

Consistency between Primary Uterine Corpus Malignancies and Their Corresponding Patient-Derived Xenograft Models
 ,
,  , ,
, ,
Abstract
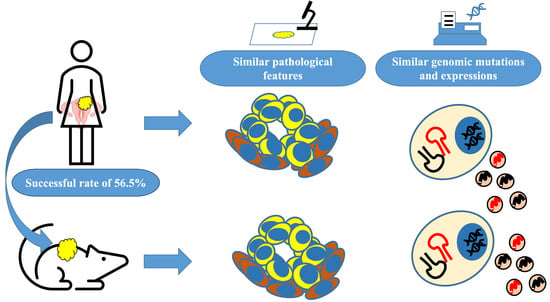
1. Introduction
2. Results
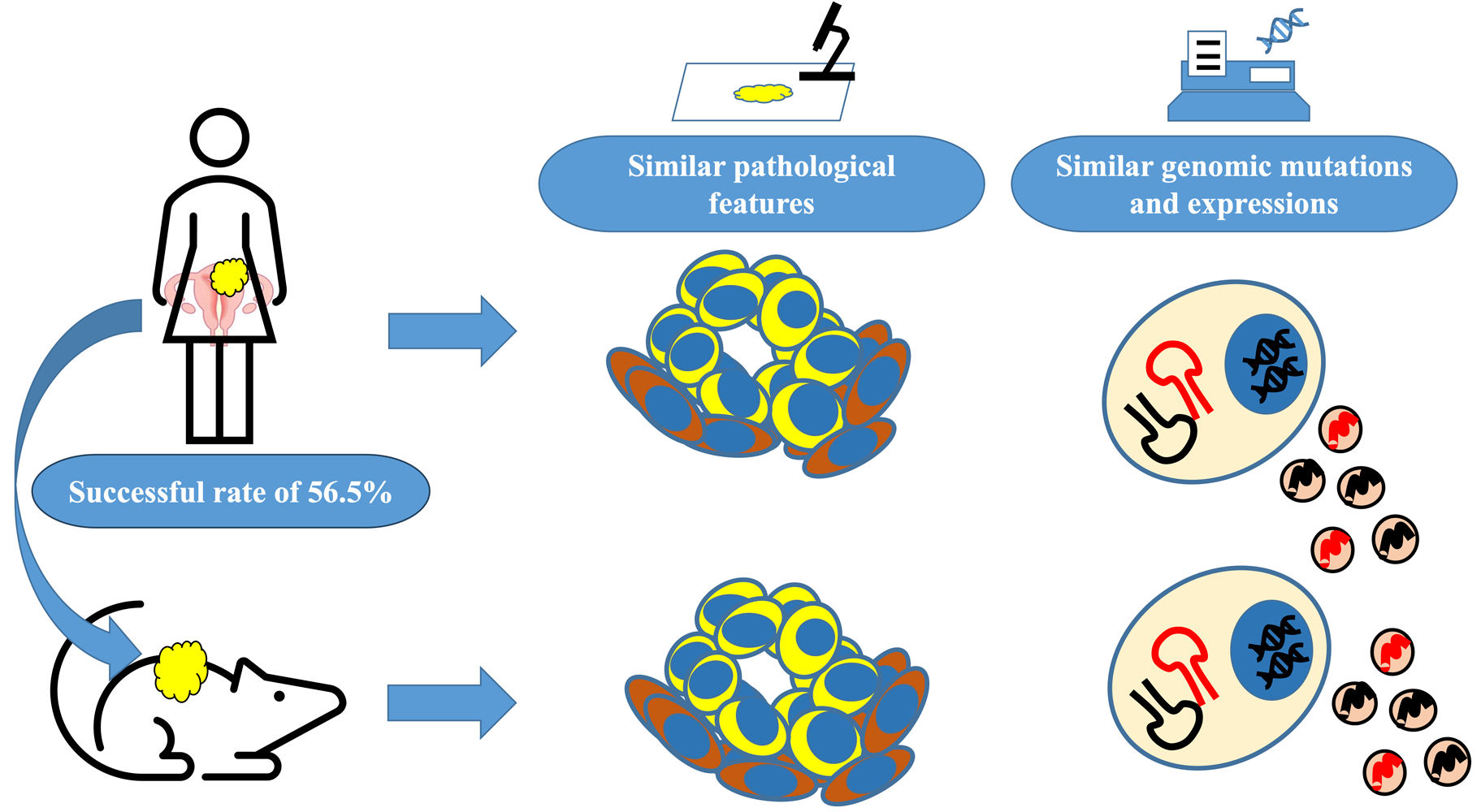
2.1. Establishment of the UC-PDX Models
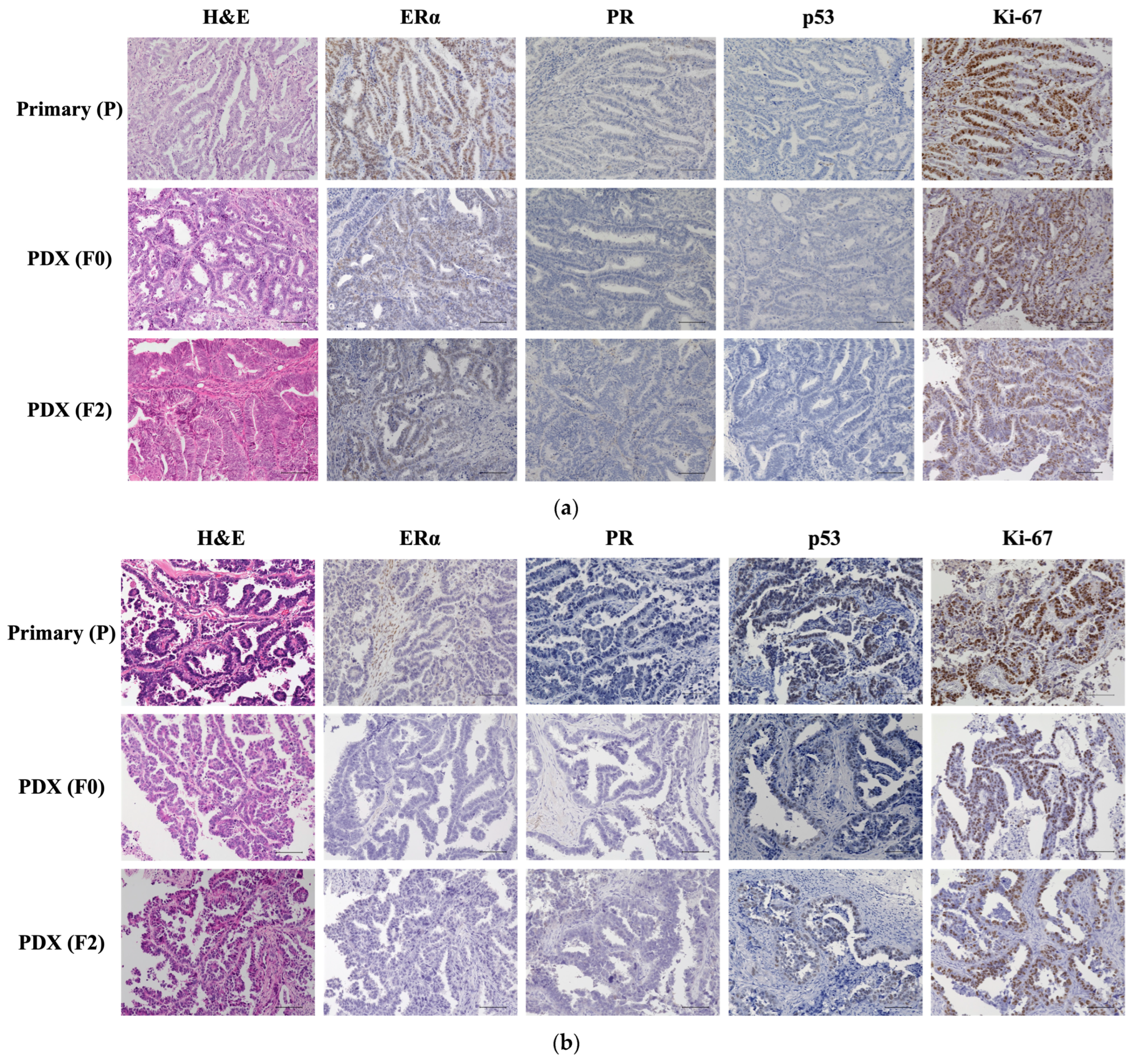
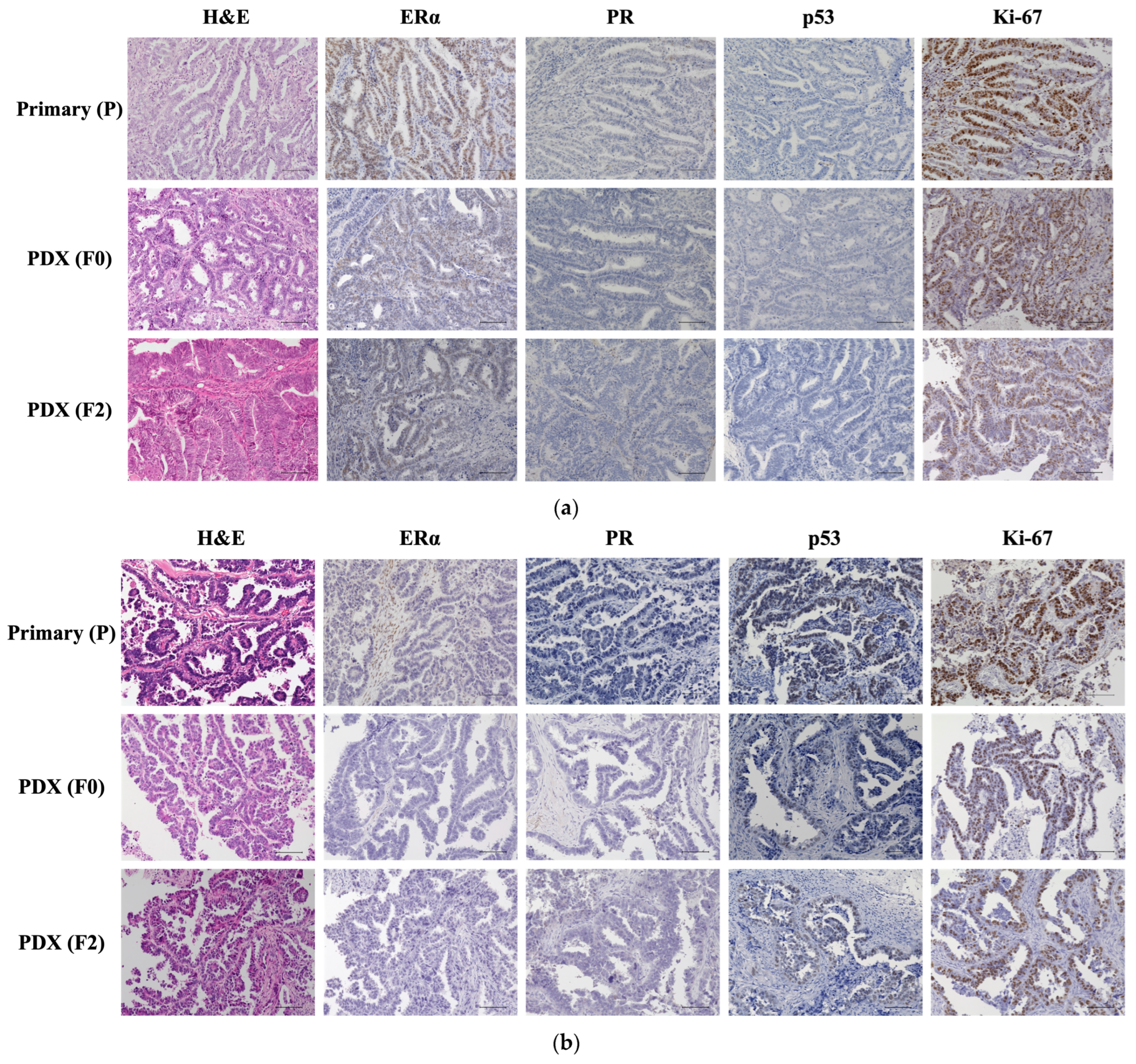
2.2. Histological Evaluation of Patient and PDX Tumors
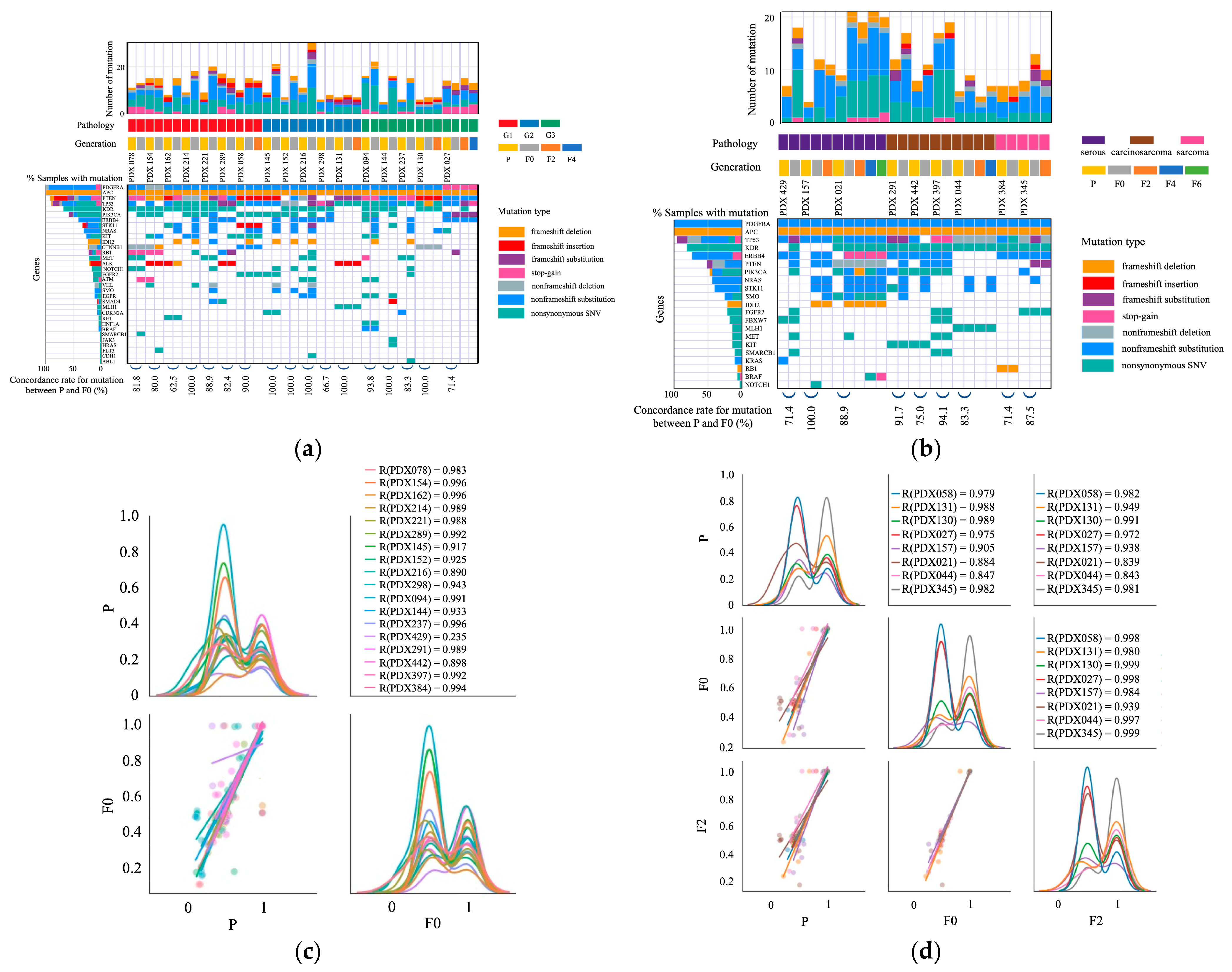
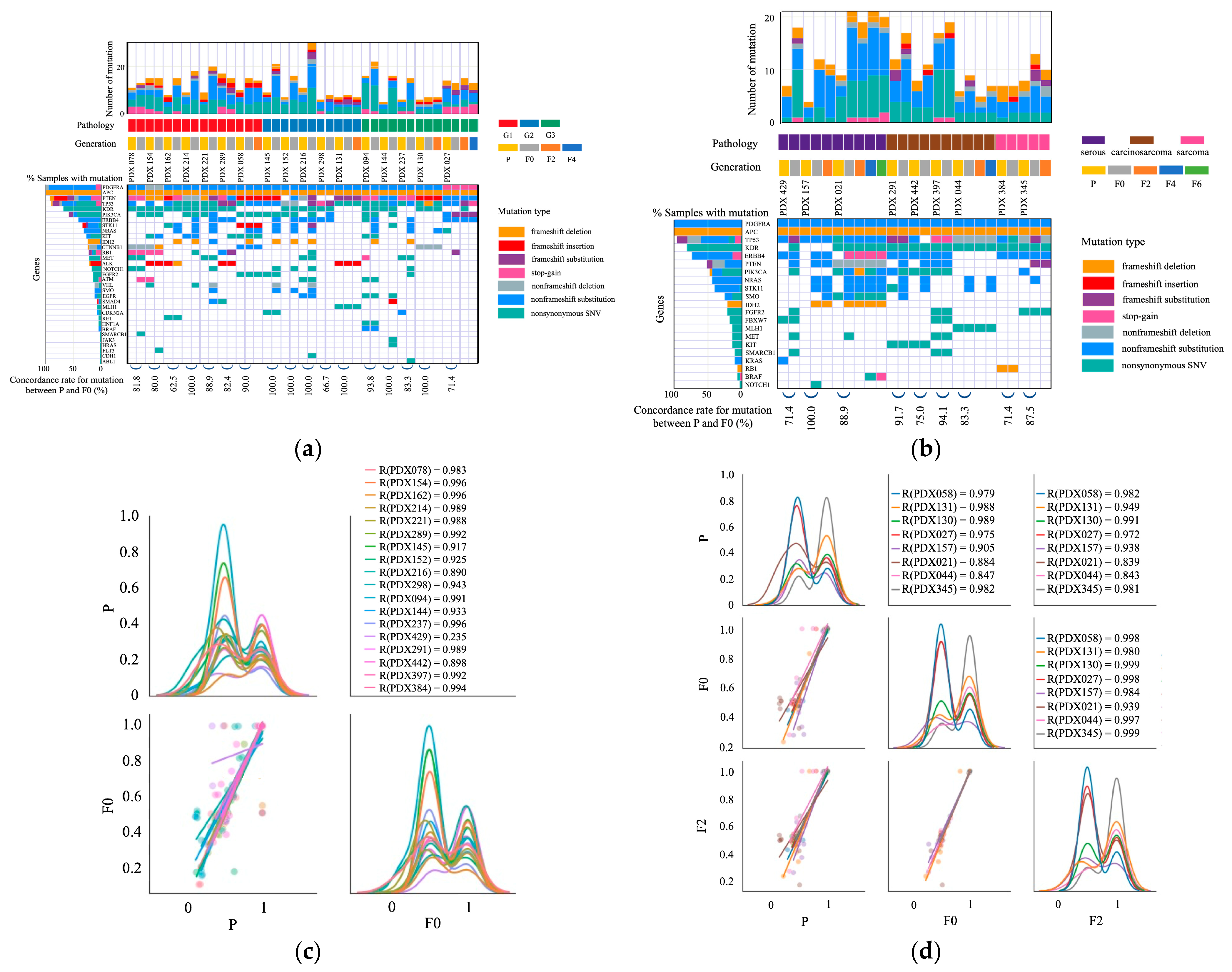
2.3. Mutations in Primary and PDX Tumors
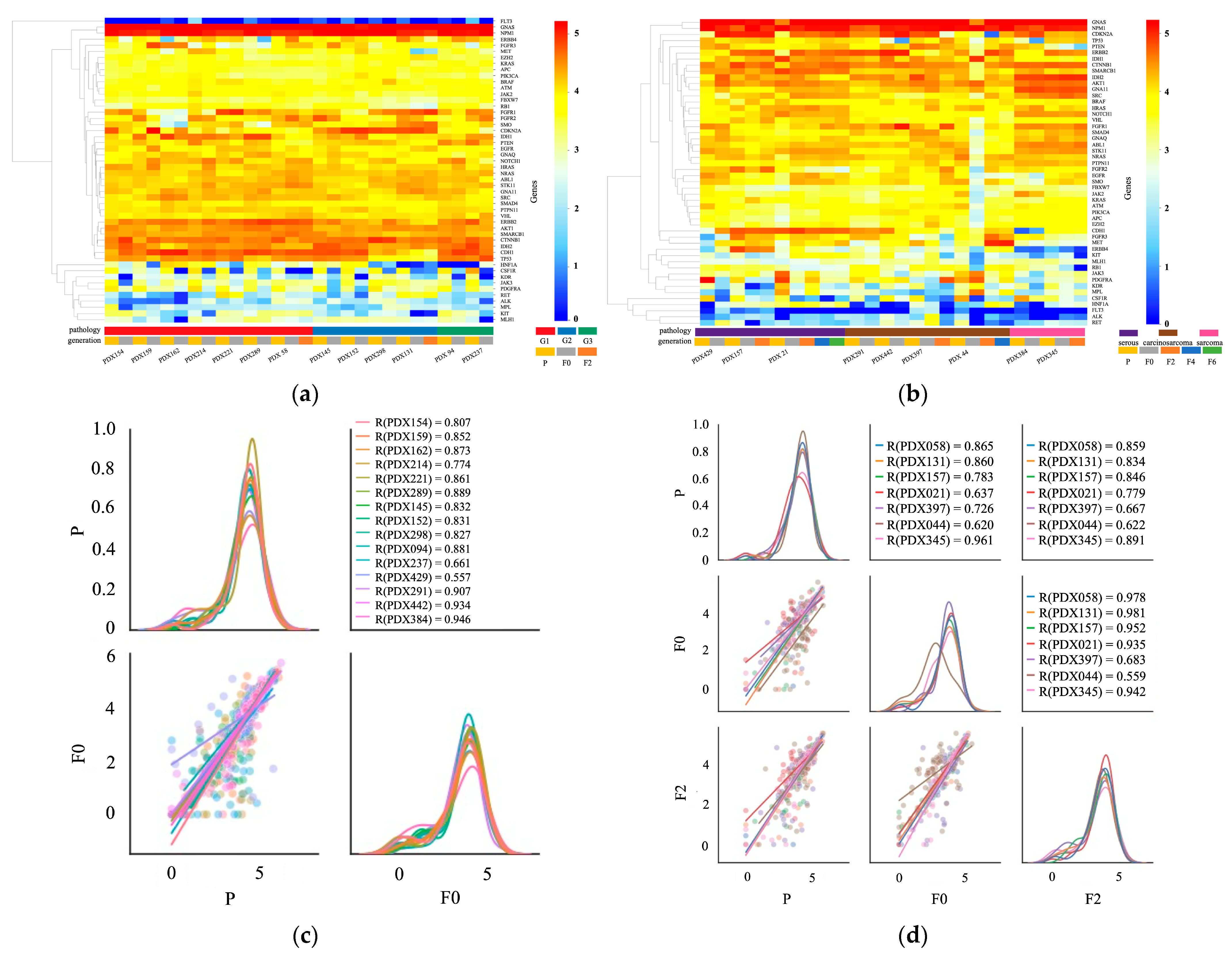
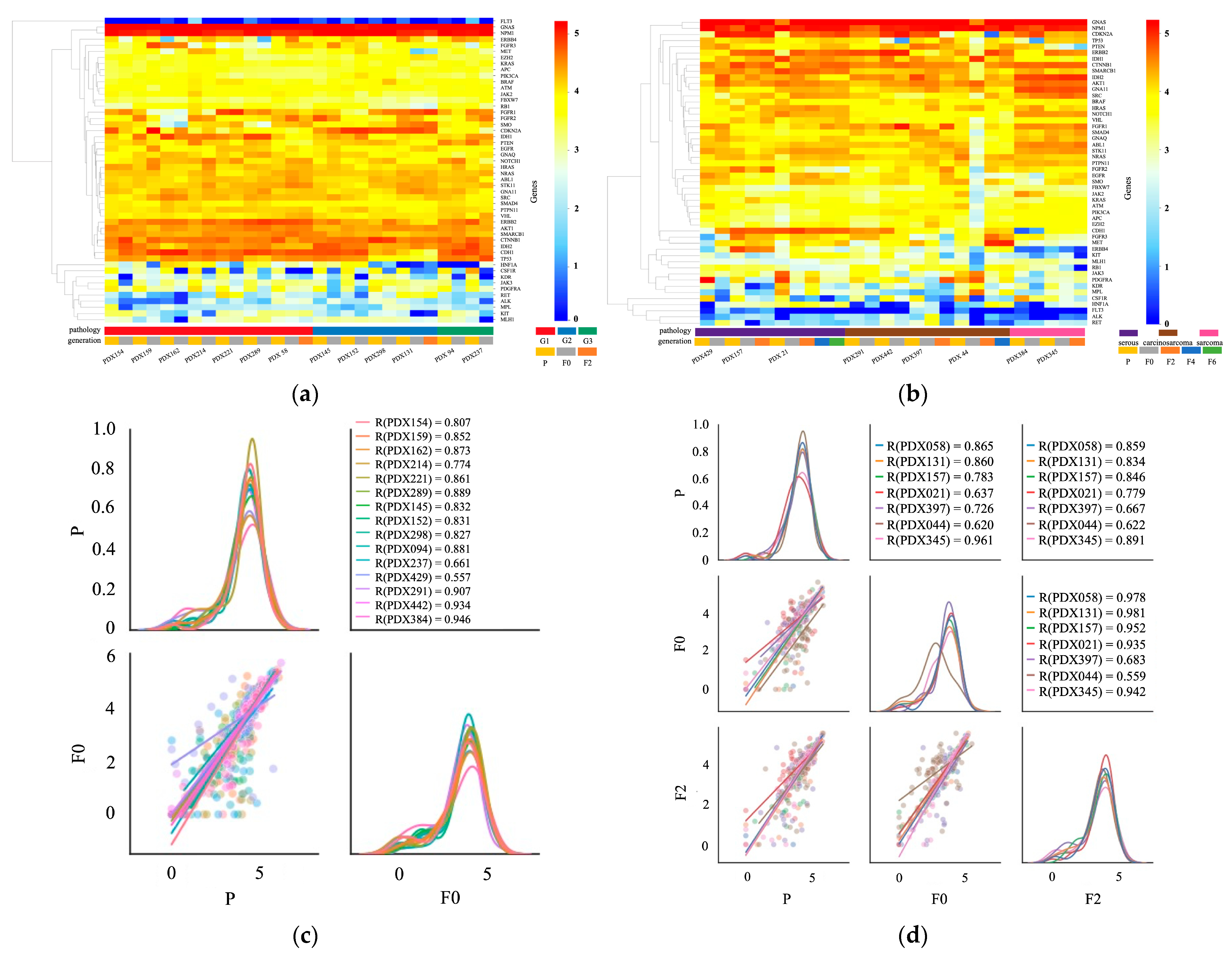
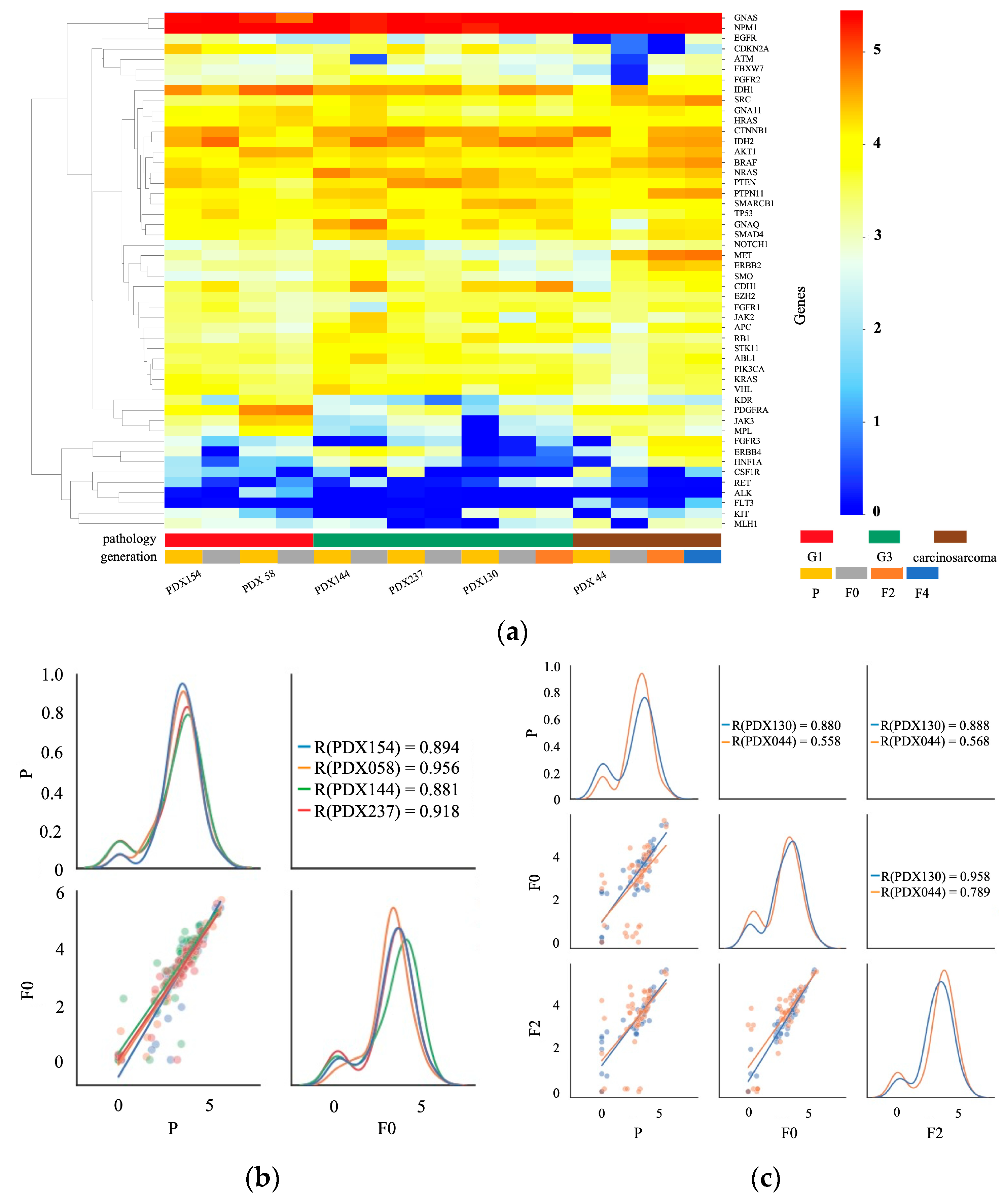
2.4. Transcription Profile of Primary and PDX Tumors
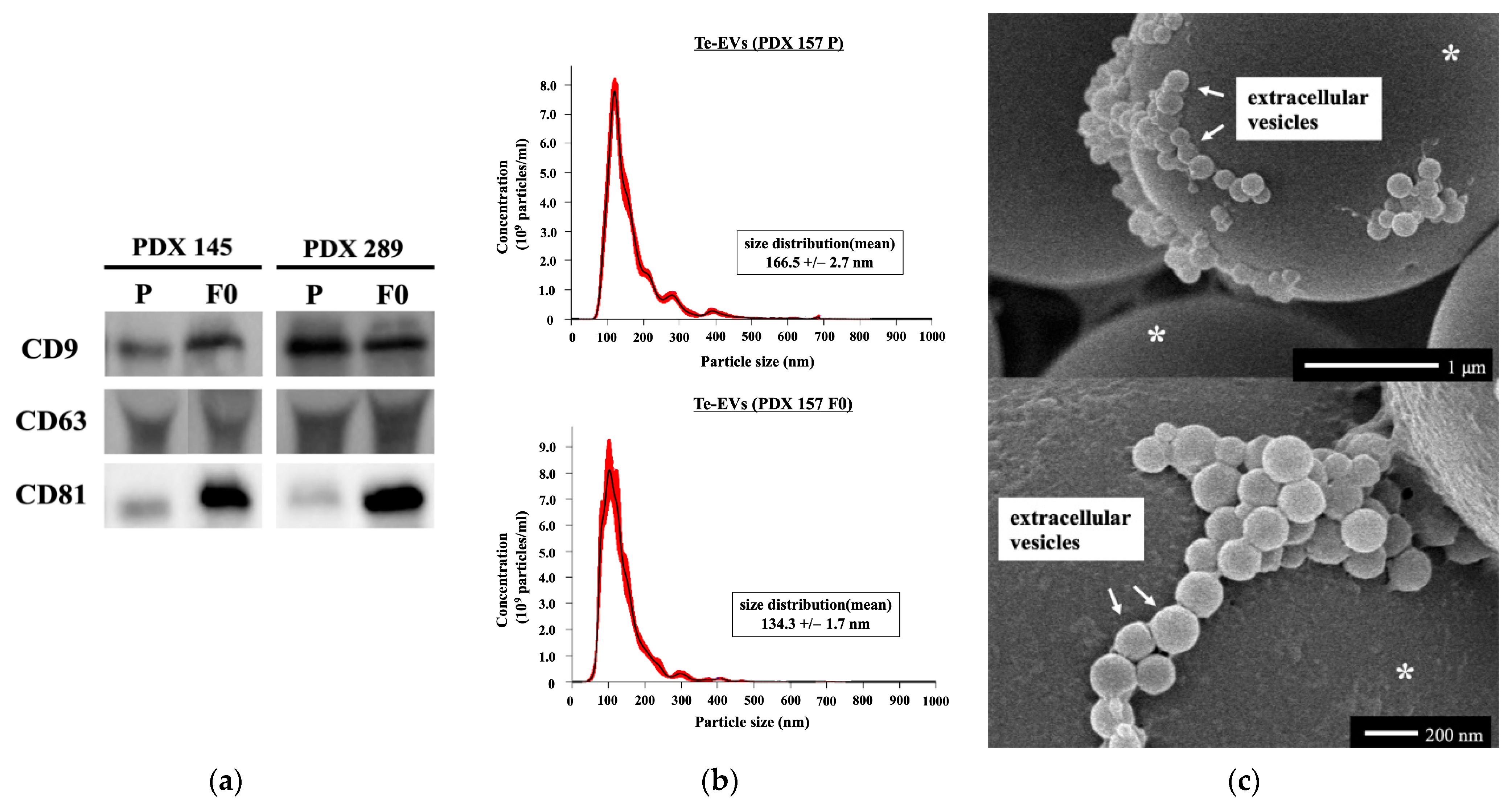
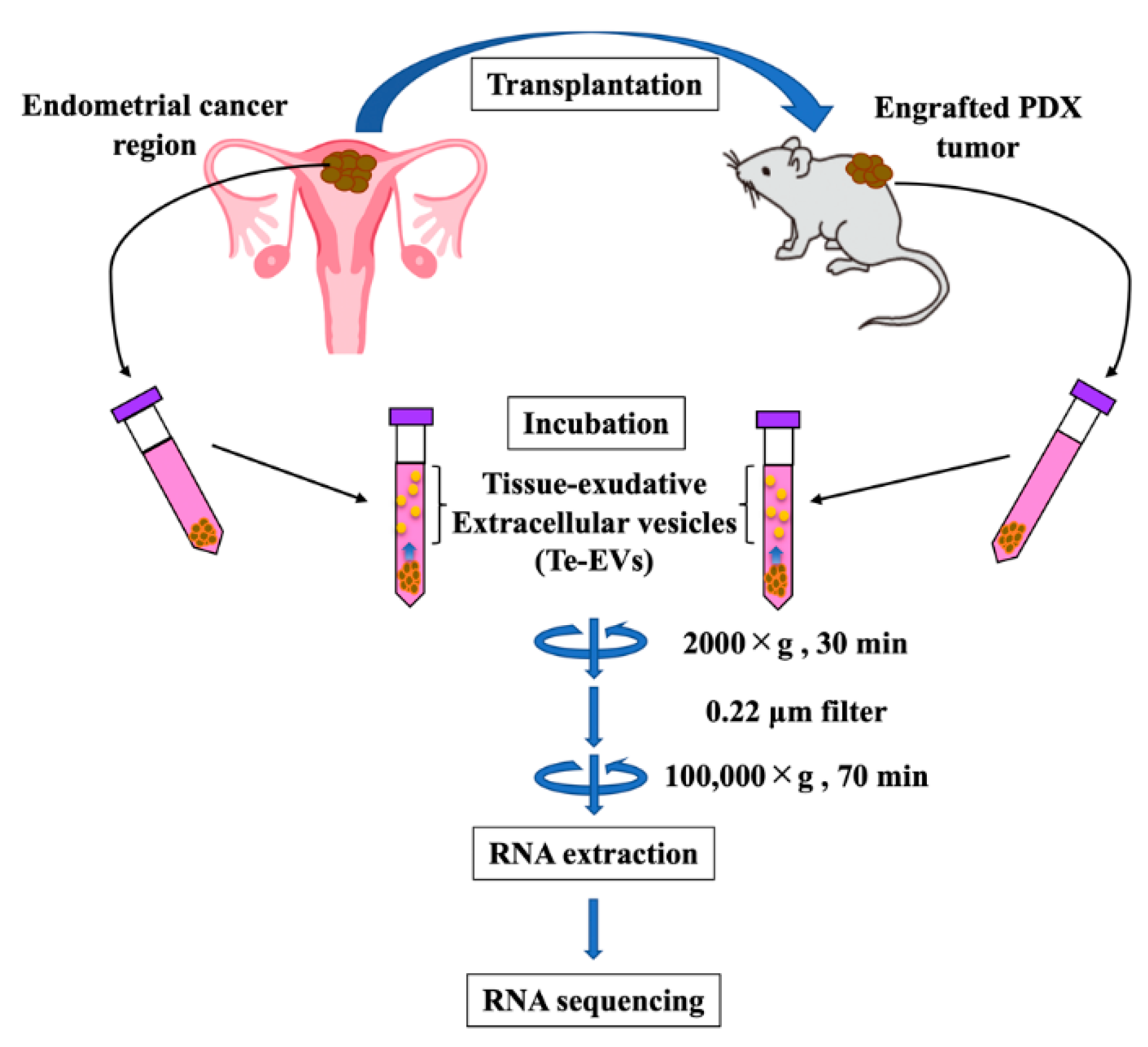
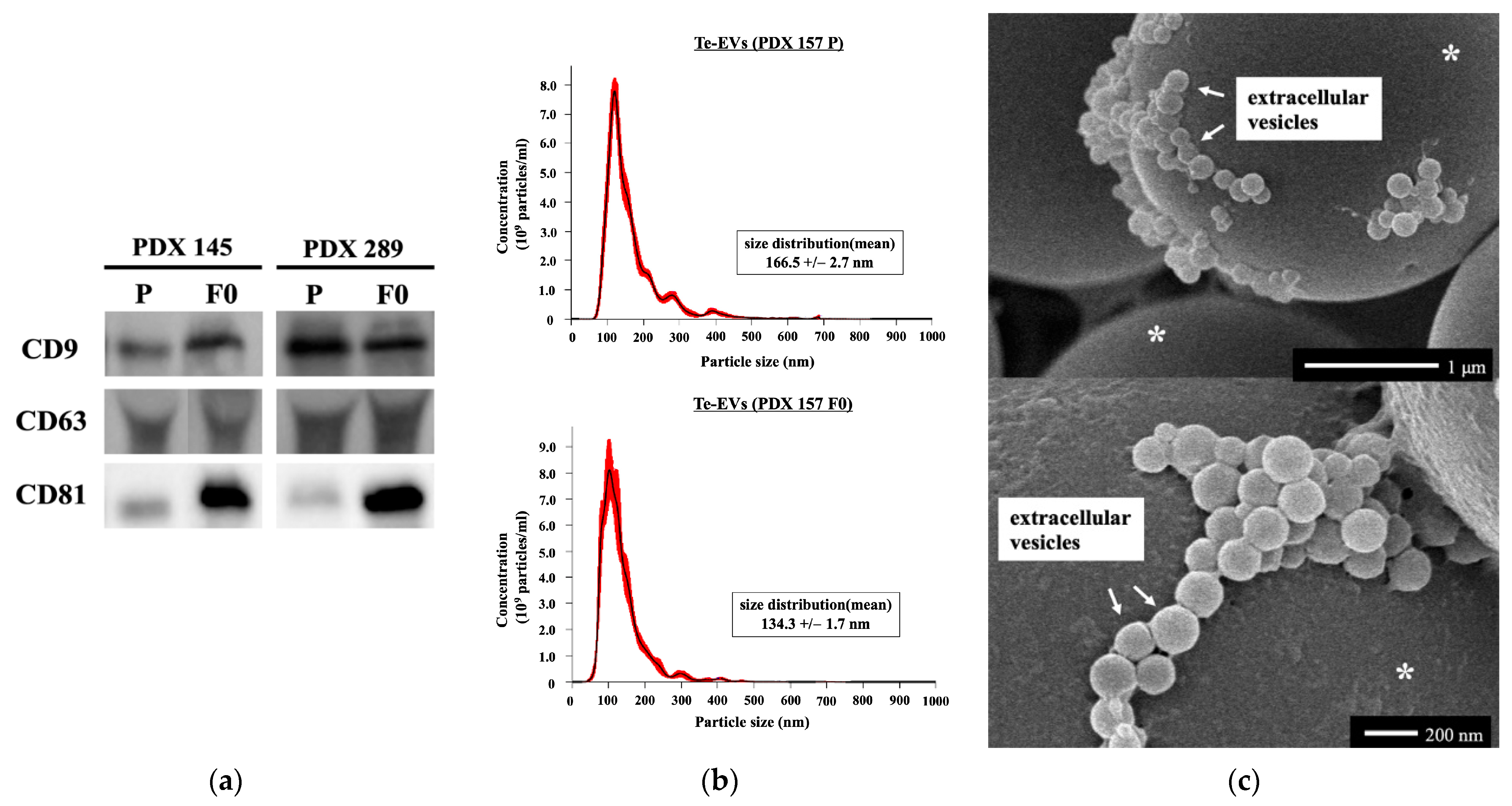
2.5. RNA Expression of Tissue-Exudative Extracellular Vesicles in Primary and PDX Tumors
3. Discussion
3.1. Success Rate and Transplantation Method
3.2. Comparison of Original Tumors and PDXs
3.3. Clinical Applications of the PDX Model
3.4. Problems in the PDX Model
4. Materials and Methods
4.1. Patients and Tissue Samples
4.2. Animals
4.3. Establishment of PDX
4.4. Pathological Analysis Using Immunohistochemistry
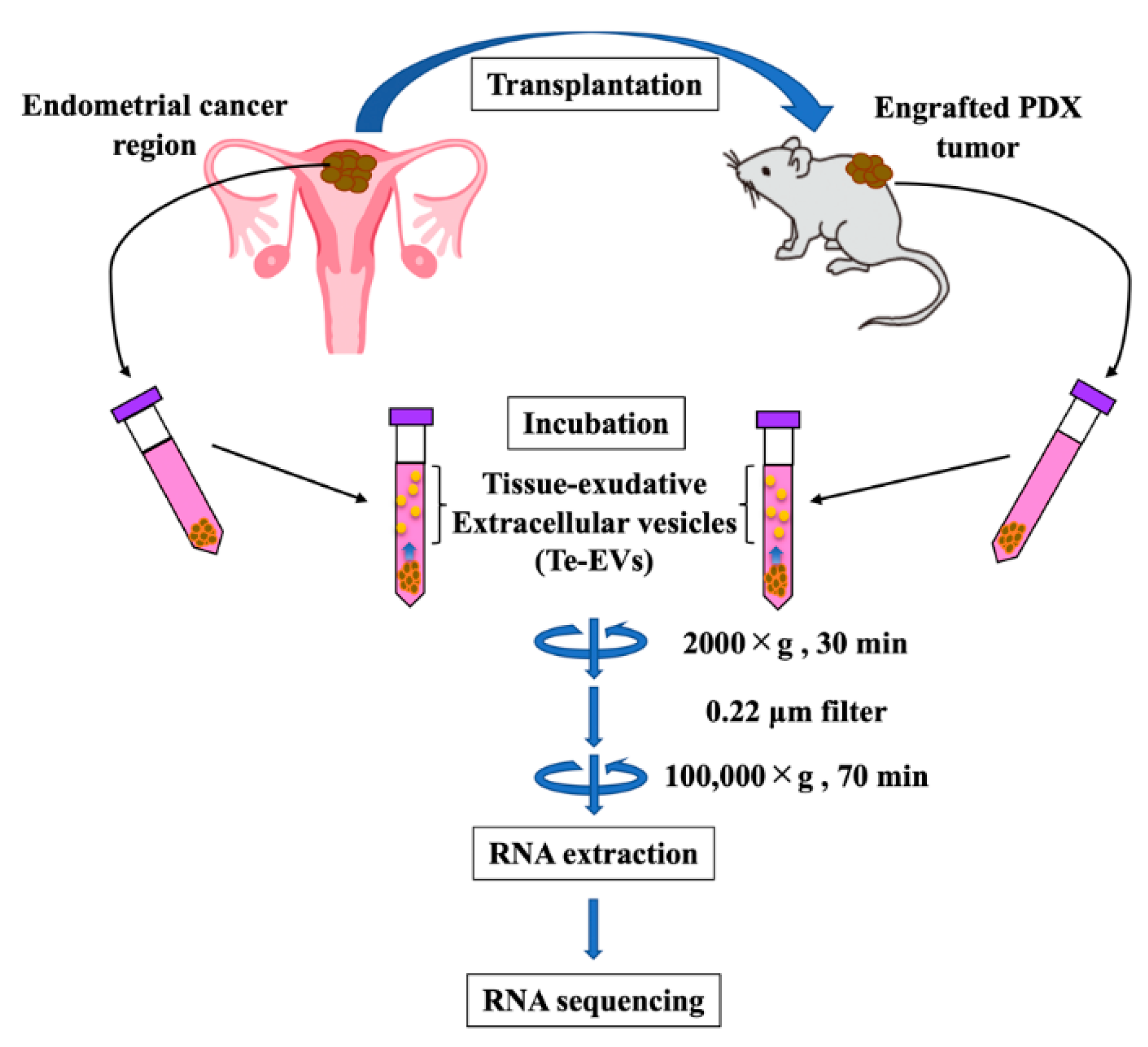
4.5. Purification of Te-EVs
4.6. Western Blot Analysis
4.7. NTA
4.8. Scanning Electron Microscopy
4.9. DNA and RNA Extraction
4.10. Amplicon Sequencing
4.11. RNA Sequencing
4.12. Bioinformatics Analysis
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lewin, S.N.; Herzog, T.J.; Barrena Medel, N.I.; Deutsch, I.; Burke, W.M.; Sun, X.; Wright, J.D. Comparative performance of the 2009 international Federation of gynecology and obstetrics’ staging system for uterine corpus cancer. Obstet. Gynecol. 2010, 116, 1141–1149. [Google Scholar] [CrossRef]
- Carey, M.S.; O’Connell, G.J.; Johanson, C.R.; Goodyear, M.D.; Murphy, K.J.; Daya, D.M.; Schepansky, A.; Peloquin, A.; Lumsden, B.J. Good outcome associated with a standardized treatment protocol using selective postoperative radiation in patients with clinical stage I adenocarcinoma of the endometrium. Gynecol. Oncol. 1995, 57, 138–144. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Daniel, V.C.; Marchionni, L.; Hierman, J.S.; Rhodes, J.T.; Devereux, W.L.; Rudin, C.M.; Yung, R.; Parmigiani, G.; Dorsch, M.; Peacock, C.D.; et al. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009, 69, 3364–3373. [Google Scholar] [CrossRef]
- Gillet, J.P.; Calcagno, A.M.; Varma, S.; Marino, M.; Green, L.J.; Vora, M.I.; Patel, C.; Orina, J.N.; Eliseeva, T.A.; Singal, V.; et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 18708–18713. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.I.; Decker, S.; Zaharevitz, D.; Rubinstein, L.V.; Venditti, J.M.; Schepartz, S.; Kalyandrug, S.; Christian, M.; Arbuck, S.; Hollingshead, M.; et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br. J. Cancer 2001, 84, 1424–1431. [Google Scholar] [CrossRef]
