphingolipid-enriched membranes but not to lipid rafts/caveolae. Mol. Biol. Cell 2001, 12, 3601–3617. [Google Scholar] [CrossRef] [PubMed]
- Sato, I.; Obata, Y.; Kasahara, K.; Nakayama, Y.; Fukumoto, Y.; Yamasaki, T.; Yokoyama, K.K.; Saito, T.; Yamaguchi, N. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J. Cell Sci. 2009, 122, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Oneyama, C.; Iino, T.; Saito, K.; Suzuki, K.; Ogawa, A.; Okada, M. Transforming potential of Src family kinases is limited by the cholesterol-enriched membrane microdomain. Mol. Cell. Biol. 2009, 29, 6462–6472. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wu, J.; Liang, Z.; Zhang, Y.; Huang, Z. Protein S-nitrosation: Biochemistry, identification, molecular mechanisms, and therapeutic applications. J. Med. Chem. 2022, 65, 5902–5925. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Senga, T.; Ito, S.; Hyodo, T.; Hasegawa, H.; Hamaguchi, M. S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J. Biol. Chem. 2010, 285, 3806–3814. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Sharma, S.V.; Settleman, J. Oncogene addiction: Setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007, 21, 3214–3231. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Riely, G.J.; Balak, M.; Marks, J.L.; Ladanyi, M.; Miller, V.A.; Pao, W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 7519–7525. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007, 4, 1669–1679, discussion 1680. [Google Scholar] [CrossRef] [PubMed]
- Vander Velde, R.; Yoon, N.; Marusyk, V.; Durmaz, A.; Dhawan, A.; Miroshnychenko, D.; Lozano-Peral, D.; Desai, B.; Balynska, O.; Poleszhuk, J.; et al. Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures. Nat. Commun. 2020, 11, 2393. [Google Scholar] [CrossRef]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015, 16, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Barnekow, A.; Paul, E.; Schartl, M. Expression of the c-src protooncogene in human skin tumors. Cancer Res. 1987, 47, 235–240. [Google Scholar] [PubMed]
- Lee, J.H.; Pyon, J.K.; Kim, D.W.; Lee, S.H.; Nam, H.S.; Kim, C.H.; Kang, S.G.; Lee, Y.J.; Park, M.Y.; Jeong, D.J.; et al. Elevated c-Src and c-Yes expression in malignant skin cancers. J. Exp. Clin. Cancer Res. 2010, 29, 116. [Google Scholar] [CrossRef]
- Marchetti, D.; Parikh, N.; Sudol, M.; Gallick, G.E. Stimulation of the protein tyrosine kinase c-Yes but not c-Src by neurotrophins in human brain-metastatic melanoma cells. Oncogene 1998, 16, 3253–3260. [Google Scholar] [CrossRef]
- Loganzo, F., Jr.; Dosik, J.S.; Zhao, Y.; Vidal, M.J.; Nanus, D.M.; Sudol, M.; Albino, A.P. Elevated expression of protein tyrosine kinase c-Yes, but not c-Src, in human malignant melanoma. Oncogene 1993, 8, 2637–2644. [Google Scholar]
- Liu, W.; Monahan, K.B.; Pfefferle, A.D.; Shimamura, T.; Sorrentino, J.; Chan, K.T.; Roadcap, D.W.; Ollila, D.W.; Thomas, N.E.; Castrillon, D.H.; et al. LKB1/STK11 inactivation leads to expansion of a prometastatic tumor subpopulation in melanoma. Cancer Cell 2012, 21, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Lun, X.K.; Szklarczyk, D.; Gabor, A.; Dobberstein, N.; Zanotelli, V.R.T.; Saez-Rodriguez, J.; von Mering, C.; Bodenmiller, B. Analysis of the human kinome and phosphatome by mass cytometry reveals overexpression-induced effects on cancer-related signaling. Mol. Cell 2019, 74, 1086–1102.e5. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Bilal, E.; Alexe, G.; Yao, M.; Cong, L.; Kulkarni, A.; Ginjala, V.; Toppmeyer, D.; Ganesan, S.; Bhanot, G. Identification of the YES1 kinase as a therapeutic target in basal-like breast cancers. Genes Cancer 2010, 1, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Yamamoto, H.; Kanzaki, H.; Suzawa, K.; Yoshioka, T.; Tomida, S.; Cui, X.; Murali, R.; Namba, K.; Sato, H.; et al. Yes1 signaling mediates the resistance to Trastuzumab/Lap atinib in breast cancer. PLoS ONE 2017, 12, e0171356. [Google Scholar] [CrossRef]
