
The Analysis of Embryoid Body Formation and Its Role in Retinal Organoid Development

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Comparison of the Events That Influence Retinal Organoid Development in Previously Published Systematic Reviews
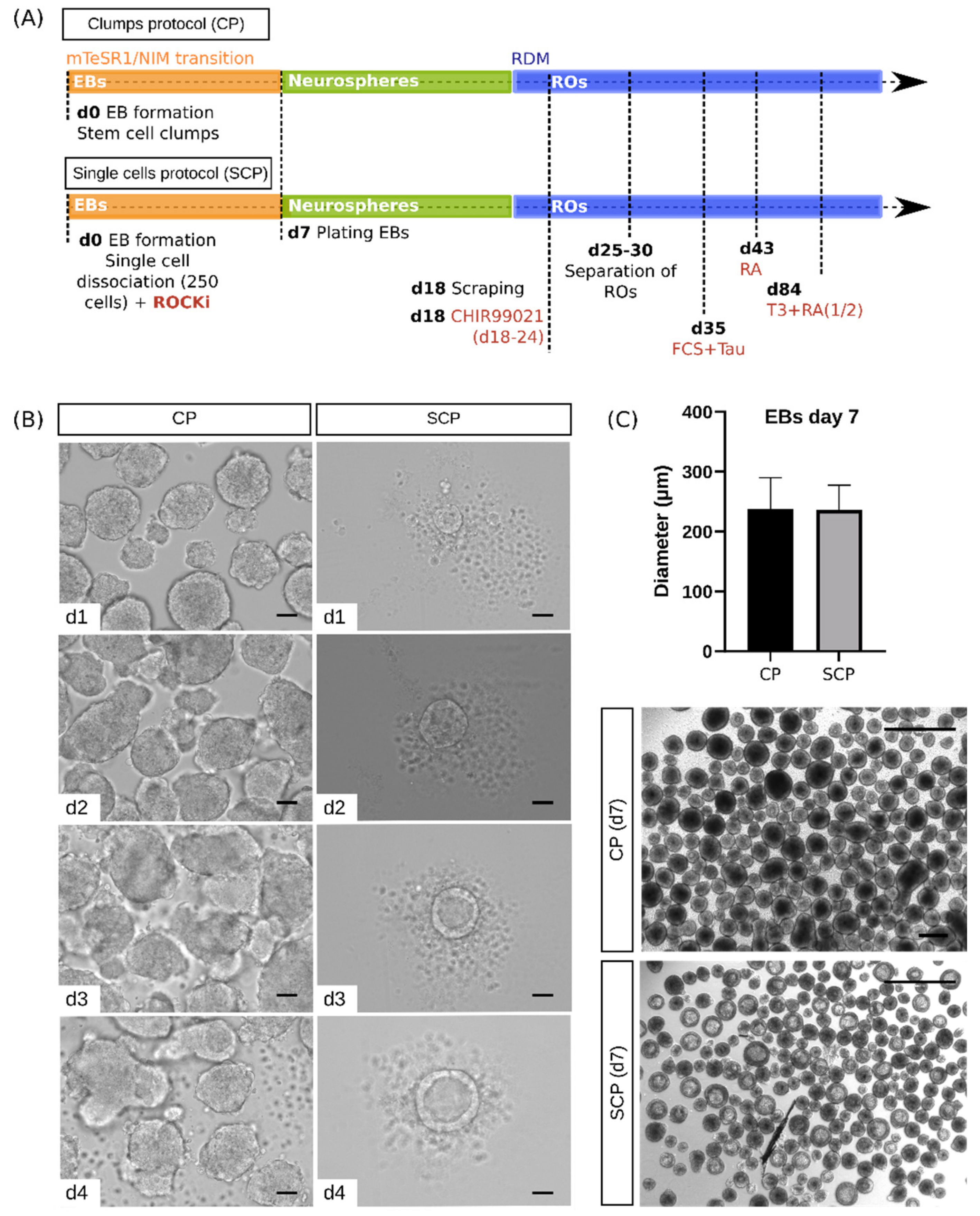
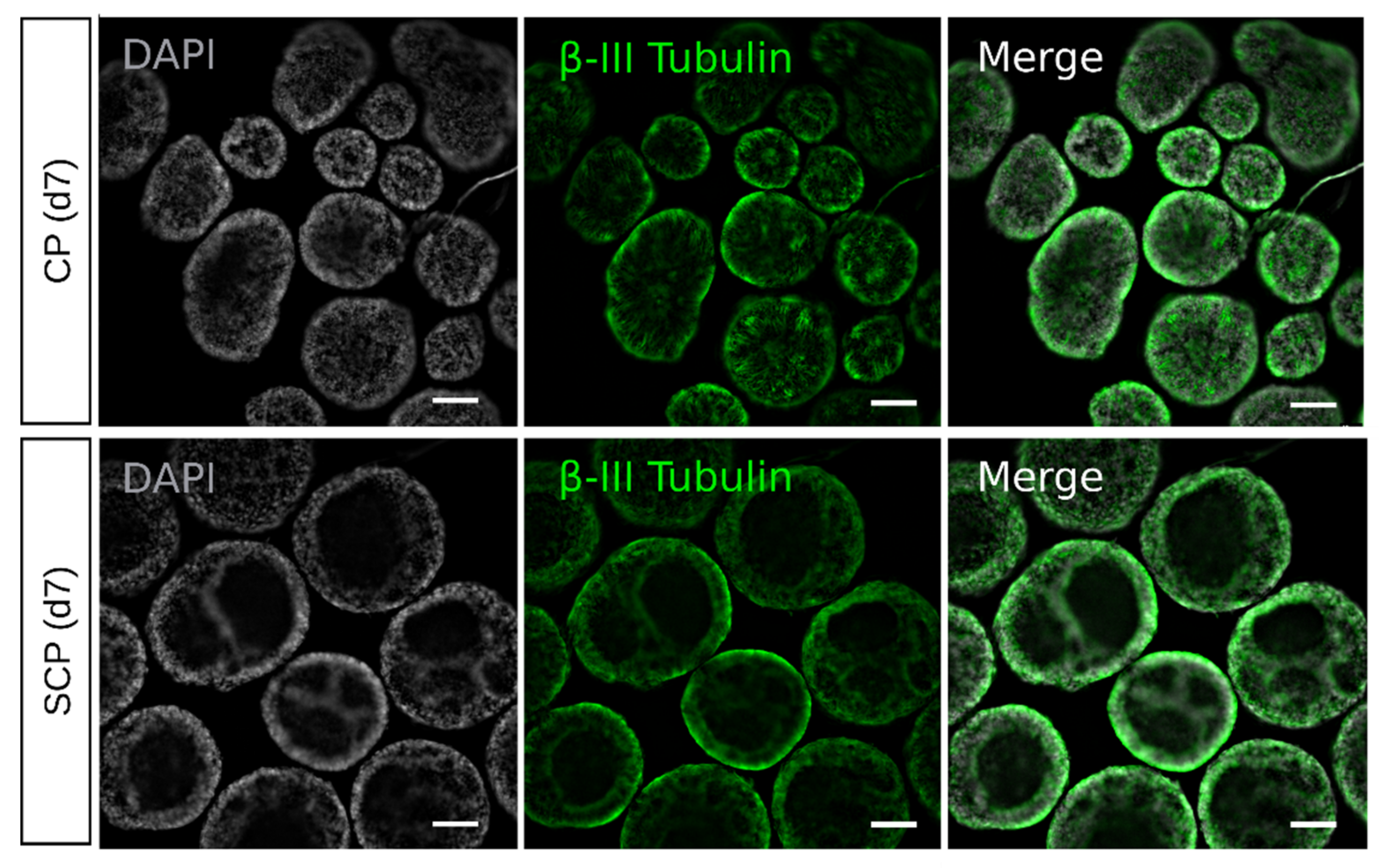
2.2. Morphological Characterization Reveals Differences between Protocols at the Embryoid Body Stage
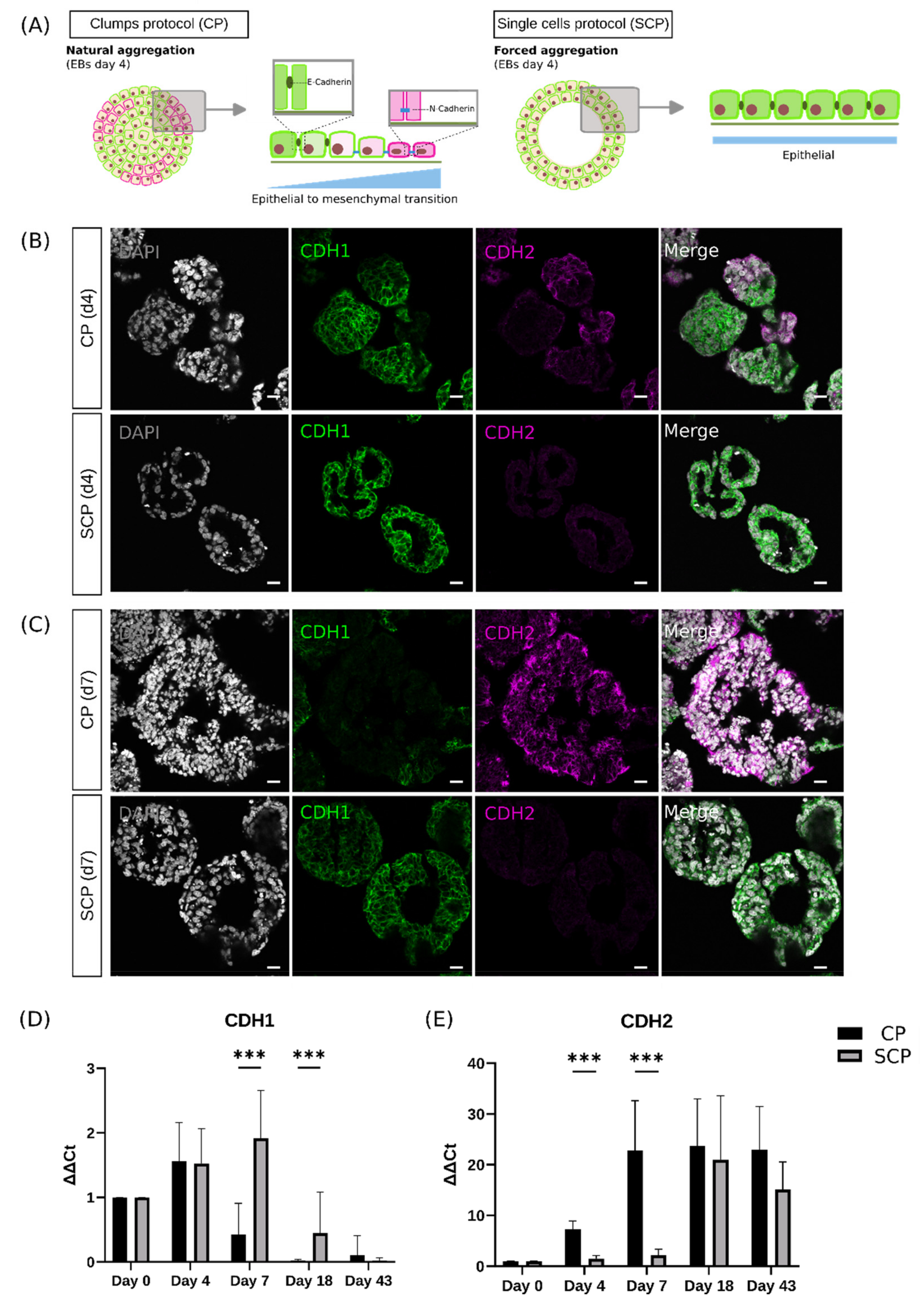
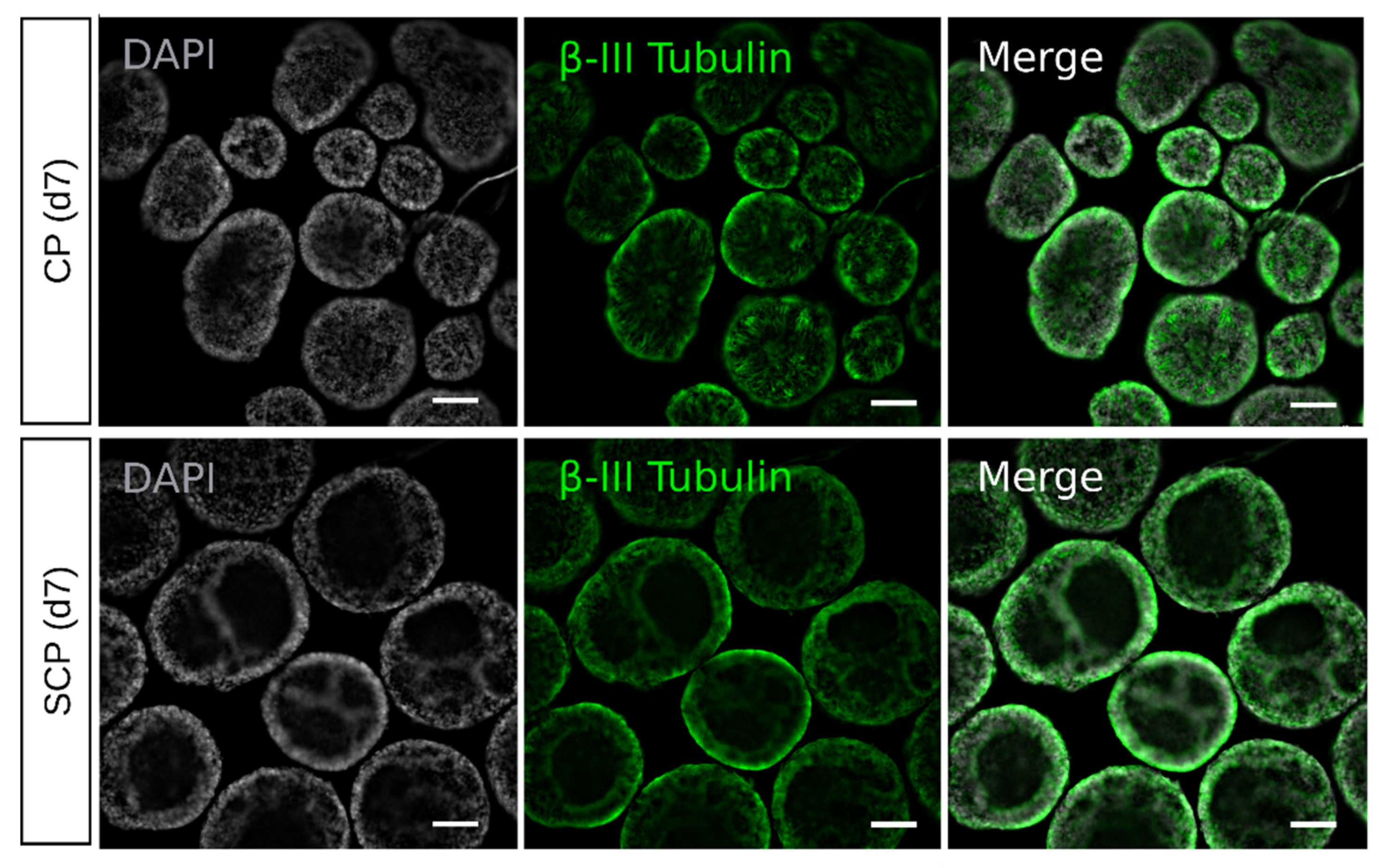
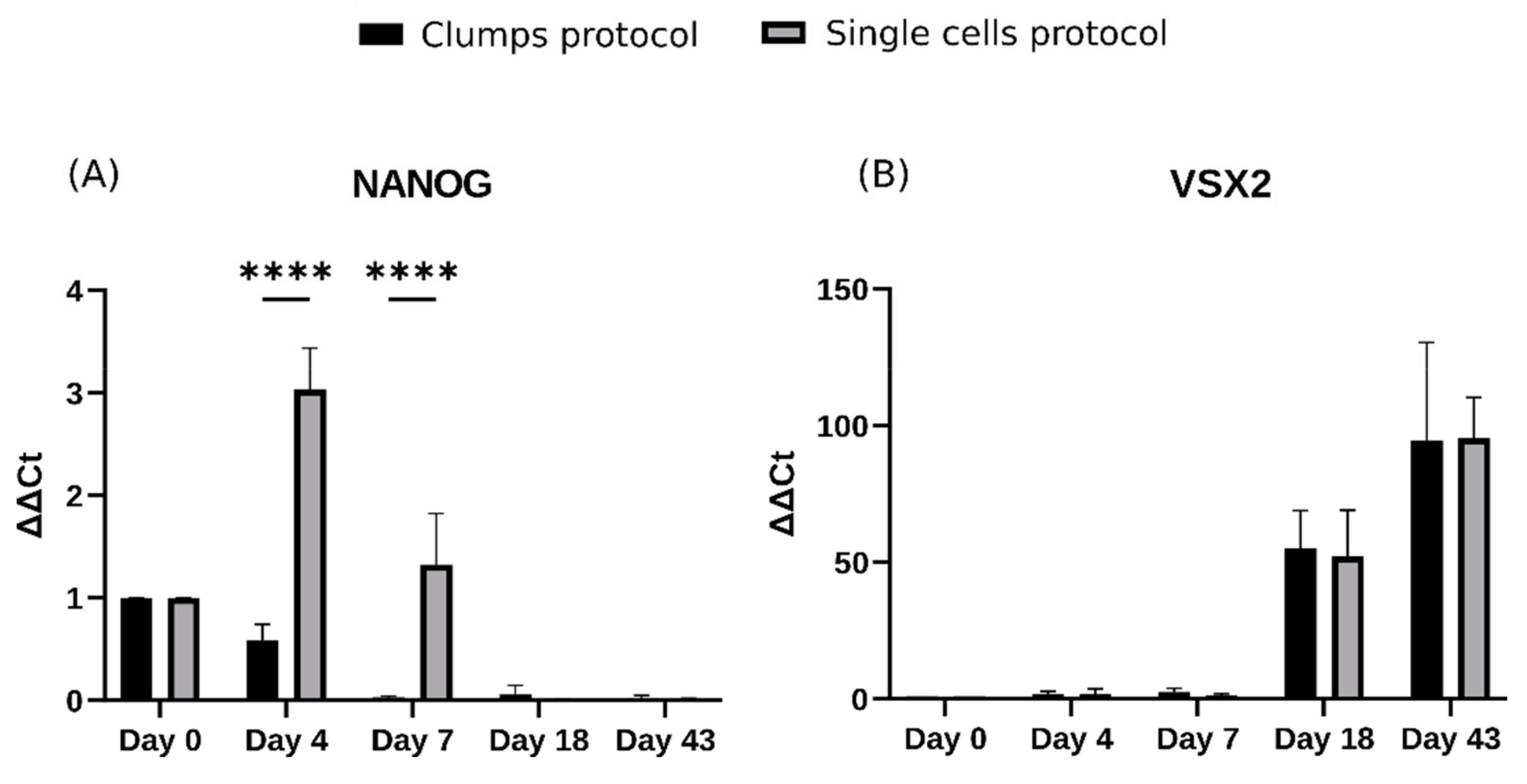
2.3. Single Cells Protocol Generates Embryoid Bodies That Delay the Epithelial-to-Mesenchymal Transition
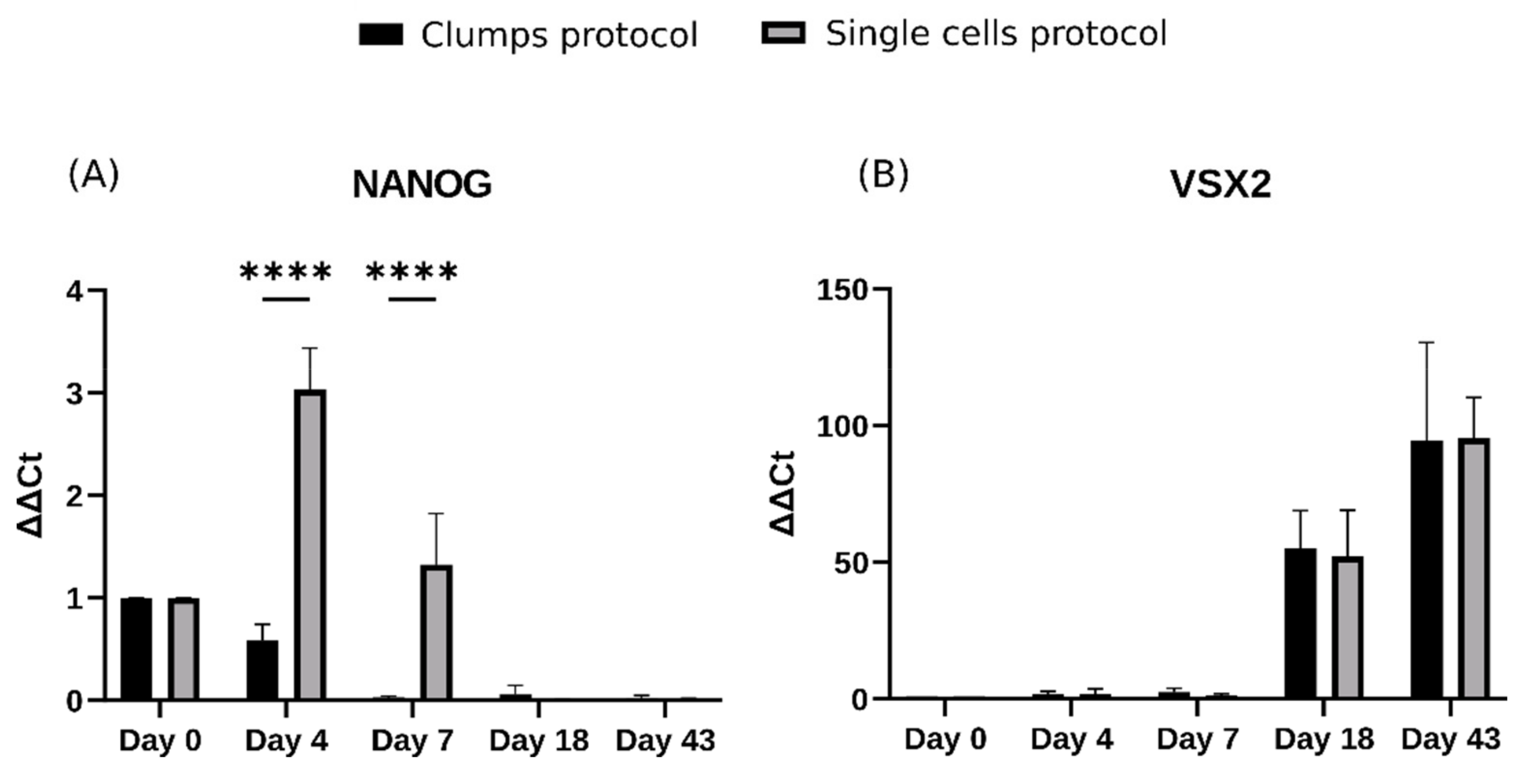
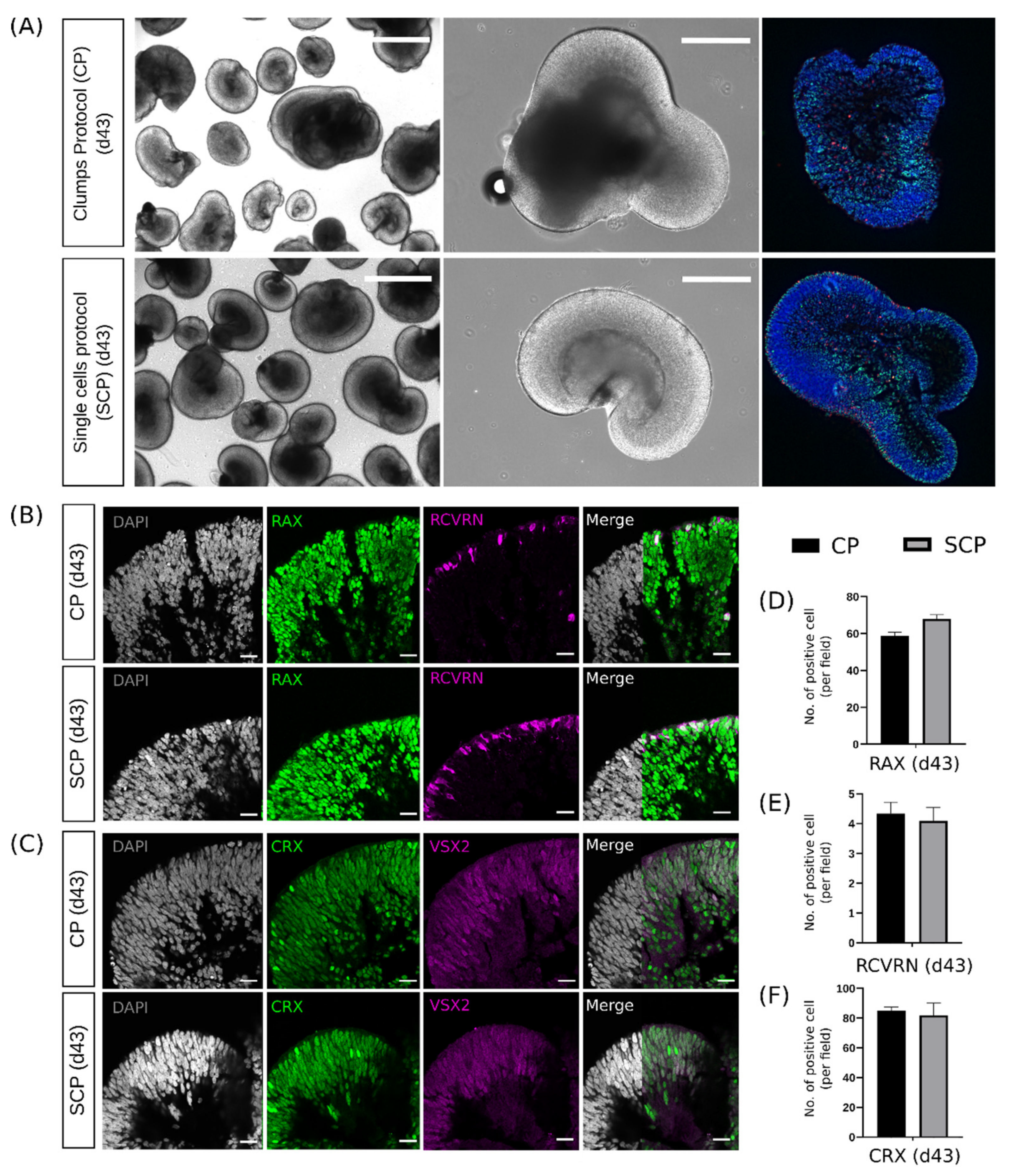
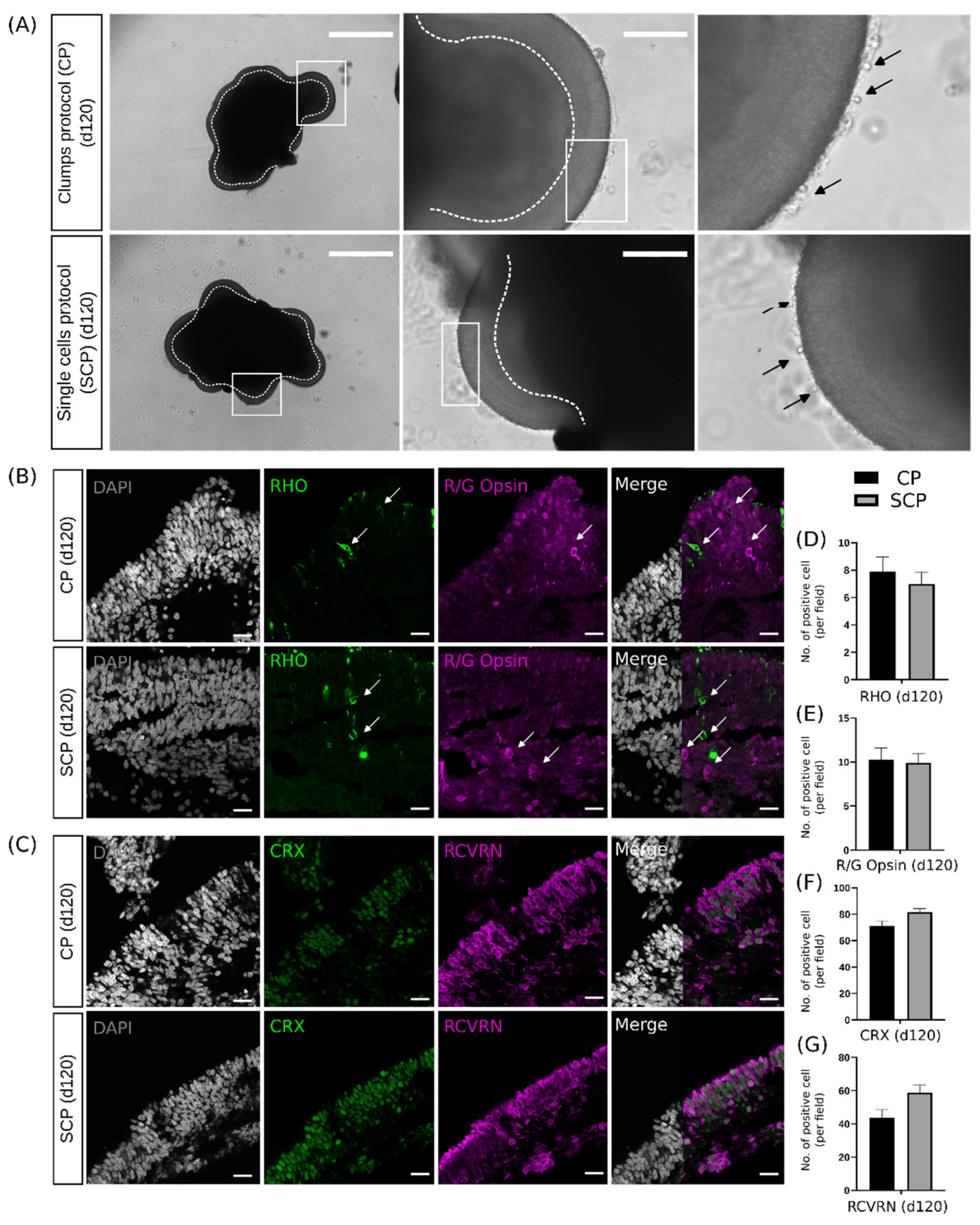
2.4. Early Differences during Embryoid Body Development Do Not Interfere with Retinal Organoid Development
3. Discussion
4. Materials and Methods
4.1. hESC Culture
4.2. Description of Embryoid Body Formation Protocols
4.3. Tissue Preparation and Immunohistochemistry
4.4. Microscopy, Quantification, and Analysis
4.5. PCR and Quantitative (q) PCR Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonnier, F.; Keating, M.E.; Wróbel, T.P.; Majzner, K.; Baranska, M.; Garcia-Munoz, A.; Blanco, A.; Byrne, H.J. Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models. Toxicol. In Vitro 2015, 29, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A look into retinal organoids: Methods, analytical techniques, and applications. Cell. Mol. Life Sci. 2021, 78, 6505–6532. [Google Scholar] [CrossRef] [PubMed]
