
Telomere Length, Mitochondrial DNA, and Micronucleus Yield in Response to Oxidative Stress in Peripheral Blood Mononuclear Cells

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
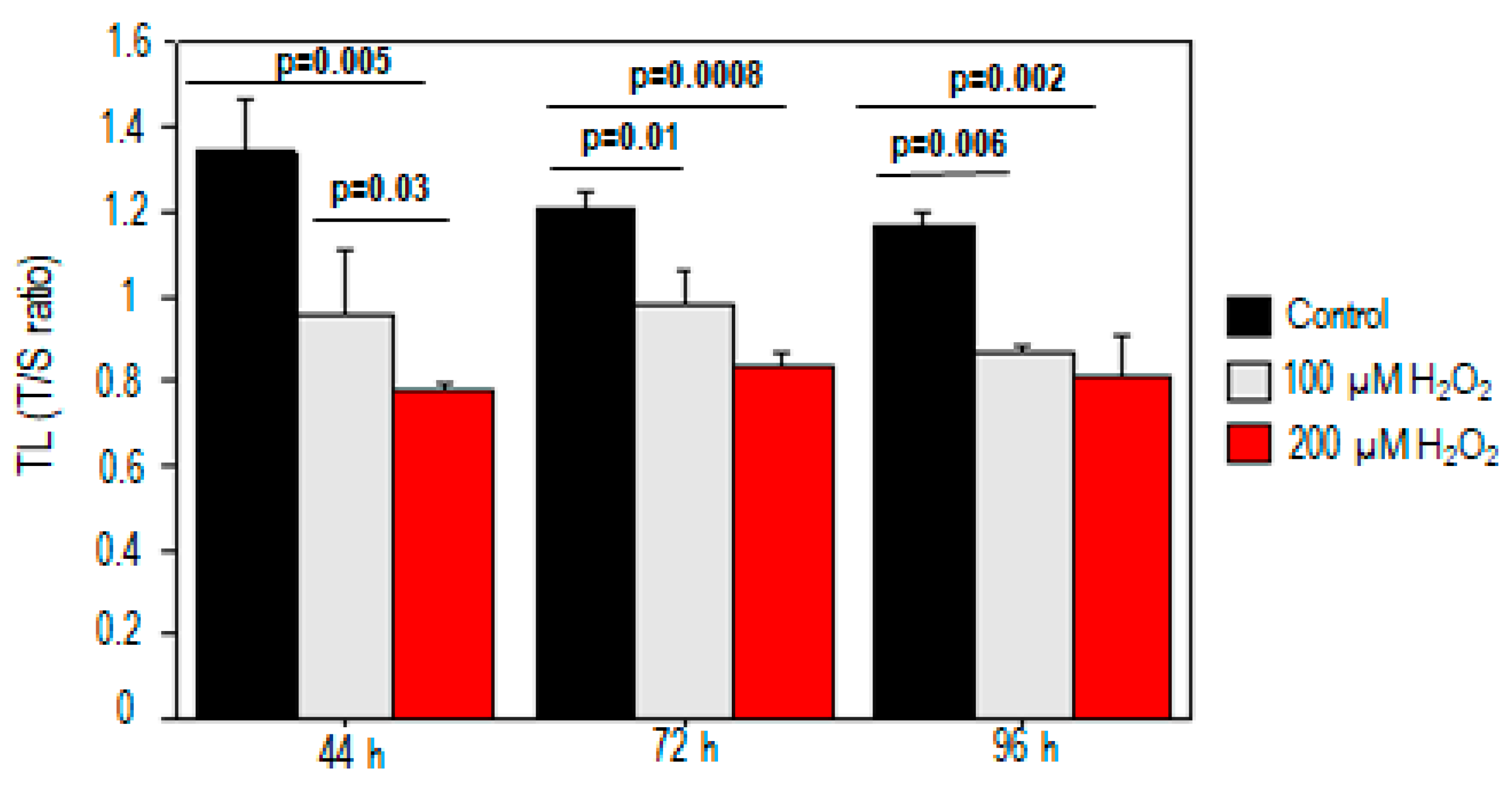
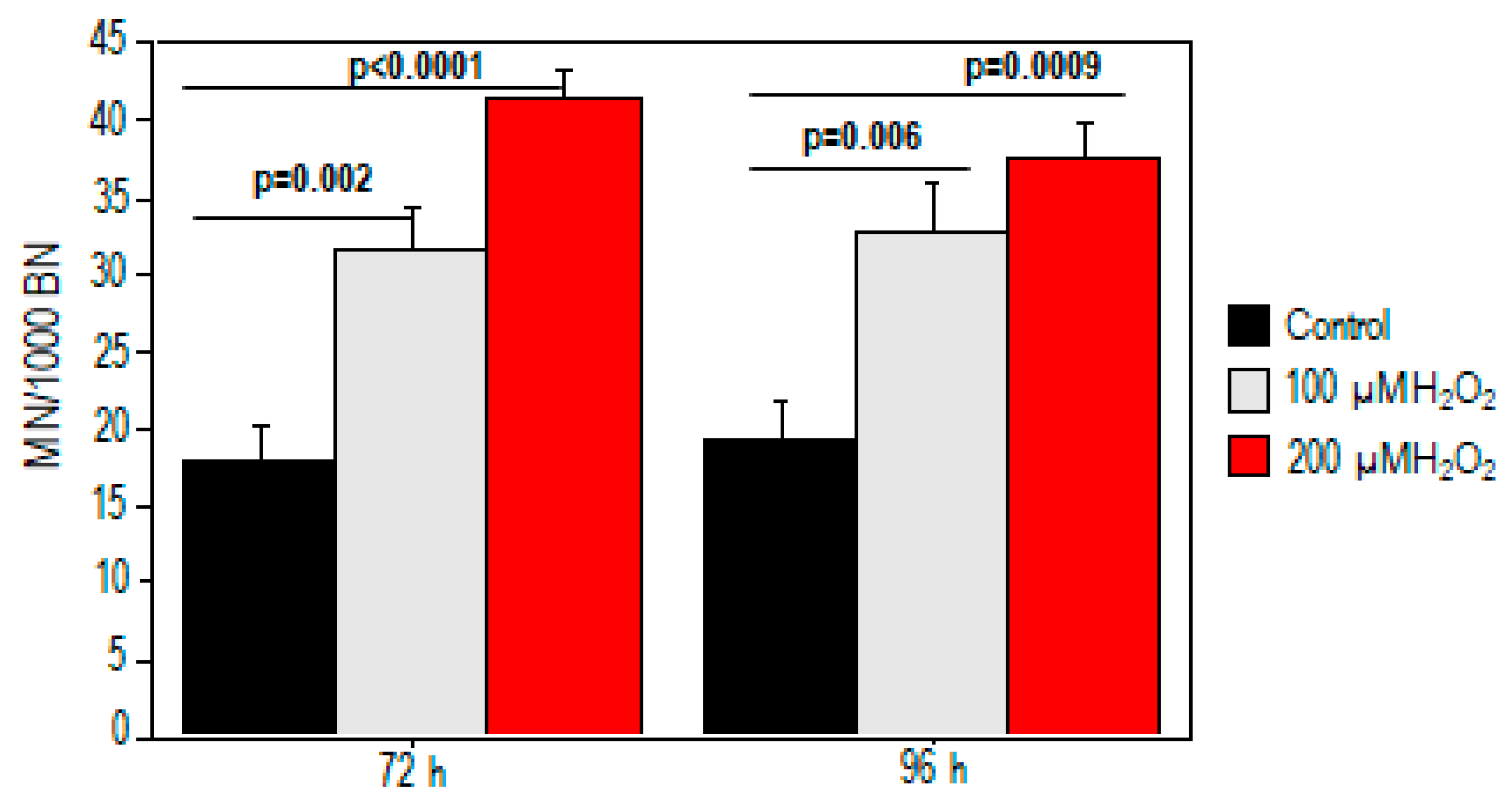
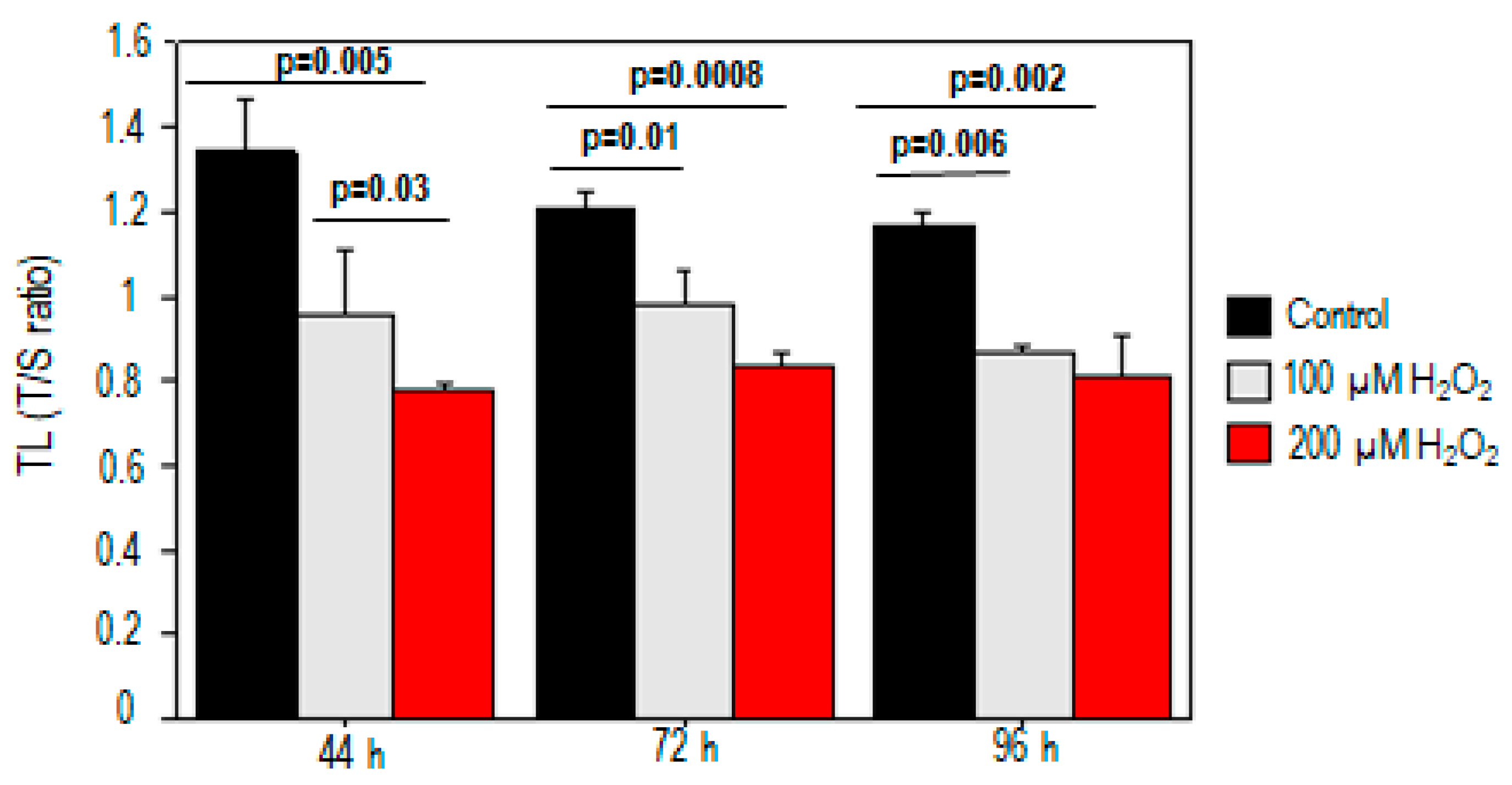
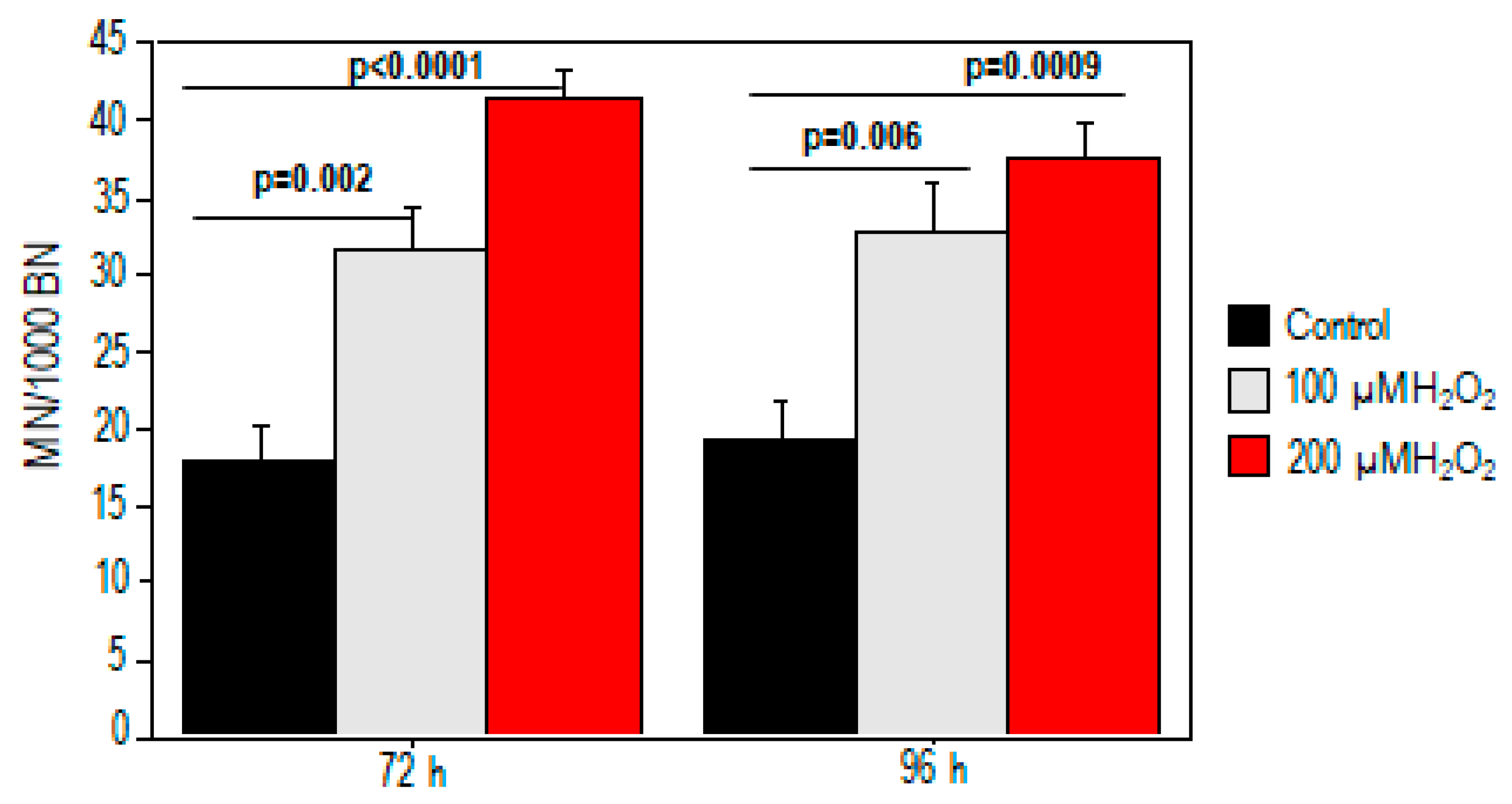
2.1. Effect of Oxidative Stress on Telomere Length and Chromosomal DNA Damage
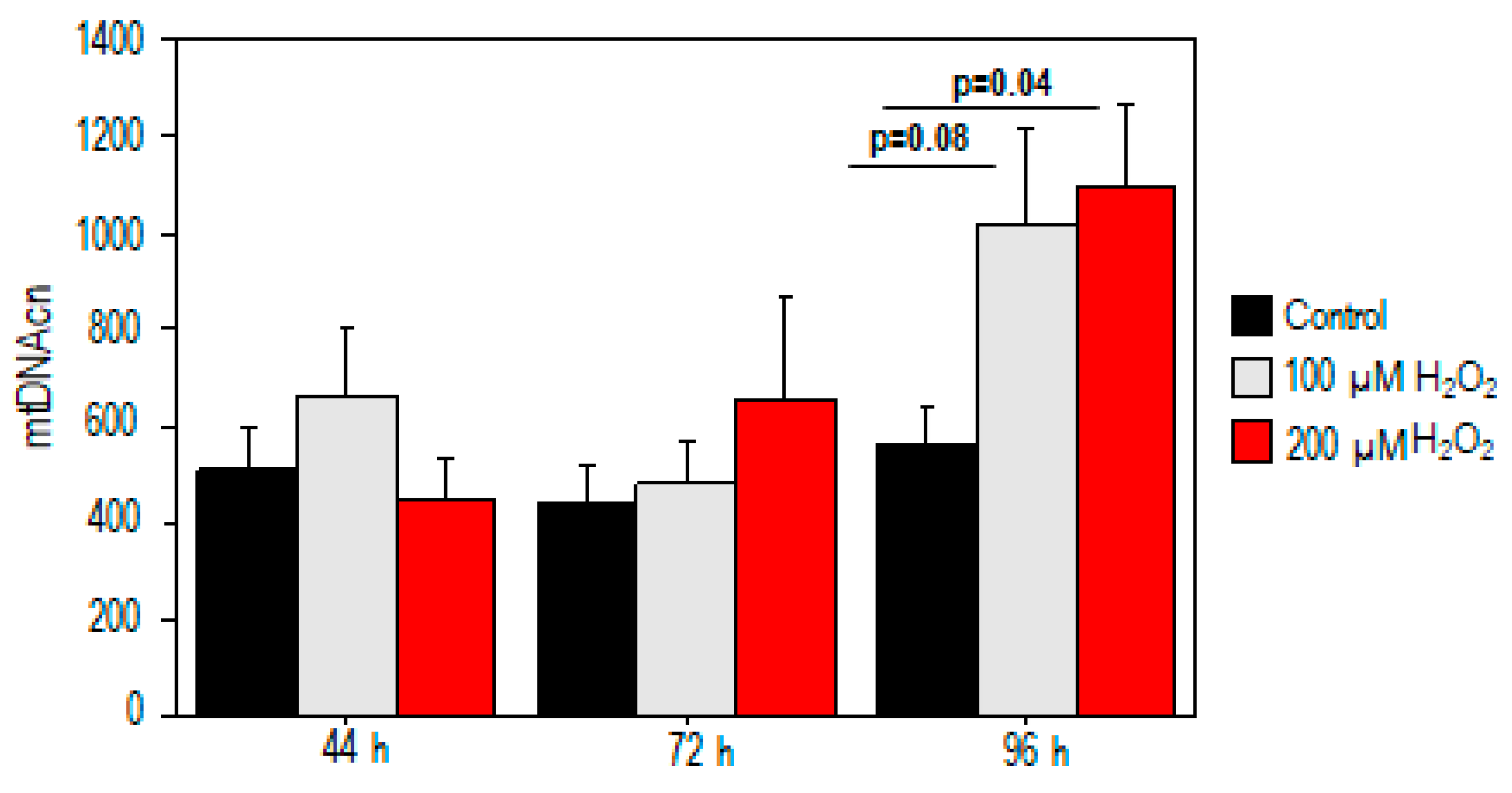
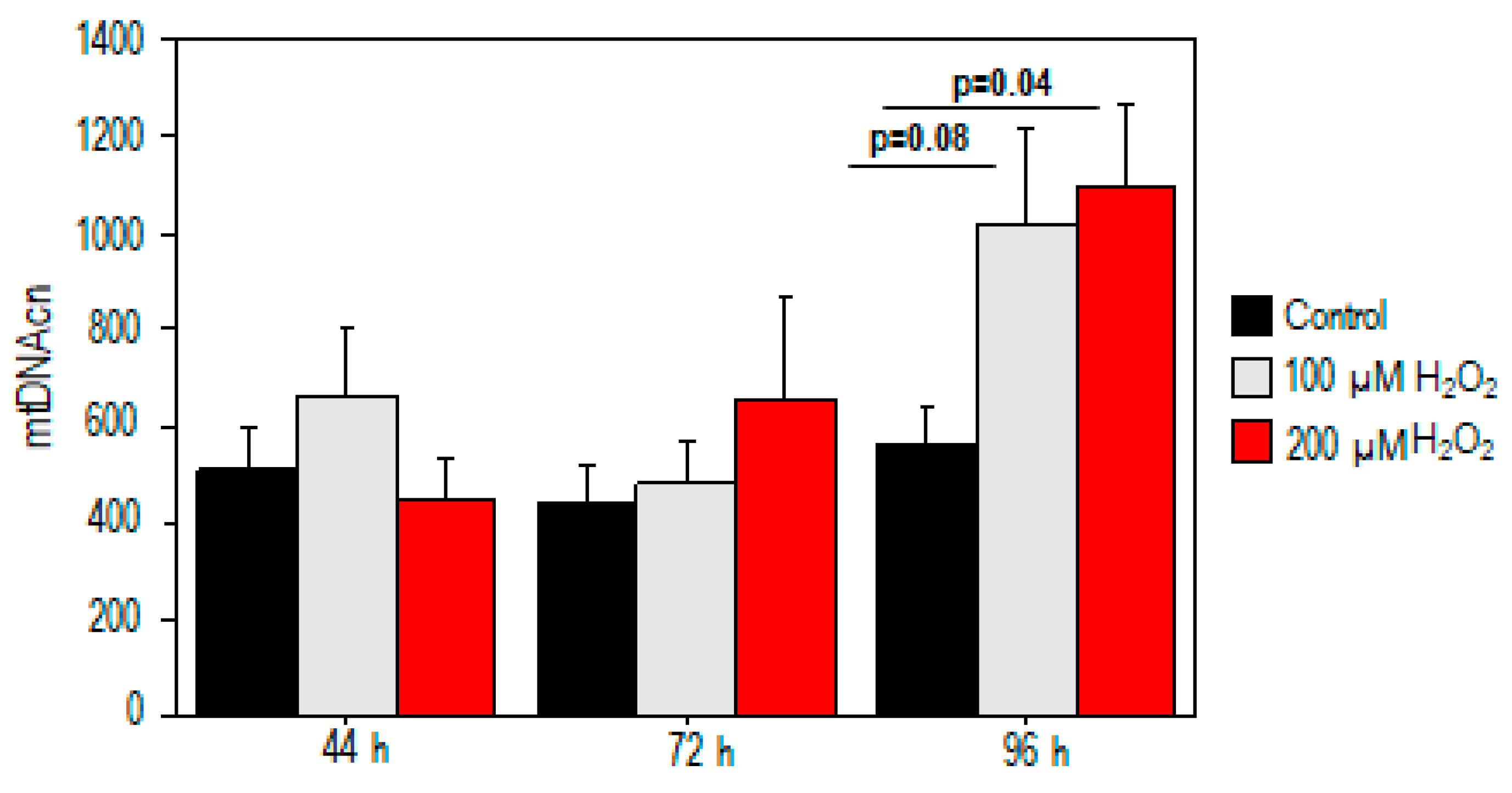
2.2. Effect of Oxidative Stress on Mitochondrial DNA Copy Number
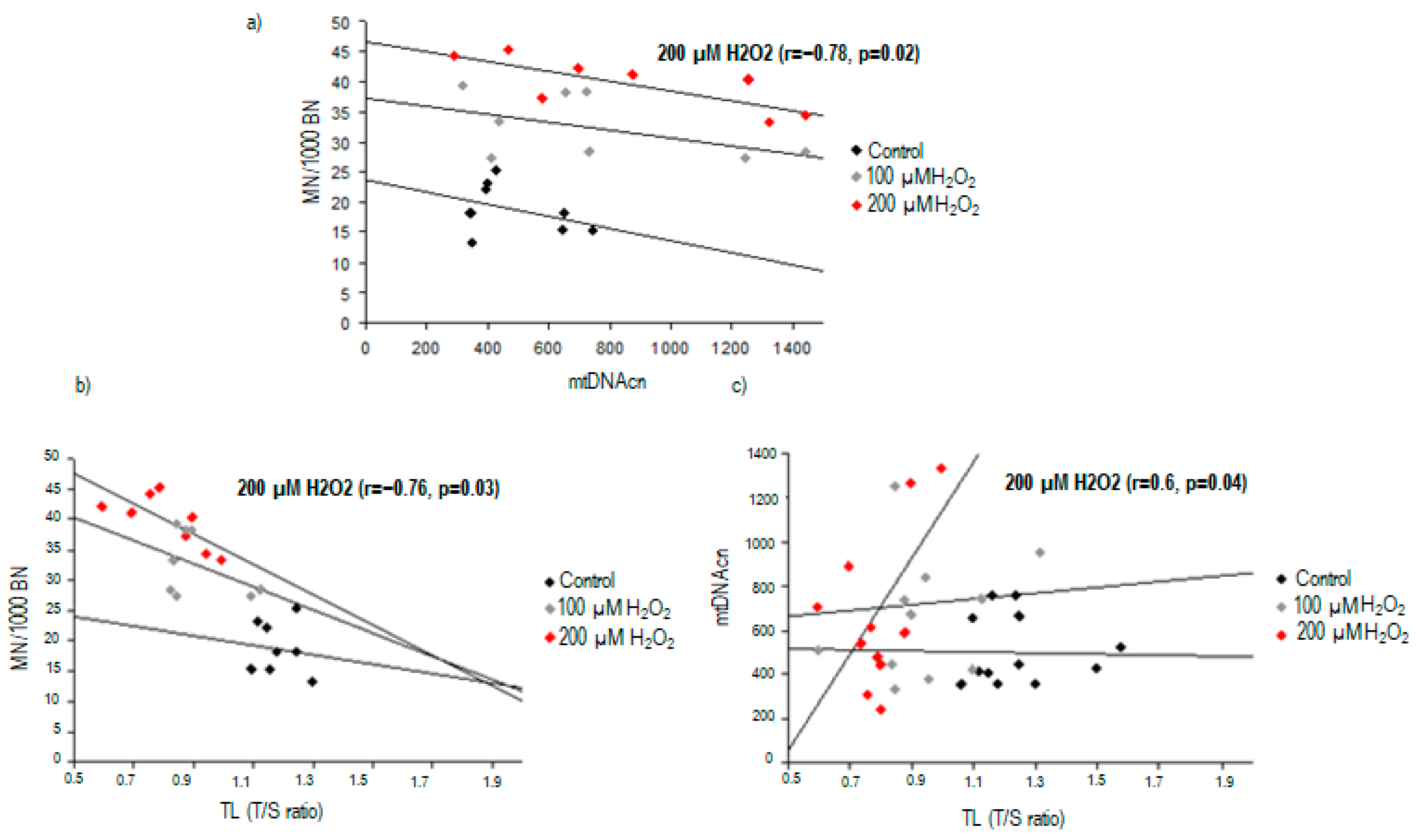
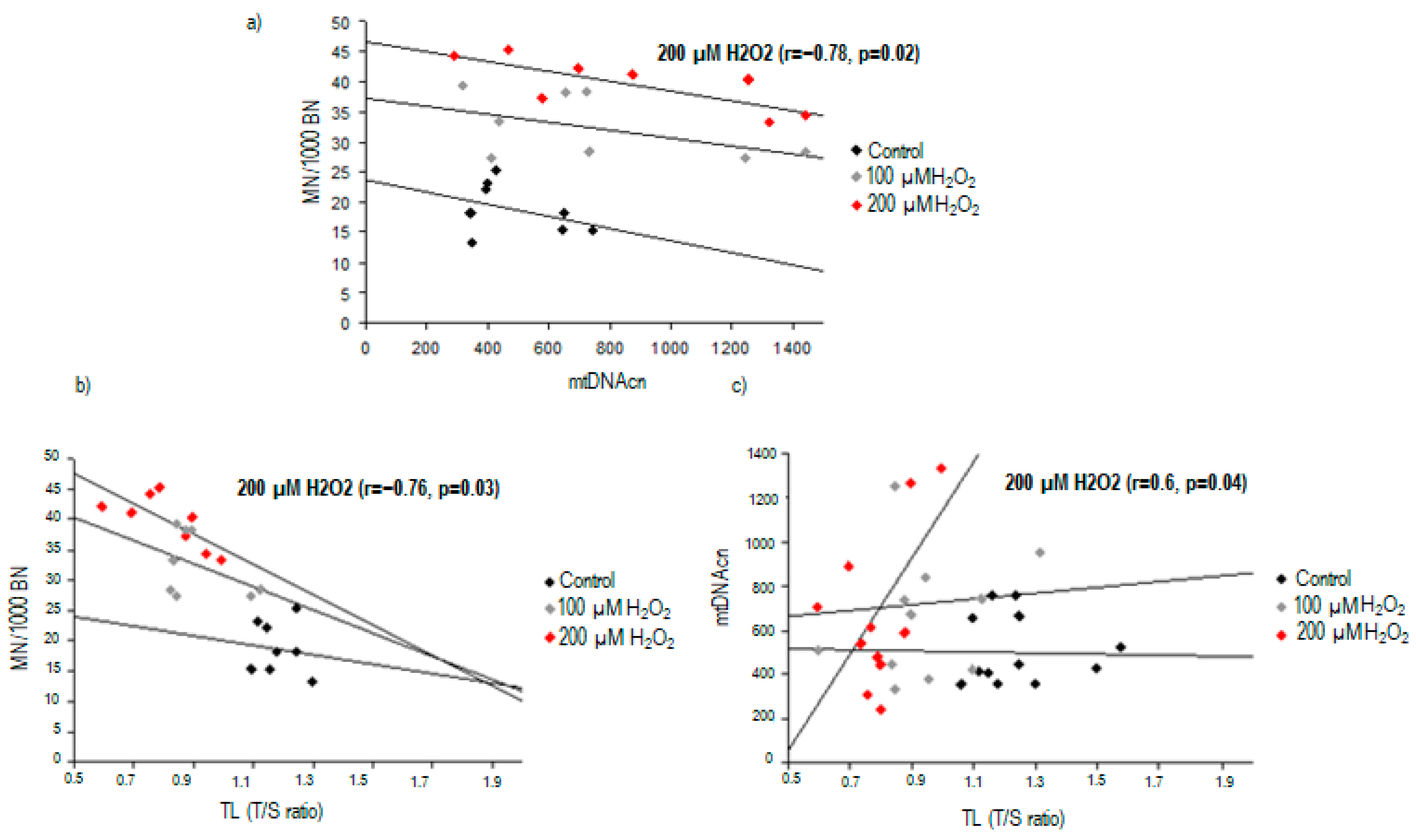
2.3. Correlation between Nuclear DNA Damage and Mitochondrial DNA Copy Number
3. Discussion
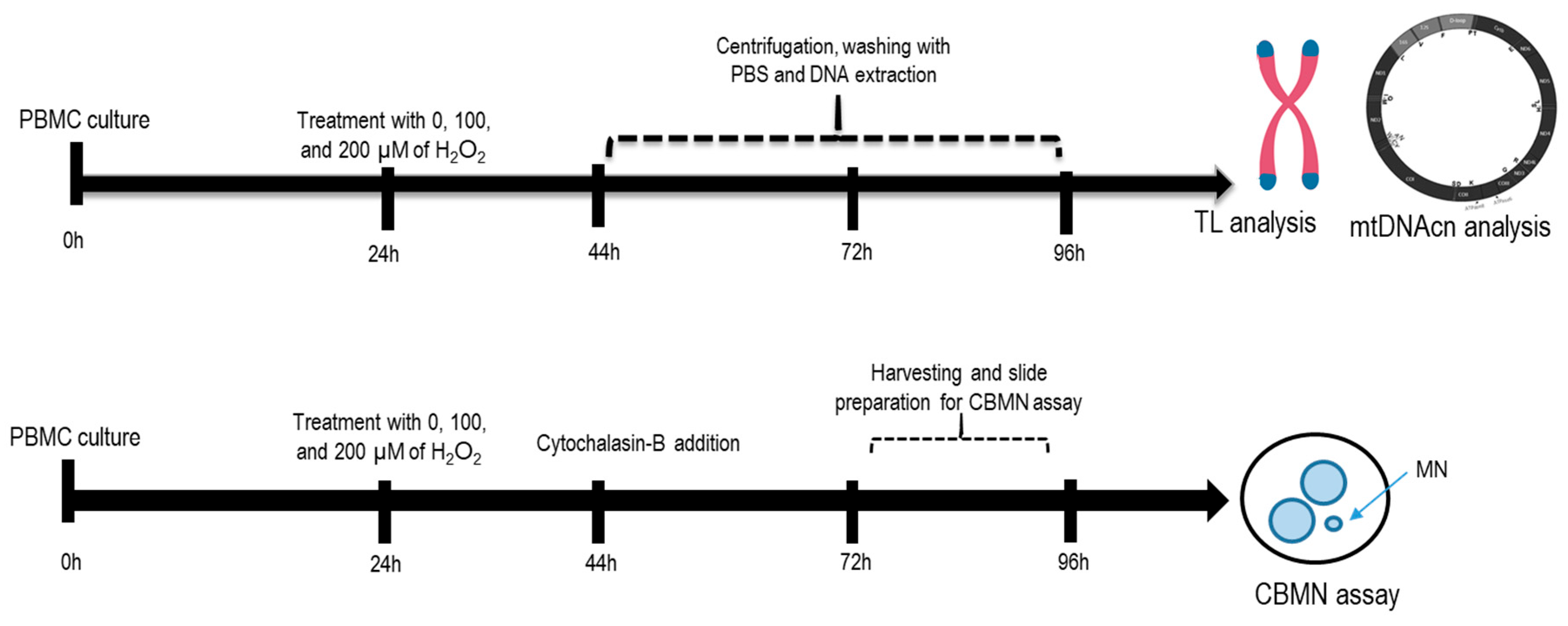
4. Materials and Methods
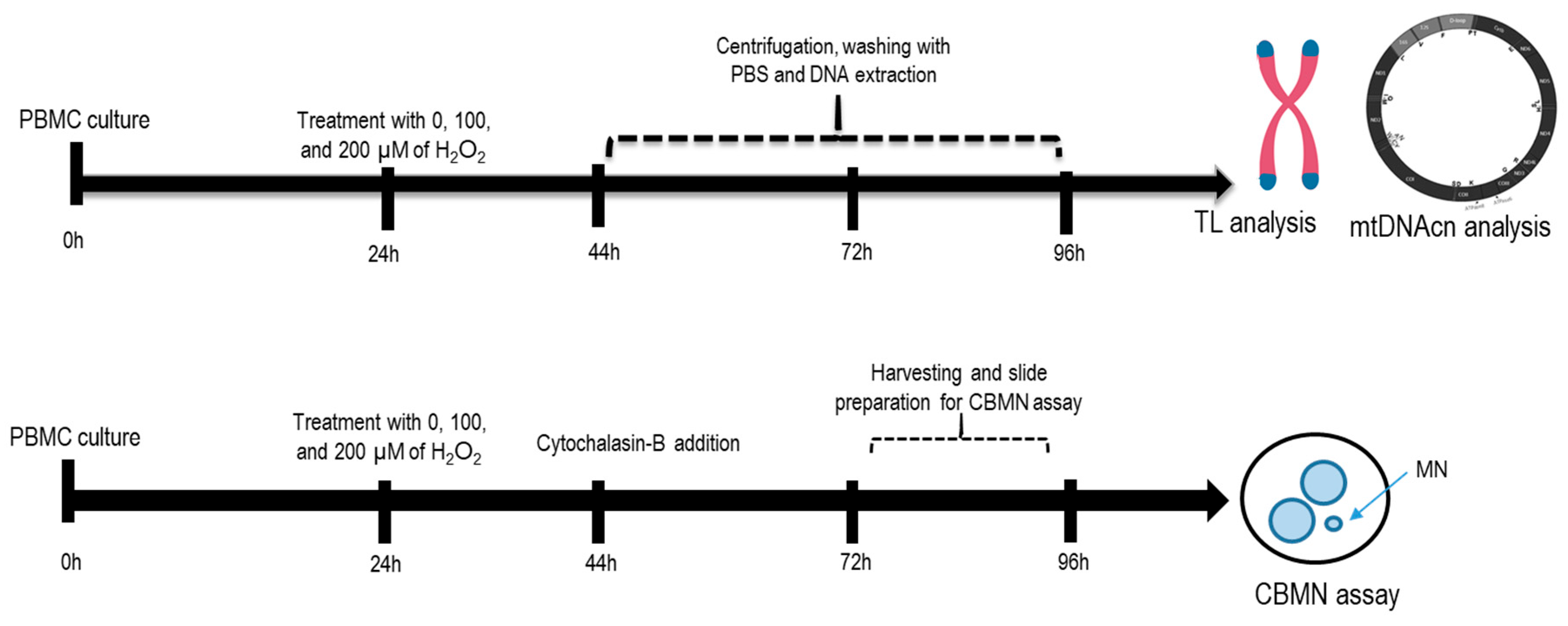
4.1. H2O2 Experiments with Peripheral Blood Mononuclear Cells
4.2. Telomere Length (TL) and Mitochondrial DNA Copy Number (mtDNAcn) Analysis