- Tiwari, S.R.; Mishra, P.; Abraham, J. Neratinib, a novel HER2-targeted tyrosine kinase inhibitor. Clin. Breast Cancer 2016, 16, 344–348. [Google Scholar] [CrossRef]
- Takeda, T.; Yamamoto, H.; Suzawa, K.; Tomida, S.; Miyauchi, S.; Araki, K.; Nakata, K.; Miura, A.; Namba, K.; Shien, K.; et al. YES1 activation induces acquired resistance to neratinib in HER2-amplified breast and lung cancers. Cancer Sci. 2020, 111, 849–856. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Q.; Xu, P.; Fu, L.; Li, Y.; Fu, H.; Quan, H.; Lou, L. YES1 amplification confers trastuzumab-emtansine (T-DM1) resistance in HER2-positive cancer. Br. J. Cancer 2020, 123, 1000–1011. [Google Scholar] [CrossRef]
- Fujihara, M.; Shien, T.; Shien, K.; Suzawa, K.; Takeda, T.; Zhu, Y.; Mamori, T.; Otani, Y.; Yoshioka, R.; Uno, M.; et al. YES1 as a therapeutic target for HER2-positive breast cancer after trastuzumab and trastuzumab-emtansine (T-DMI) resistance development. Int. J. Mol. Sci. 2021, 22, 12809. [Google Scholar] [CrossRef]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, E.; Westover, D.; Meador, C.B.; Yan, Y.; Bauer, J.A.; Lu, P.; Ye, F.; Kulick, A.; de Stanchina, E.; McEwen, R.; et al. SFK/FAK signaling attenuates osimertinib efficacy in both drug-sensitive and drug-resistant models of EGFR-mutant lung cancer. Cancer Res. 2017, 77, 2990–3000. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.D.; Narzisi, G.; Jayaprakash, A.D.; Venturini, E.; Robine, N.; Smibert, P.; Germer, S.; Yu, H.A.; Jordan, E.J.; Paik, P.K.; et al. YES1 amplification is a mechanism of acquired resistance to EGFR inhibitors identified by transposon mutagenesis and clinical genomics. Proc. Natl. Acad. Sci. USA 2018, 115, E6030–E6038. [Google Scholar] [CrossRef]
- Sato, H.; Kubota, D.; Qiao, H.; Jungbluth, A.; Rekhtman, N.; Schoenfeld, A.J.; Yu, H.A.; Riely, G.J.; Toyooka, S.; Lovly, C.M.; et al. SRC family kinase inhibition targets YES1 and YAP1 as Primary drivers of lung cancer and as mediators of acquired resistance to ALK and epidermal growth factor receptor inhibitors. JCO Precis. Oncol. 2022, 6, e2200088. [Google Scholar] [CrossRef]
- Garmendia, I.; Pajares, M.J.; Hermida-Prado, F.; Ajona, D.; Bertolo, C.; Sainz, C.; Lavin, A.; Remirez, A.B.; Valencia, K.; Moreno, H.; et al. YES1 drives lung cancer growth and progression and predicts sensitivity to dasatinib. Am. J. Respir. Crit. Care Med. 2019, 200, 888–899. [Google Scholar] [CrossRef]
- Minari, R.; Valentini, S.; Madeddu, D.; Cavazzoni, A.; La Monica, S.; Lagrasta, C.A.M.; Bertorelli, R.; De Sanctis, V.; Fassan, P.; Azzoni, C.; et al. YES1 and MYC amplifications as synergistic resistance mechanisms to different generation ALK tyrosine kinase inhibitors in advanced NSCLC: Brief report of clinical and preclinical proofs. JTO Clin. Res. Rep. 2022, 3, 100278. [Google Scholar] [CrossRef]
- Hamanaka, N.; Nakanishi, Y.; Mizuno, T.; Horiguchi-Takei, K.; Akiyama, N.; Tanimura, H.; Hasegawa, M.; Satoh, Y.; Tachibana, Y.; Fujii, T.; et al. YES1 is a targetable oncogene in cancers harboring YES1 gene amplification. Cancer Res. 2019, 79, 5734–5745. [Google Scholar] [CrossRef]