- O’Hara-Wright, M.; Gonzalez-Cordero, A. Retinal organoids: A window into human retinal development. Development 2020, 147, dev189746. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, E.L.; Ten Asbroek, A.L.M.A.; Ten Brink, J.B.; Jansonius, N.M.; Bergen, A.A.B. An alternative approach to produce versatile retinal organoids with accelerated ganglion cell development. Sci. Rep. 2021, 11, 1101. [Google Scholar] [CrossRef] [PubMed]
- Achberger, K.; Haderspeck, J.C.; Kleger, A.; Liebau, S. Stem cell-based retina models. Adv. Drug Deliv. Rev. 2019, 140, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Oswald, J.; Baranov, P. Regenerative medicine in the retina: From stem cells to cell replacement therapy. Ther. Adv. Ophthalmol. 2018, 10, 251584141877443. [Google Scholar] [CrossRef] [PubMed]
- Mellough, C.B.; Collin, J.; Queen, R.; Hilgen, G.; Dorgau, B.; Zerti, D.; Felemban, M.; White, K.; Sernagor, E.; Lako, M. Systematic Comparison of Retinal Organoid Differentiation from Human Pluripotent Stem Cells Reveals Stage Specific, Cell Line, and Methodological Differences. Stem Cells Transl. Med. 2019, 8, 694–706. [Google Scholar] [CrossRef]
- Radojevic, B.; Conley, S.M.; Bennett, L.D. Adherent but Not Suspension-Cultured Embryoid Bodies Develop into Laminated Retinal Organoids. J. Dev. Biol. 2021, 9, 38. [Google Scholar] [CrossRef]
- Sanjurjo-Soriano, C.; Erkilic, N.; Damodar, K.; Boukhaddaoui, H.; Diakatou, M.; Garita-Hernandez, M.; Mamaeva, D.; Dubois, G.; Jazouli, Z.; Jimenez-Medina, C.; et al. Retinoic acid delays initial photoreceptor differentiation and results in a highly structured mature retinal organoid. Stem Cell Res. Ther. 2022, 13, 478. [Google Scholar] [CrossRef]