- Jiang, W.; Xie, S.; Liu, Y.; Zou, S.; Zhu, X. The Application of Patient-Derived Xenograft Models in Gynecologic Cancers. J. Cancer 2020, 11, 5478–5489. [Google Scholar] [CrossRef]
- Depreeuw, J.; Hermans, E.; Schrauwen, S.; Annibali, D.; Coenegrachts, L.; Thomas, D.; Luyckx, M.; Gutierrez-Roelens, I.; Debruyne, D.; Konings, K.; et al. Characterization of patient-derived tumor xenograft models of endometrial cancer for preclinical evaluation of targeted therapies. Gynecol. Oncol. 2015, 139, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nishie, R.; Ueda, S.; Miyamoto, S.; Hashida, S.; Konishi, H.; Terada, S.; Kogata, Y.; Sasaki, H.; Tsunetoh, S.; et al. Endometrial Cancer Patient-Derived Xenograft Models: A Systematic Review. J. Clin. Med. 2022, 11, 2606. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Cho, Y.J.; Ryu, J.Y.; Choi, J.J.; Hwang, J.R.; Kim, B.; Lee, Y.Y.; Kim, H.S.; Lee, J.W. Establishment and preclinical application of a patient-derived xenograft model for uterine cancer. Gynecol. Oncol. 2021, 162, 173–181. [Google Scholar] [CrossRef]
- Zhu, M.; Jia, N.; Nie, Y.; Chen, J.; Jiang, Y.; Lv, T.; Li, Y.; Yao, L.; Feng, W. Establishment of Patient-Derived Tumor Xenograft Models of High-Risk Endometrial Cancer. Int. J. Gynecol. Cancer 2018, 28, 1812–1820. [Google Scholar] [CrossRef]
- Villafranca-Magdalena, B.; Masferrer-Ferragutcasas, C.; Lopez-Gil, C.; Coll-de la Rubia, E.; Rebull, M.; Parra, G.; Garcia, A.; Reques, A.; Cabrera, S.; Colas, E.; et al. Genomic Validation of Endometrial Cancer Patient-Derived Xenograft Models as a Preclinical Tool. Int. J. Mol. Sci. 2022, 23, 6266. [Google Scholar] [CrossRef]
- Miyamoto, S.; Tanaka, T.; Hirosuna, K.; Nishie, R.; Ueda, S.; Hashida, S.; Terada, S.; Konishi, H.; Kogata, Y.; Taniguchi, K.; et al. Validation of a Patient-Derived Xenograft Model for Cervical Cancer Based on Genomic and Phenotypic Characterization. Cancers 2022, 14, 2969. [Google Scholar] [CrossRef]
- Groeneweg, J.W.; Hernandez, S.F.; Byron, V.F.; DiGloria, C.M.; Lopez, H.; Scialabba, V.; Kim, M.; Zhang, L.; Borger, D.R.; Tambouret, R.; et al. Dual HER2 targeting impedes growth of HER2 gene-amplified uterine serous carcinoma xenografts. Clin. Cancer Res. 2014, 20, 6517–6528. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, C.; Wei, Z.; Zhang, R.; Wang, Y.; Jiang, L.; Chen, K.; Qiu, S.; Zhang, Y.; Zhang, T.; et al. Patient-derived non-small cell lung cancer xenograft mirrors complex tumor heterogeneity. Cancer Biol. Med. 2021, 18, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-derived xenograft (PDX) models, applications and challenges in cancer research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef] [PubMed]
- Bonazzi, V.F.; Kondrashova, O.; Smith, D.; Nones, K.; Sengal, A.T.; Ju, R.; Packer, L.M.; Koufariotis, L.T.; Kazakoff, S.H.; Davidson, A.L.; et al. Patient-derived xenograft models capture genomic heterogeneity in endometrial cancer. Genome Med. 2022, 14, 3. [Google Scholar] [CrossRef]