- Redin, E.; Garrido-Martin, E.M.; Valencia, K.; Redrado, M.; Solorzano, J.L.; Carias, R.; Echepare, M.; Exposito, F.; Serrano, D.; Ferrer, I.; et al. YES1 is a druggable oncogenic target in SCLC. J. Thorac. Oncol. 2022, 17, 1387–1403. [Google Scholar] [CrossRef]
- Ozawa, Y.; Williams, A.H.; Estes, M.L.; Matsushita, N.; Boschelli, F.; Jove, R.; List, A.F. Src family kinases promote AML cell survival through activation of signal transducers and activators of transcription (STAT). Leuk. Res. 2008, 32, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, R.A.; Pettengell, R.; Pandha, H.S.; Morgan, R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 2013, 27, 1000–1008. [Google Scholar] [CrossRef]
- Vegi, N.M.; Klappacher, J.; Oswald, F.; Mulaw, M.A.; Mandoli, A.; Thiel, V.N.; Bamezai, S.; Feder, K.; Martens, J.H.A.; Rawat, V.P.S.; et al. MEIS2 is an oncogenic partner in AML1-ETO-positive AML. Cell Rep. 2016, 16, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Ha, S.A.; Kim, H.K.; Yoo, J.; Kim, S.; Yim, S.H.; Jung, S.H.; Kim, D.W.; Chung, Y.J.; Kim, J.W. Gene expression signatures associated with the in vitro resistance to two tyrosine kinase inhibitors, nilotinib and imatinib. Blood Cancer J. 2011, 1, e32. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Okada, M.; Tokuda, M.; Shiratori, Y.; Hatase, O.; Shirai, M.; Nishioka, M.; Omata, M. Reduced C-terminal Src kinase (Csk) activities in hepatocellular carcinoma. Hepatology 1999, 29, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Nonomura, T.; Masaki, T.; Morishita, A.; Jian, G.; Uchida, N.; Himoto, T.; Izuishi, K.; Iwama, H.; Yoshiji, H.; Watanabe, S.; et al. Identification of c-Yes expression in the nuclei of hepatocellular carcinoma cells: Involvement in the early stages of hepatocarcinogenesis. Int. J. Oncol. 2007, 30, 105–111. [Google Scholar] [CrossRef]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.M.; Gladson, C.L. Targeting SRC in glioblastoma tumors and brain metastases: Rationale and preclinical studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar] [CrossRef]
- Han, X.; Zhang, W.; Yang, X.; Wheeler, C.G.; Langford, C.P.; Wu, L.; Filippova, N.; Friedman, G.K.; Ding, Q.; Fathallah-Shaykh, H.M.; et al. The role of Src family kinases in growth and migration of glioma stem cells. Int. J. Oncol. 2014, 45, 302–310. [Google Scholar] [CrossRef]
- Indovina, P.; Casini, N.; Forte, I.M.; Garofano, T.; Cesari, D.; Iannuzzi, C.A.; Del Porro, L.; Pentimalli, F.; Napoliello, L.; Boffo, S.; et al. SRC family kinase inhibition in ewing sarcoma cells induces p38 MAP kinase-mediated cytotoxicity and reduces cell migration. J. Cell. Physiol. 2017, 232, 129–135. [Google Scholar] [CrossRef]
- Bertin, B.; Zugman, M.; Schvartsman, G. The current treatment landscape of malignant pleural mesothelioma and future directions. Cancers 2023, 15, 5808. [Google Scholar] [CrossRef]
- Menges, C.W.; Chen, Y.; Mossman, B.T.; Chernoff, J.; Yeung, A.T.; Testa, J.R. A phosphotyrosine proteomic screen identifies multiple tyrosine kinase signaling pathways aberrantly activated in malignant mesothelioma. Genes Cancer 2010, 1, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sekine, M.; Virgona, N.; Ota, M.; Yano, T. Yes is a central mediator of cell growth in malignant mesothelioma cells. Oncol. Rep. 2012, 28, 1889–1893. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.M. Crk family adaptors-signalling complex formation and biological roles. Oncogene 2001, 20, 6348–6371. [Google Scholar] [CrossRef]