- Rossi, G.; Manfrin, A.; Lutolf, M.P. Progress and potential in organoid research. Nat. Rev. Genet. 2018, 19, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, E.L.; Heredero Berzal, A.; Boon, C.J.F.; Quinn, P.M.J.; ten Asbroek, A.L.M.A.; Bergen, A.A. The Role of Small Molecules and Their Effect on the Molecular Mechanisms of Early Retinal Organoid Development. Int. J. Mol. Sci. 2021, 22, 7081. [Google Scholar] [CrossRef] [PubMed]
- Doetschman, T.C.; Eistetter, H.; Katz, M.; Schmidt, W.; Kemler, R. The in vitro development of blastocyst-derived embryonic stem cell lines: Formation of visceral yolk sac, blood islands and myocardium. Development 1985, 87, 27–45. [Google Scholar] [CrossRef]
- Brickman, J.M.; Serup, P. Properties of embryoid bodies. WIREs Dev. Biol. 2017, 6, e259. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, K.; Elsafi Mabrouk, M.H.; Wagner, W.; Goetzke, R. Cell Mechanics in Embryoid Bodies. Cells 2020, 9, 2270. [Google Scholar] [CrossRef] [PubMed]
- Ohlemacher, S.K.; Iglesias, C.L.; Sridhar, A.; Gamm, D.M.; Meyer, J.S. Generation of Highly Enriched Populations of Optic Vesicle-Like Retinal Cells from Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2015, 32, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-C.; Wang, M.-L.; Chen, S.-J.; Kuo, J.-C.; Wang, W.-J.; Nhi Nguyen, P.N.; Wahlin, K.J.; Lu, J.-F.; Tran, A.A.; Shi, M.; et al. Morphological and Molecular Defects in Human Three-Dimensional Retinal Organoid Model of X-Linked Juvenile Retinoschisis. Stem Cell Rep. 2019, 13, 906–923. [Google Scholar] [CrossRef] [PubMed]