- Shin, H.Y.; Lee, E.J.; Yang, W.; Kim, H.S.; Chung, D.; Cho, H.; Kim, J.H. Identification of Prognostic Markers of Gynecologic Cancers Utilizing Patient-Derived Xenograft Mouse Models. Cancers 2022, 14, 829. [Google Scholar] [CrossRef]
- Zanella, E.R.; Grassi, E.; Trusolino, L. Towards precision oncology with patient-derived xenografts. Nat. Rev. Clin. Oncol. 2022, 19, 719–732. [Google Scholar] [CrossRef]
- Dobrolecki, L.E.; Airhart, S.D.; Alferez, D.G.; Aparicio, S.; Behbod, F.; Bentires-Alj, M.; Brisken, C.; Bult, C.J.; Cai, S.; Clarke, R.B.; et al. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016, 35, 547–573. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.L.; Miller, A.L.; Yoon, K.J. Patient-Derived Xenograft Models of Pancreatic Cancer: Overview and Comparison with Other Types of Models. Cancers 2020, 12, 1327. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Lang, S.H. A systematic review of the validity of patient derived xenograft (PDX) models: The implications for translational research and personalised medicine. PeerJ 2018, 6, e5981. [Google Scholar] [CrossRef]
- Sueyoshi, K.; Komura, D.; Katoh, H.; Yamamoto, A.; Onoyama, T.; Chijiwa, T.; Isagawa, T.; Tanaka, M.; Suemizu, H.; Nakamura, M.; et al. Multi-tumor analysis of cancer-stroma interactomes of patient-derived xenografts unveils the unique homeostatic process in renal cell carcinomas. iScience 2021, 24, 103322. [Google Scholar] [CrossRef] [PubMed]
- Cai, E.Y.; Garcia, J.; Liu, Y.; Vakar-Lopez, F.; Arora, S.; Nguyen, H.M.; Lakely, B.; Brown, L.; Wong, A.; Montgomery, B.; et al. A bladder cancer patient-derived xenograft displays aggressive growth dynamics in vivo and in organoid culture. Sci. Rep. 2021, 11, 4609. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nishie, R.; Ueda, S.; Miyamoto, S.; Hashida, S.; Konishi, H.; Terada, S.; Kogata, Y.; Sasaki, H.; Tsunetoh, S.; et al. Patient-Derived Xenograft Models in Cervical Cancer: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 9369. [Google Scholar] [CrossRef] [PubMed]
- Cybula, M.; Wang, L.; Wang, L.; Drumond-Bock, A.L.; Moxley, K.M.; Benbrook, D.M.; Gunderson-Jackson, C.; Ruiz-Echevarria, M.J.; Bhattacharya, R.; Mukherjee, P.; et al. Patient-Derived Xenografts of High-Grade Serous Ovarian Cancer Subtype as a Powerful Tool in Pre-Clinical Research. Cancers 2021, 13, 6288. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef]
- Moiola, C.P.; Lopez-Gil, C.; Cabrera, S.; Garcia, A.; Van Nyen, T.; Annibali, D.; Fonnes, T.; Vidal, A.; Villanueva, A.; Matias-Guiu, X.; et al. Patient-Derived Xenograft Models for Endometrial Cancer Research. Int. J. Mol. Sci. 2018, 19, 2431. [Google Scholar] [CrossRef]
- Kogure, A.; Yoshioka, Y.; Ochiya, T. Extracellular Vesicles in Cancer Metastasis: Potential as Therapeutic Targets and Materials. Int. J. Mol. Sci. 2020, 21, 4463. [Google Scholar] [CrossRef]