- Yeung, C.L.; Ngo, V.N.; Grohar, P.J.; Arnaldez, F.I.; Asante, A.; Wan, X.; Khan, J.; Hewitt, S.M.; Khanna, C.; Staudt, L.M.; et al. Loss-of-function screen in rhabdomyosarcoma identifies CRKL-YES as a critical signal for tumor growth. Oncogene 2013, 32, 5429–5438. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.R.; Windham, T.C.; Estrella, V.C.; Parikh, N.U.; Thall, P.F.; Deavers, M.T.; Bast, R.C.; Mills, G.B.; Gallick, G.E. Activated SRC protein tyrosine kinase is overexpressed in late-stage human ovarian cancers. Gynecol. Oncol. 2003, 88, 73–79. [Google Scholar] [CrossRef]
- Espada, J.; Martin-Perez, J. An update on Src family of nonreceptor tyrosine kinases biology. Int. Rev. Cell Mol. Biol. 2017, 331, 83–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, X.; Zhong, M.Z.; Yang, S.; Zhou, J.; Klinkebiel, D.L.; Karpf, A.R.; Chen, Y.; Dong, J. Cyclin-dependent kinase 1-mediated phosphorylation of YES links mitotic arrest and apoptosis during antitubulin chemotherapy. Cell. Signal. 2018, 52, 137–146. [Google Scholar] [CrossRef]
- Fornari, F.; Milazzo, M.; Chieco, P.; Negrini, M.; Calin, G.A.; Grazi, G.L.; Pollutri, D.; Croce, C.M.; Bolondi, L.; Gramantieri, L. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2010, 70, 5184–5193. [Google Scholar] [CrossRef]
- Chen, L.; Cao, H.; Feng, Y. MiR-199a suppresses prostate cancer paclitaxel resistance by targeting YES1. World J. Urol. 2018, 36, 357–365. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, C.; Ding, J.; Chen, Y.; Sun, Y.; Cheng, Z. miR-133a targets YES1 to reduce cisplatin resistance in ovarian cancer by regulating cell autophagy. Cancer Cell Int. 2022, 22, 15. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Sun, D.; Hou, H. Role of YES1 amplification in EGFR mutation-positive non-small cell lung cancer: Primary resistance to afatinib in a patient. Thorac. Cancer 2020, 11, 2736–2739. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Liu, J.; Qian, J.; Meng, C.; Guo, J.; Gao, W.; Xiong, B.; Ling, C.; Zhang, Y. Recent advances in multi-target drugs targeting protein kinases and histone deacetylases in cancer therapy. Curr. Med. Chem. 2020, 27, 7264–7288. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Rao, S.; Gurbani, D.; Henning, N.J.; Jiang, J.; Che, J.; Yang, A.; Ficarro, S.B.; Marto, J.A.; Aguirre, A.J.; et al. Structure-based design of a potent and selective covalent inhibitor for SRC kinase that targets a P-Loop cysteine. J. Med. Chem. 2020, 63, 1624–1641. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kook, E.; Chun, K.-S.; Kim, D.-H. Emerging Roles of YES1 in Cancer: The Putative Target in Drug Resistance. Int. J. Mol. Sci. 2024, 25, 1450. https://doi.org/10.3390/ijms25031450
Kook E, Chun K-S, Kim D-H. Emerging Roles of YES1 in Cancer: The Putative Target in Drug Resistance. International Journal of Molecular Sciences. 2024; 25(3):1450. https://doi.org/10.3390/ijms25031450
Chicago/Turabian StyleKook, Eunjin, Kyung-Soo Chun, and Do-Hee Kim. 2024. "Emerging Roles of YES1 in Cancer: The Putative Target in Drug Resistance" International Journal of Molecular Sciences 25, no. 3: 1450. https://doi.org/10.3390/ijms25031450
APA StyleKook, E., Chun, K.-S., & Kim, D.-H. (2024). Emerging Roles of YES1 in Cancer: The Putative Target in Drug Resistance. International Journal of Molecular Sciences, 25(3), 1450. https://doi.org/10.3390/ijms25031450