- Berber, P.; Milenkovic, A.; Michaelis, L.; Weber, B.H.F. Retinal organoid differentiation methods determine organoid cellular composition. J. Transl. Genet. Genomics 2021, 5, 292–303. [Google Scholar] [CrossRef]
- Choy Buentello, D.; Koch, L.S.; Trujillo-de Santiago, G.; Alvarez, M.M.; Broersen, K. Use of standard U-bottom and V-bottom well plates to generate neuroepithelial embryoid bodies. PLoS ONE 2022, 17, e0262062. [Google Scholar] [CrossRef]
- Chichagova, V.; Hilgen, G.; Ghareeb, A.; Georgiou, M.; Carter, M.; Sernagor, E.; Lako, M.; Armstrong, L. Human iPSC differentiation to retinal organoids in response to IGF1 and BMP4 activation is line- and method-dependent. Stem Cells 2020, 38, 195–201. [Google Scholar] [CrossRef]
- Van Winkle, A.P.; Gates, I.D.; Kallos, M.S. Mass Transfer Limitations in Embryoid Bodies during Human Embryonic Stem Cell Differentiation. Cells Tissues Organs 2012, 196, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Rothová, M.; Hölzenspies, J.J.; Livigni, A.; Villegas, S.N.; Brickman, J.M. Differentiation of Mouse Embryonic Stem Cells into Ventral Foregut Precursors. Curr. Protoc. Stem Cell Biol. 2016, 36, 1G.3.1–1G.3.12. [Google Scholar] [CrossRef] [PubMed]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of Human Embryonic Stem Cells into Embryoid Bodies Comprising the Three Embryonic Germ Layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Pekkanen-Mattila, M.; Pelto-Huikko, M.; Kujala, V.; Suuronen, R.; Skottman, H.; Aalto-Setälä, K.; Kerkelä, E. Spatial and temporal expression pattern of germ layer markers during human embryonic stem cell differentiation in embryoid bodies. Histochem. Cell Biol. 2010, 133, 595–606. [Google Scholar] [CrossRef]