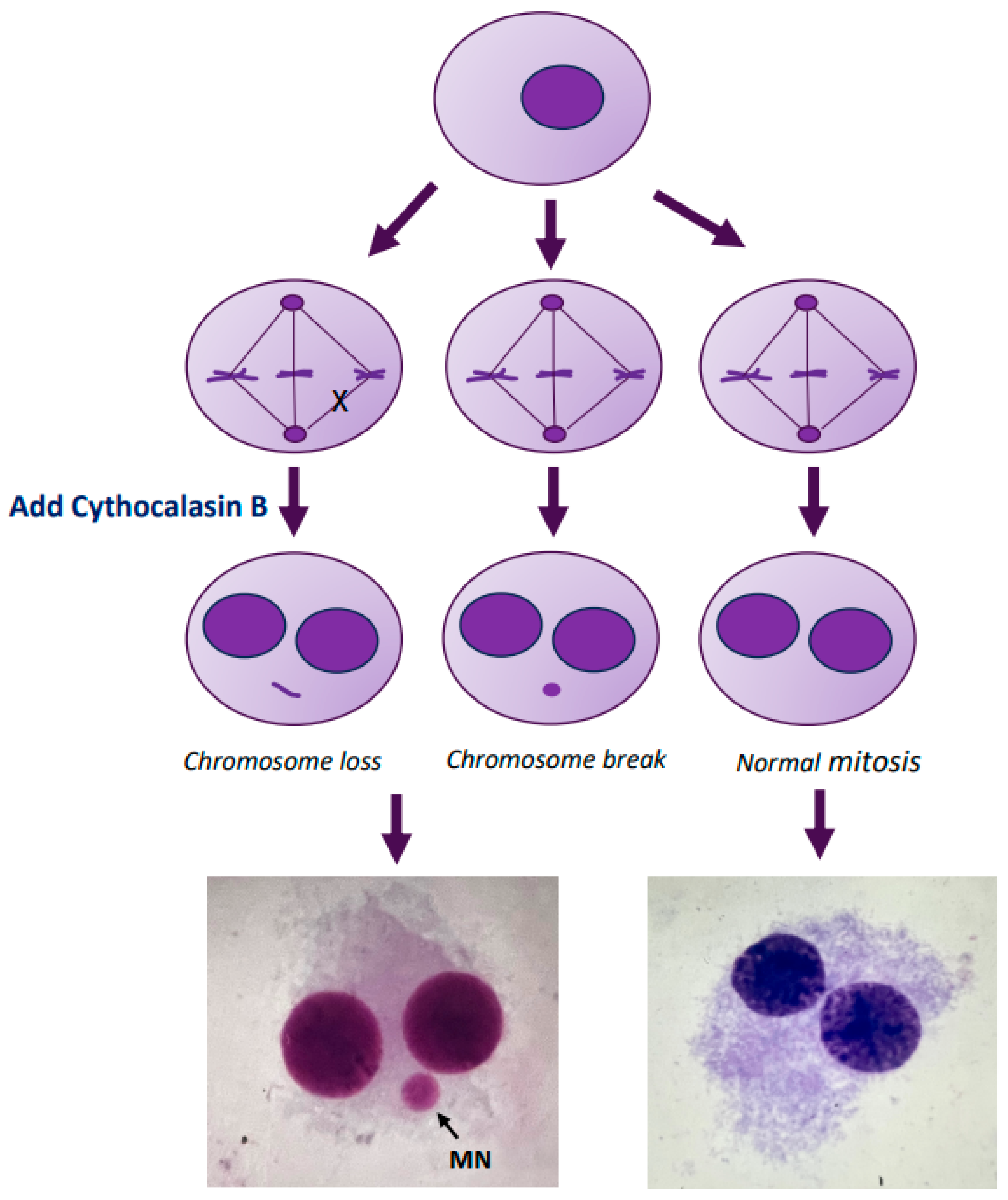
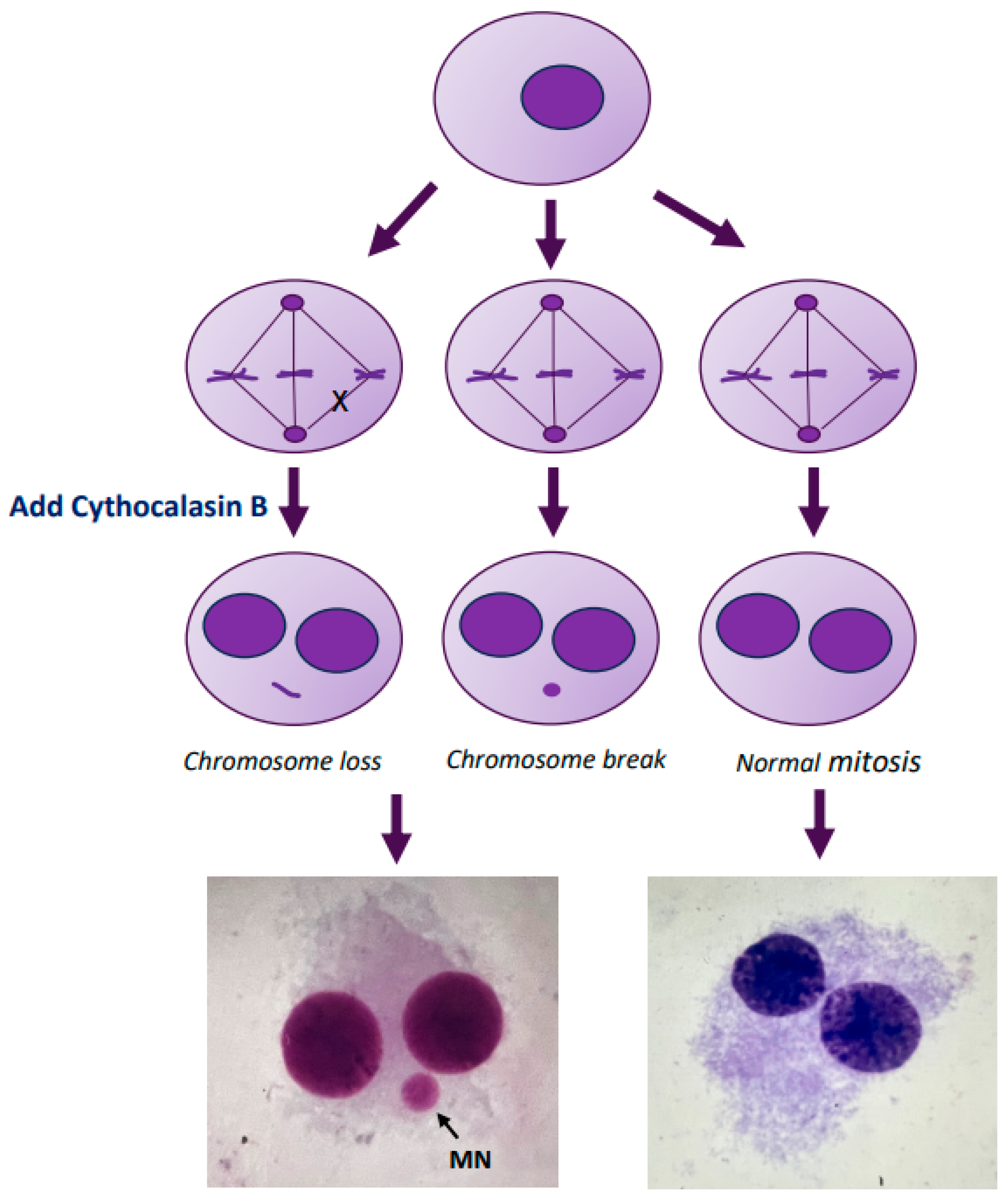
4.3. Micronucleus Assay
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laffon, B.; Bonassi, S.; Costa, S.; Valdiglesia, V. Genomic instability as a main driving factor of unsuccessful ageing: Potential for translating the use of micronuclei into clinical practice. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108359. [Google Scholar] [CrossRef]
- Wolters, S.; Schumacher, B. Genome maintenance and transcription integrity in aging and disease. Front. Genet. 2013, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M.; Knasmueller, S.; Knudsen, L.E.; Kirsch-Volders, M.; Deo, P.; Franzke, B.; Stopper, H.; Andreassi, M.G.; Bolognesi, C.; Dhillon, V.S.; et al. “Micronuclei and Disease” special issue: Aims, scope, and synthesis of outcomes. Mutat. Res. Rev. Mutat. Res. 2021, 788, 108384. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomeres and telomerase: Their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005, 579, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Chang, F.W.; Tsai, J.D.; Lin, K.M.; Chen, C.M.; Lin, S.Z.; Liu, C.A.; Harn, H.J. Telomeres and Cancer. Life 2021, 11, 1405. [Google Scholar] [CrossRef] [PubMed]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere dysfunction in ageing and age-related diseases. Nat. Cell Biol. 2022, 24, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Vecoli, C.; Borghini, A.; Andreassi, M.G. The molecular biomarkers of vascular aging and atherosclerosis: Telomere length and mitochondrial DNA4977 common deletion. Mutat. Res. Rev. Mutat. Res. 2020, 784, 108309. [Google Scholar] [CrossRef]
- Patel, J.; Baptiste, B.A.; Kim, E.; Hussain, M.; Croteau, D.L.; Bohr, V.A. DNA damage and mitochondria in cancer and aging. Carcinogenesis 2020, 41, 1625–1634. [Google Scholar] [CrossRef]
- Gao, X.; Yu, X.; Zhang, C.; Wang, Y.; Sun, Y.; Sun, H.; Zhang, H.; Shi, Y.; He, X. Telomeres and Mitochondrial Metabolism: Implications for Cellular Senescence and Age-related Diseases. Stem Cell Rev. Rep. 2022, 18, 2315–2327. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Pilger, R.; Sitte, N. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic. Biol. Med. 2000, 28, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, S.; Kawanishi, S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999, 453, 365–368. [Google Scholar] [CrossRef]
- Murnane, J.P. Telomere dysfunction and chromosome instability. Mutat. Res. 2012, 730, 28–36. [Google Scholar] [CrossRef]
- Gisselsson, D.; Jonson, T.; Petersen, A.; Strombeck, B.; Dal Cin, P.; Höglund, M.; Mitelman, F.; Mertens, F.; Mandahl, N. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 12683–12688. [Google Scholar] [CrossRef]
- Pampalona, J.; Soler, D.; Genesca, A.; Tusell, L. Telomere dysfunction and chromosome structure modulate the contribution of individual chromosomes in abnormal nuclear morphologies. Mutat. Res. 2010, 683, 16–22. [Google Scholar] [CrossRef]
- Fenech, M. The in vitro micronucleus technique. Mutat. Res. 2000, 455, 81–95. [Google Scholar] [CrossRef]
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE 2014, 9, e110963. [Google Scholar] [CrossRef]
- Nowell, P.C. Phytohemagglutinin: An initiator of mitosis in cultures of normal human leukocytes. Cancer Res. 1960, 20, 462–466. [Google Scholar]
- von Andrian, U.H.; Mackay, C.R. Advances in immunology: T-cell function and migration—Two sides of the same coin. N. Engl. J. Med. 2000, 343, 1020–10332. [Google Scholar] [CrossRef] [PubMed]
- Minato, N.; Hattori, M.; Hamazaki, Y. Physiology and pathology of T-cell aging. Int. Immunol. 2020, 32, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A Dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Sahin, S.; Colla, M.; Liesa, J.; Moslehi, F.L.; Müller, M.; Guo, M.; Cooper, D.; Kotton, A.J.; Fabian, C.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P. Cell biology: Aging theories unified. Nature 2011, 470, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Yin, P.H.; Lu, C.Y.; Chi, C.W.; Wei, Y.H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 