- Santos, M.F.; Rappa, G.; Karbanová, J.; Fontana, S.; Bella, M.A.D.; Pope, M.R.; Parrino, B.; Cascioferro, S.M.; Vistoli, G.; Diana, P.; et al. Itraconazole inhibits nuclear delivery of extracellular vesicle cargo by disrupting the entry of late endosomes into the nucleoplasmic reticulum. J. Extracell. Vesicles 2021, 10, e12132. [Google Scholar] [CrossRef]
- Santos, M.F.; Rappa, G.; Karbanová, J.; Diana, P.; Cirrincione, G.; Carbone, D.; Manna, D.; Aalam, F.; Wang, D.; Vanier, C.; et al. HIV-1-induced nuclear invaginations mediated by VAP-A, ORP3, and Rab7 complex explain infection of activated T cells. Nat. Commun. 2023, 14, 4588. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, R.; Wang, L.; Correa, A.M.; Pataer, A.; Xu, Y.; Zhang, X.; Ren, C.; Wu, S.; Meng, Q.H.; et al. Tumor characteristics associated with engraftment of patient-derived non-small cell lung cancer xenografts in immunocompromised mice. Cancer 2019, 125, 3738–3748. [Google Scholar] [CrossRef]
- Cabrera, S.; Llauradó, M.; Castellví, J.; Fernandez, Y.; Alameda, F.; Colás, E.; Ruiz, A.; Doll, A.; Schwartz, S., Jr.; Carreras, R.; et al. Generation and characterization of orthotopic murine models for endometrial cancer. Clin. Exp. Metastasis 2012, 29, 217–227. [Google Scholar] [CrossRef]
- Haldorsen, I.S.; Popa, M.; Fonnes, T.; Brekke, N.; Kopperud, R.; Visser, N.C.; Rygh, C.B.; Pavlin, T.; Salvesen, H.B.; McCormack, E.; et al. Multimodal Imaging of Orthotopic Mouse Model of Endometrial Carcinoma. PLoS ONE 2015, 10, e0135220. [Google Scholar] [CrossRef]
- Abel, L.; Durmaz, A.; Hu, R.; Longhurst, C.; Baschnagel, A.M.; Wheeler, D.; Scott, J.G.; Kimple, R.J. Impact of immediate cryopreservation on the establishment of patient derived xenografts from head and neck cancer patients. J. Transl. Med. 2021, 19, 180. [Google Scholar] [CrossRef]
- Alkema, N.G.; Tomar, T.; Duiker, E.W.; Jan Meersma, G.; Klip, H.; van der Zee, A.G.; Wisman, G.B.; de Jong, S. Biobanking of patient and patient-derived xenograft ovarian tumour tissue: Efficient preservation with low and high fetal calf serum based methods. Sci. Rep. 2015, 5, 14495. [Google Scholar] [CrossRef]
- Cui, J.H.; Krueger, U.; Henne-Bruns, D.; Kremer, B.; Kalthoff, H. Orthotopic transplantation model of human gastrointestinal cancer and detection of micrometastases. World J. Gastroenterol. 2001, 7, 381–386. [Google Scholar] [CrossRef]
- Linnebacher, M.; Maletzki, C.; Ostwald, C.; Klier, U.; Krohn, M.; Klar, E.; Prall, F. Cryopreservation of human colorectal carcinomas prior to xenografting. BMC Cancer 2010, 10, 362. [Google Scholar] [CrossRef]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells 2019, 8, 889. [Google Scholar] [CrossRef]
- Xu, C.; Li, X.; Liu, P.; Li, M.; Luo, F. Patient-derived xenograft mouse models: A high fidelity tool for individualized medicine. Oncol. Lett. 2019, 17, 3–10. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.; Lee, J.E.; Shin, S.J.; Oh, S.; Kwon, G.; Kim, H.; Choi, Y.Y.; White, M.A.; Paik, S.; et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 Toward Gastric Cancer Cells With Markers of the Epithelial-Mesenchymal Transition, Due to Loss of NAPRT. Gastroenterology 2018, 155, 799–814.e713. [Google Scholar] [CrossRef]