- Isla-Magrané, H.; Veiga, A.; García-Arumí, J.; Duarri, A. Multiocular organoids from human induced pluripotent stem cells displayed retinal, corneal, and retinal pigment epithelium lineages. Stem Cell Res. Ther. 2021, 12, 581. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Ouyang, S.; Ma, J.; Sun, H.; Luo, L.; Chen, S.; Liu, Y. Generation and Staging of Human Retinal Organoids Based on Self-Formed Ectodermal Autonomous Multi-Zone System. Front. Cell Dev. Biol. 2021, 9, 732382. [Google Scholar] [CrossRef]
- Berber, P.; Bondarenko, S.; Michaelis, L.; Weber, B.H.F. Transient Retention of Photoreceptor Outer Segments in Matrigel-Embedded Retinal Organoids. Int. J. Mol. Sci. 2022, 23, 14893. [Google Scholar] [CrossRef]
- Kurosawa, H. Methods for inducing embryoid body formation: In vitro differentiation system of embryonic stem cells. J. Biosci. Bioeng. 2007, 103, 389–398. [Google Scholar] [CrossRef]
- Qi, Y.; Liu, J.; Saadat, S.; Tian, X.; Han, Y.; Fong, G.-H.; Pandolfi, P.P.; Lee, L.Y.; Li, S. PTEN induces apoptosis and cavitation via HIF-2-dependent Bnip3 upregulation during epithelial lumen formation. Cell Death Differ. 2015, 22, 875–884. [Google Scholar] [CrossRef]
- Pedroza, M.; Gassaloglu, S.I.; Dias, N.; Zhong, L.; Hou, T.-C.J.; Kretzmer, H.; Smith, Z.D.; Sozen, B. Self-patterning of human stem cells into post-implantation lineages. Nature 2023, 622, 574–583. [Google Scholar] [CrossRef]