2000, 348, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 2012, 13, 397–404. [Google Scholar] [CrossRef]
- Schank, M.; Zhao, J.; Wang, L.; Li, Z.; Cao, D.; Nguyen, L.N.; Yao, Z.Q. Telomeric injury by KML001 in human T cells induces mitochondrial dysfunction through the p53-PGC-1alpha pathway. Cell Death Dis. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, J.; Wu, F.; Lai, C.; Li, Y.; Zhang, G.; Peng, X.; Yu, S.; Yang, J.; Wang, W.; et al. Biological and epigenetic alterations of mitochondria involved in cellular replicative and hydrogen peroxide-induced premature senescence of human embryonic lung fibroblasts. Ecotoxicol. Environ. Saf. 2021, 216, 112204. [Google Scholar] [CrossRef]
- Borghini, A.; Mercuri, A.; Campolo, C.; Parolini, M.; Ndreu, R.; Turchi, S.; Andreassi, M.G. Influence of Chromosome 9p21.3 rs1333049 Variant on Telomere Length and Their Interactive Impact on the Prognosis of Coronary Artery Disease. J. Cardiovasc. Dev. Dis. 2023, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Vecoli, C.; Borghini, A.; Pulignani, S.; Mercuri, A.; Turchi, S.; Carpeggiani, C.; Picano, E.; Andreassi, M.G. Prognostic value of mitochondrial DNA4977 deletion and mitochondrial DNA copy number in patients with stable coronary artery disease. Atherosclerosis 2018, 217, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Balendra, V.; Esposto, J.; Obaid, A.A.; Maccioni, R.B.; Jha, N.K.; Perry, G.; Moustafa, M.; Al-Shehri, M.; Singh, M.P.; et al. Therapeutic Antiaging Strategies. Biomedicines 2022, 10, 2515. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borghini, A.; Ndreu, R.; Canale, P.; Campolo, J.; Marinaro, I.; Mercuri, A.; Turchi, S.; Andreassi, M.G. Telomere Length, Mitochondrial DNA, and Micronucleus Yield in Response to Oxidative Stress in Peripheral Blood Mononuclear Cells. Int. J. Mol. Sci. 2024, 25, 1428. https://doi.org/10.3390/ijms25031428