- Reddavid, R.; Corso, S.; Moya-Rull, D.; Giordano, S.; Degiuli, M. Patient-Derived Orthotopic Xenograft models in gastric cancer: A systematic review. Updates Surg. 2020, 72, 951–966. [Google Scholar] [CrossRef]
- Butler, K.A.; Hou, X.; Becker, M.A.; Zanfagnin, V.; Enderica-Gonzalez, S.; Visscher, D.; Kalli, K.R.; Tienchaianada, P.; Haluska, P.; Weroha, S.J. Prevention of Human Lymphoproliferative Tumor Formation in Ovarian Cancer Patient-Derived Xenografts. Neoplasia 2017, 19, 628–636. [Google Scholar] [CrossRef]
- Taurozzi, A.J.; Beekharry, R.; Wantoch, M.; Labarthe, M.C.; Walker, H.F.; Seed, R.I.; Simms, M.; Rodrigues, G.; Bradford, J.; van der Horst, G.; et al. Spontaneous development of Epstein-Barr Virus associated human lymphomas in a prostate cancer xenograft program. PLoS ONE 2017, 12, e0188228. [Google Scholar] [CrossRef]
- Choi, Y.Y.; Lee, J.E.; Kim, H.; Sim, M.H.; Kim, K.K.; Lee, G.; Kim, H.I.; An, J.Y.; Hyung, W.J.; Kim, C.B.; et al. Establishment and characterisation of patient-derived xenografts as paraclinical models for gastric cancer. Sci. Rep. 2016, 6, 22172. [Google Scholar] [CrossRef]
- Unno, K.; Ono, M.; Winder, A.D.; Maniar, K.P.; Paintal, A.S.; Yu, Y.; Wei, J.J.; Lurain, J.R.; Kim, J.J. Establishment of human patient-derived endometrial cancer xenografts in NOD scid gamma mice for the study of invasion and metastasis. PLoS ONE 2014, 9, e116064. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef]
- Jingushi, K.; Uemura, M.; Ohnishi, N.; Nakata, W.; Fujita, K.; Naito, T.; Fujii, R.; Saichi, N.; Nonomura, N.; Tsujikawa, K.; et al. Extracellular vesicles isolated from human renal cell carcinoma tissues disrupt vascular endothelial cell morphology via azurocidin. Int. J. Cancer 2018, 142, 607–617. [Google Scholar] [CrossRef]
- Crescitelli, R.; Lässer, C.; Lötvall, J. Isolation and characterization of extracellular vesicle subpopulations from tissues. Nat. Protoc. 2021, 16, 1548–1580. [Google Scholar] [CrossRef] [PubMed]
- Li, S.R.; Man, Q.W.; Gao, X.; Lin, H.; Wang, J.; Su, F.C.; Wang, H.Q.; Bu, L.L.; Liu, B.; Chen, G. Tissue-derived extracellular vesicles in cancers and non-cancer diseases: Present and future. J. Extracell. Vesicles 2021, 10, e12175. [Google Scholar] [CrossRef]
- Hannafon, B.N.; Trigoso, Y.D.; Calloway, C.L.; Zhao, Y.D.; Lum, D.H.; Welm, A.L.; Zhao, Z.J.; Blick, K.E.; Dooley, W.C.; Ding, W.Q. Plasma exosome microRNAs are indicative of breast cancer. Breast Cancer Res. 2016, 18, 90. [Google Scholar] [CrossRef]
- Lasota, J.; Miettinen, M. KIT and PDGFRA mutations in gastrointestinal stromal tumors (GISTs). Semin. Diagn. Pathol. 2006, 23, 91–102. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef]
- Johnson, D.B.; Puzanov, I. Treatment of NRAS-mutant melanoma. Curr. Treat. Options Oncol. 2015, 16, 15. [Google Scholar] [CrossRef]