- Pettinato, G.; Wen, X.; Zhang, N. Engineering Strategies for the Formation of Embryoid Bodies from Human Pluripotent Stem Cells. Stem Cells Dev. 2015, 24, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.; Harris, R.; Bhansali, P.; Cvekl, A.; Liu, W. Intercellular Adhesion-Dependent Cell Survival and ROCK-Regulated Actomyosin-Driven Forces Mediate Self-Formation of a Retinal Organoid. Stem Cell Rep. 2016, 6, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Buonpane, C.; Yuan, C.; Wood, D.; Ares, G.; Klonoski, S.C.; Hunter, C.J. ROCK1 inhibitor stabilizes E-cadherin and improves barrier function in experimental necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G781–G792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, X.; Hu, C.; Wang, P.; Li, X. Rho kinase inhibitor Y-27632 and Accutase dramatically increase mouse embryonic stem cell derivation. Vitro Cell. Dev. Biol. Anim. 2012, 48, 30–36. [Google Scholar] [CrossRef]
- Cowan, C.S.; Renner, M.; De Gennaro, M.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; Munz, M.; Rodrigues, T.M.; Krol, J.; Szikra, T.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e34. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Study Design | Cell Line | Category | ||
|---|---|---|---|---|---|
| Radojevic et al., 2021 [9] | Suspension protocol | Plating protocol | hiPSC | Physical features of the culture environment | |
| Choy Buentello et al., 2022 [19] | U-bottom plate | V-bottom plate | hESC | Physical features of the culture environment | |
| Berber et al., 2021 [18] | 3D protocol | 3D-2D-3D protocol | 3D-2D-3D + BMP4 protocol | hiPSC | Physical features of the culture environment |