Borghini A, Ndreu R, Canale P, Campolo J, Marinaro I, Mercuri A, Turchi S, Andreassi MG. Telomere Length, Mitochondrial DNA, and Micronucleus Yield in Response to Oxidative Stress in Peripheral Blood Mononuclear Cells. International Journal of Molecular Sciences. 2024; 25(3):1428. https://doi.org/10.3390/ijms25031428
Chicago/Turabian StyleBorghini, Andrea, Rudina Ndreu, Paola Canale, Jonica Campolo, Irene Marinaro, Antonella Mercuri, Stefano Turchi, and Maria Grazia Andreassi. 2024. "Telomere Length, Mitochondrial DNA, and Micronucleus Yield in Response to Oxidative Stress in Peripheral Blood Mononuclear Cells" International Journal of Molecular Sciences 25, no. 3: 1428. https://doi.org/10.3390/ijms25031428
APA StyleBorghini, A., Ndreu, R., Canale, P., Campolo, J., Marinaro, I., Mercuri, A., Turchi, S., & Andreassi, M. G. (2024). Telomere Length, Mitochondrial DNA, and Micronucleus Yield in Response to Oxidative Stress in Peripheral Blood Mononuclear Cells. International Journal of Molecular Sciences, 25(3), 1428. https://doi.org/10.3390/ijms25031428