- Lim, W.; Olschwang, S.; Keller, J.J.; Westerman, A.M.; Menko, F.H.; Boardman, L.A.; Scott, R.J.; Trimbath, J.; Giardiello, F.M.; Gruber, S.B.; et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 2004, 126, 1788–1794. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef]
- Wang, D.; Pham, N.A.; Tong, J.; Sakashita, S.; Allo, G.; Kim, L.; Yanagawa, N.; Raghavan, V.; Wei, Y.; To, C.; et al. Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int. J. Cancer 2017, 140, 662–673. [Google Scholar] [CrossRef]
- Kim, J.; Rhee, H.; Kim, J.; Lee, S. Validity of patient-derived xenograft mouse models for lung cancer based on exome sequencing data. Genomics Inform. 2020, 18, e3. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Tosi, F.; Sartore-Bianchi, A.; Lonardi, S.; Amatu, A.; Leone, F.; Ghezzi, S.; Martino, C.; Bencardino, K.; Bonazzina, E.; Bergamo, F.; et al. Long-term Clinical Outcome of Trastuzumab and Lapatinib for HER2-positive Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2020, 19, 256–262.e252. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [PubMed]
- Hinze, L.; Labrosse, R.; Degar, J.; Han, T.; Schatoff, E.M.; Schreek, S.; Karim, S.; McGuckin, C.; Sacher, J.R.; Wagner, F.; et al. Exploiting the Therapeutic Interaction of WNT Pathway Activation and Asparaginase for Colorectal Cancer Therapy. Cancer Discov. 2020, 10, 1690–1705. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Kavuri, S.M.; Jain, N.; Galimi, F.; Cottino, F.; Leto, S.M.; Migliardi, G.; Searleman, A.C.; Shen, W.; Monsey, J.; Trusolino, L.; et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015, 5, 832–841. [Google Scholar] [CrossRef]
- Schütte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Stebbing, J.; Paz, K.; Schwartz, G.K.; Wexler, L.H.; Maki, R.; Pollock, R.E.; Morris, R.; Cohen, R.; Shankar, A.; Blackman, G.; et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014, 120, 2006–2015. [Google Scholar] [CrossRef]
- Hidalgo, M.; Bruckheimer, E.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Oliveira, E.; Rubio-Viqueira, B.; Strawn, S.; Wick, M.J.; Martell, J.; Sidransky, D. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol. Cancer Ther. 2011, 10, 1311–1316. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Kanikarla Marie, P.; Bitner, L.; Syed, M.; Woods, M.; Manyam, G.; Kwong, L.N.; Johnson, B.; Morris, V.K.; Jones, P.; et al. Targeting RAS Mutant Colorectal Cancer with Dual Inhibition of MEK and CDK4/6. Cancer Res. 2022, 82, 3335–3344. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.; Vrignaud, P.; Vacher, S.; Richon, S.; Lièvre, A.; Cacheux, W.; Weiswald, L.B.; Massonnet, G.; Chateau-Joubert, S.; Nicolas, A.; et al. Evaluating patient-derived colorectal cancer xenografts as preclinical models by comparison with patient clinical data. Cancer Res. 2015, 75, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.M.; Donoho, G.P.; Iversen, P.W.; Zhang, Y.; Van Horn, R.D.; Forest, A.; Novosiadly, R.D.; Webster, Y.W.; Ebert, P.; Bray, S.; et al. Mouse PDX Trial Suggests Synergy of Concurrent Inhibition of RAF and EGFR in Colorectal Cancer with BRAF or KRAS Mutations. Clin. Cancer Res. 2017, 23, 5547–5560. [Google Scholar] [CrossRef] [PubMed]