| Sanjurjo-Soriano et al., 2022 [10] | Without retinoic acid | With retinoic acid | hiPSC | Exogenous signals | |
| Wagstaff et al., 2021 [12] | RI, IGF1, IWR1e, SB431542/LDN193189, CHIR99021, SU5402, CHIR99021/SU5402, DAPT | hESC | Exogenous signals | ||
| Chichagova et al., 2020 [20] | BMP4, IGF1 | hiPSC | Exogenous signals | ||
| Mellough et al., 2019 [8] | Mechanical, enzymatic, or dissociation aggregation approaches to generate embryonic bodies | hESC, hiPSC | Starting cell conditions | ||
| Antibodies | Dilution | Catalog Number | Host | Manufacturer |
|---|---|---|---|---|
| CDH1 | 1:200 | 610182 | Mouse | BD Biosciences (Becton, NJ, USA) |
| CDH2 | 1:200 | 22018-1-AP | Rabbit | Proteintech (Rosemont, IL, USA) |
| CRX | 1:200 | H00001406-M02 | Mouse | Abnova (Taipei City, Taiwan) |
| GATA4 | 1:200 | sc-25310 | Mouse | Santa Cruz (Dallas, TX, USA) |
| RCVRN | 1:200 | AB5585 | Rabbit | Merck Millipore (Burlington, MA, USA) |
| RHO | 1:200 | MAB5356 | Mouse | Merck |
| R/G opsin | 1:200 | AB5405 | Rabbit | Merck |
| SOX2 | 1:200 | ab75627 | Rabbit | Abcam (Cambridge, UK) |
| VSX2 | 1:200 | ab16141 | Sheep | Abcam |
| Antibodies | Dilution | Catalogue Number | Manufacturer |
|---|---|---|---|
| Donkey anti-mouse, Alexa Fluor 488 | 1:200 | 1509983 | Jackson Immuno Research (West Grove, PA, USA) |
| Goat anti-mouse, Alexa Fluor 488 | 1:200 | 115546006 | Jackson Immuno Research |
| Donkey anti-rabbit, Alexa Fluor 594 | 1:200 | A21207 | Thermo Fisher Scientific |
| Donkey anti-sheep, Alexa Fluor 594 | 1:200 | A11016 | Thermo Fisher Scientific |
| Gene | Forward | Reverse | Amplicon Size (bp) |
|---|---|---|---|
| CDH1 | ATTTTTCCCTCGACACCCGAT | TCCCAGGCGTAGACCAAGA | 109 |
| CDH2 | GTGCATGAAGGACAGCCTCT | TGGAAAGCTTCTCACGGCAT | 138 |
| EF1α | CAAAGCGACCCAAAGGTGGAT | AAATAAGCGCCGGCTATGCC | 219 |
| NANOG | TTTGGAAGCTGCTGGGGAAG | GATGGGAGGAGGGGAGAGGA | 194 |
| VSX2 | CGGCGACACAGGACAATCTT | CCTCCAGCGACTTTTTGTGC | 182 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heredero Berzal, A.; Wagstaff, E.L.; ten Asbroek, A.L.M.A.; ten Brink, J.B.; Bergen, A.A.; Boon, C.J.F. The Analysis of Embryoid Body Formation and Its Role in Retinal Organoid Development. Int. J. Mol. Sci. 2024, 25, 1444. https://doi.org/10.3390/ijms25031444
Heredero Berzal A, Wagstaff EL, ten Asbroek ALMA, ten Brink JB, Bergen AA, Boon CJF. The Analysis of Embryoid Body Formation and Its Role in Retinal Organoid Development. International Journal of Molecular Sciences. 2024; 25(3):1444. https://doi.org/10.3390/ijms25031444
Chicago/Turabian StyleHeredero Berzal, Andrea, Ellie L. Wagstaff, Anneloor L. M. A. ten Asbroek, Jacoline B. ten Brink, Arthur A. Bergen, and Camiel J. F. Boon. 2024. "The Analysis of Embryoid Body Formation and Its Role in Retinal Organoid Development" International Journal of Molecular Sciences 25, no. 3: 1444. https://doi.org/10.3390/ijms25031444
APA StyleHeredero Berzal, A., Wagstaff, E. L., ten Asbroek, A. L. M. A., ten Brink, J. B., Bergen, A. A., & Boon, C. J. F. (2024). The Analysis of Embryoid Body Formation and Its Role in Retinal Organoid Development. International Journal of Molecular Sciences, 25(3), 1444. https://doi.org/10.3390/ijms25031444