- Chuprin, J.; Buettner, H.; Seedhom, M.O.; Greiner, D.L.; Keck, J.G.; Ishikawa, F.; Shultz, L.D.; Brehm, M.A. Humanized mouse models for immuno-oncology research. Nat. Rev. Clin. Oncol. 2023, 20, 192–206. [Google Scholar] [CrossRef]
- Yin, L.; Wang, X.J.; Chen, D.X.; Liu, X.N.; Wang, X.J. Humanized mouse model: A review on preclinical applications for cancer immunotherapy. Am. J. Cancer Res. 2020, 10, 4568–4584. [Google Scholar] [PubMed]
- Wege, A.K. Humanized Mouse Models for the Preclinical Assessment of Cancer Immunotherapy. BioDrugs 2018, 32, 245–266. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, J.; Li, D.; Zhou, W.; Yan, F.; Wang, W. Humanized mouse models: A valuable platform for preclinical evaluation of human cancer. Biotechnol. Bioeng. 2023, 27, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, W.; Cai, C.; Zhang, H.; Shen, H.; Han, Y. Patient-derived xenograft models in cancer therapy: Technologies and applications. Signal Transduct. Target. Ther. 2023, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Nebhan, C.A.; Saleh, N.; Shattuck-Brandt, R.; Chen, S.C.; Ayers, G.D.; Weiss, V.; Richmond, A.; Vilgelm, A.E. Generation of Orthotopic Patient-Derived Xenografts in Humanized Mice for Evaluation of Emerging Targeted Therapies and Immunotherapy Combinations for Melanoma. Cancers 2023, 15, 3695. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Takeya, M. CAFs and TAMs: Maestros of the tumour microenvironment. J. Pathol. 2017, 241, 313–315. [Google Scholar] [CrossRef]
- Liu, Y.; Chanana, P.; Davila, J.I.; Hou, X.; Zanfagnin, V.; McGehee, C.D.; Goode, E.L.; Polley, E.C.; Haluska, P.; Weroha, S.J.; et al. Gene expression differences between matched pairs of ovarian cancer patient tumors and patient-derived xenografts. Sci. Rep. 2019, 9, 6314. [Google Scholar] [CrossRef]
- Shi, J.; Li, Y.; Jia, R.; Fan, X. The fidelity of cancer cells in PDX models: Characteristics, mechanism and clinical significance. Int. J. Cancer 2020, 146, 2078–2088. [Google Scholar] [CrossRef]
- Braekeveldt, N.; von Stedingk, K.; Fransson, S.; Martinez-Monleon, A.; Lindgren, D.; Axelson, H.; Levander, F.; Willforss, J.; Hansson, K.; Øra, I.; et al. Patient-Derived Xenograft Models Reveal Intratumor Heterogeneity and Temporal Stability in Neuroblastoma. Cancer Res. 2018, 78, 5958–5969. [Google Scholar] [CrossRef]
- Leitao, M.M., Jr.; Hensley, M.L.; Barakat, R.R.; Aghajanian, C.; Gardner, G.J.; Jewell, E.L.; O’Cearbhaill, R.; Soslow, R.A. Immunohistochemical expression of estrogen and progesterone receptors and outcomes in patients with newly diagnosed uterine leiomyosarcoma. Gynecol. Oncol. 2012, 124, 558–562. [Google Scholar] [CrossRef]
- Alkushi, A.; Lim, P.; Coldman, A.; Huntsman, D.; Miller, D.; Gilks, C.B. Interpretation of p53 immunoreactivity in endometrial carcinoma: Establishing a clinically relevant cut-off level. Int. J. Gynecol. Pathol. 2004, 23, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; Kitajima, M.; Hiraki, K.; Fujishita, A.; Nakashima, M.; Ishimaru, T.; Masuzaki, H. Cell proliferation effect of GnRH agonist on pathological lesions of women with endometriosis, adenomyosis and uterine myoma. Hum. Reprod. 2010, 25, 2878–2890. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total | Engrafted | Failed | Engraftment Rate (%) | p Value |
|---|---|---|---|---|---|
| Number of patients | 92 | 52 | 40 | 56.5 | |
| Age (years) * | 60.9 ± 10.8 | 61.2 ± 9.4 | 60.6 ± 12.5 | 0.782 | |
| CA19-9 (U/mL) * | 53.0 ± 152.4 | 59.4 ± 188.4 | 44.6 ± 84.9 | 0.647 | |
| CA125 (U/mL) * | 135.4 ± 375.7 | 192.5 ± 465.9 | 61.1 ± 204.6 | 0.100 | |
| Histology | 0.017 | ||||
| Endometrioid carcinoma G1 | 47 | 19 | 28 | 40.4 | |
| Endometrioid carcinoma G2 | 13 | 8 | 5 | 61.5 | |
| Endometrioid carcinoma G3 | 9 | 8 | 1 | 88.9 | |
| serous carcinoma | 11 | 7 | 4 | 63.6 | |
| carcinosarcoma | 8 | 6 | 2 | 75.0 | |
| sarcoma | 3 | 3 | 0 | 100.0 | |
| small cell carcinoma | 1 | 1 | 0 | 100.0 | |
| FIGO Stage | 0.002 | ||||
| I | 55 | 23 | 32 | 41.8 | |
| II | 1 | 1 | 1 | 100.0 | |
| III | 25 | 21 | 4 | 84.0 | |
| IV | 11 | 7 | 4 | 63.6 | |
| Lympho-vascular invasion | 40 | 30 | 10 | 75.0 | 0.002 |
| Lymph node metastasis | 21 | 14 | 7 | 66.7 | 0.281 |
| Peritoneal cytology positive | 23 | 15 | 8 | 65.2 | 0.328 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueda, S.; Tanaka, T.; Hirosuna, K.; Miyamoto, S.; Murakami, H.; Nishie, R.; Tsuchihashi, H.; Toji, A.; Morita, N.; Hashida, S.; et al. Consistency between Primary Uterine Corpus Malignancies and Their Corresponding Patient-Derived Xenograft Models. Int. J. Mol. Sci. 2024, 25, 1486. https://doi.org/10.3390/ijms25031486
Ueda S, Tanaka T, Hirosuna K, Miyamoto S, Murakami H, Nishie R, Tsuchihashi H, Toji A, Morita N, Hashida S, et al. Consistency between Primary Uterine Corpus Malignancies and Their Corresponding Patient-Derived Xenograft Models. International Journal of Molecular Sciences. 2024; 25(3):1486. https://doi.org/10.3390/ijms25031486
Chicago/Turabian StyleUeda, Shoko, Tomohito Tanaka, Kensuke Hirosuna, Shunsuke Miyamoto, Hikaru Murakami, Ruri Nishie, Hiromitsu Tsuchihashi, Akihiko Toji, Natsuko Morita, Sousuke Hashida, and et al. 2024. "Consistency between Primary Uterine Corpus Malignancies and Their Corresponding Patient-Derived Xenograft Models" International Journal of Molecular Sciences 25, no. 3: 1486. https://doi.org/10.3390/ijms25031486
APA StyleUeda, S., Tanaka, T., Hirosuna, K., Miyamoto, S., Murakami, H., Nishie, R., Tsuchihashi, H., Toji, A., Morita, N., Hashida, S., Daimon, A., Terada, S., Maruoka, H., Kogata, Y., Taniguchi, K., Komura, K., & Ohmichi, M. (2024). Consistency between Primary Uterine Corpus Malignancies and Their Corresponding Patient-Derived Xenograft Models. International Journal of Molecular Sciences, 25(3), 1486. https://doi.org/10.3390/ijms25031486