
Genome-Wide Identification, Functional Characterization, and Stress-Responsive Expression Profiling of Subtilase (SBT) Gene Family in Peanut (Arachis hypogaea L.)

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Protein Physicochemical Property Analysis and Identification of AhSBTs in the STQ Genome
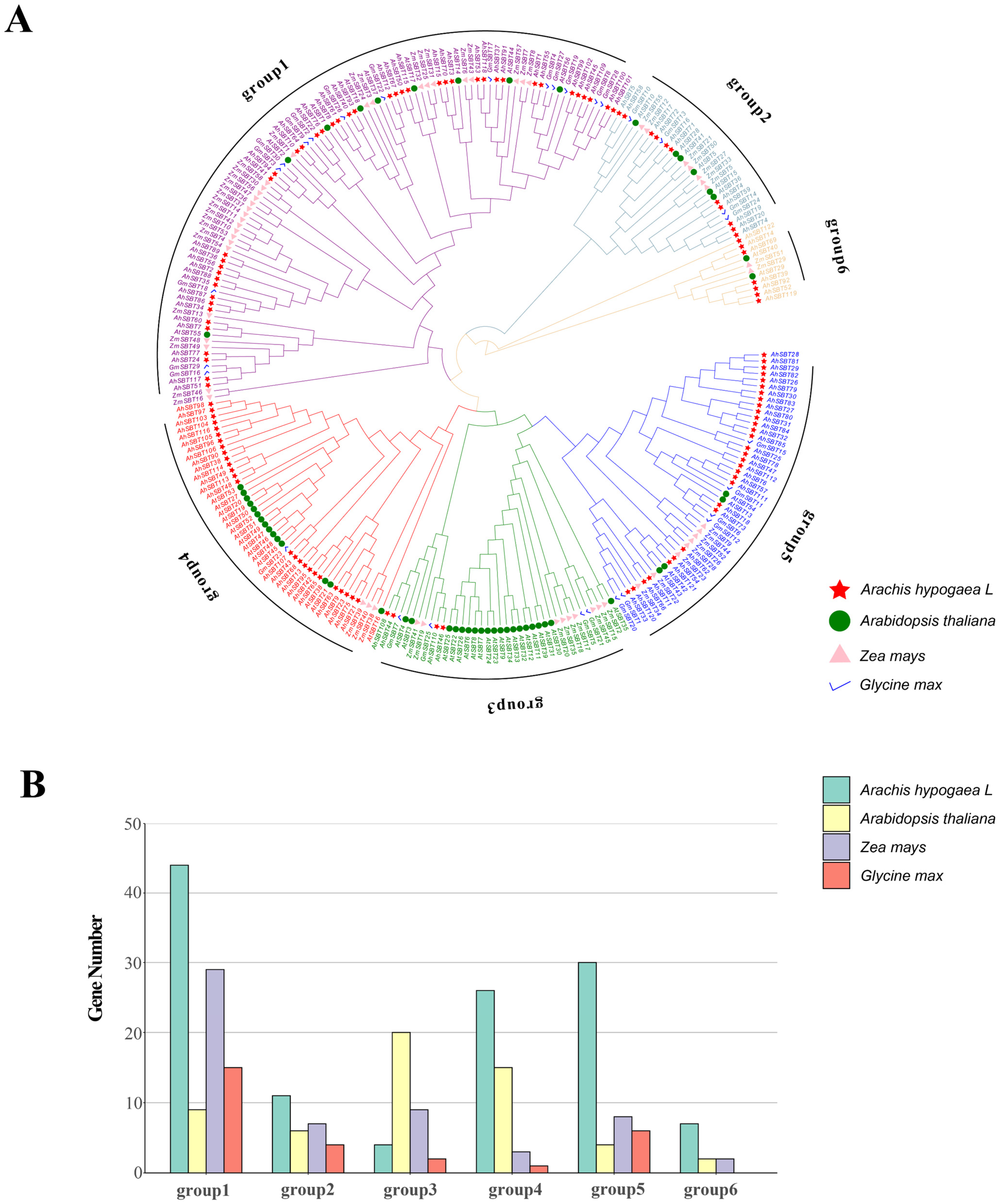
2.2. AhSBTs Phylogenetic Tree Analysis
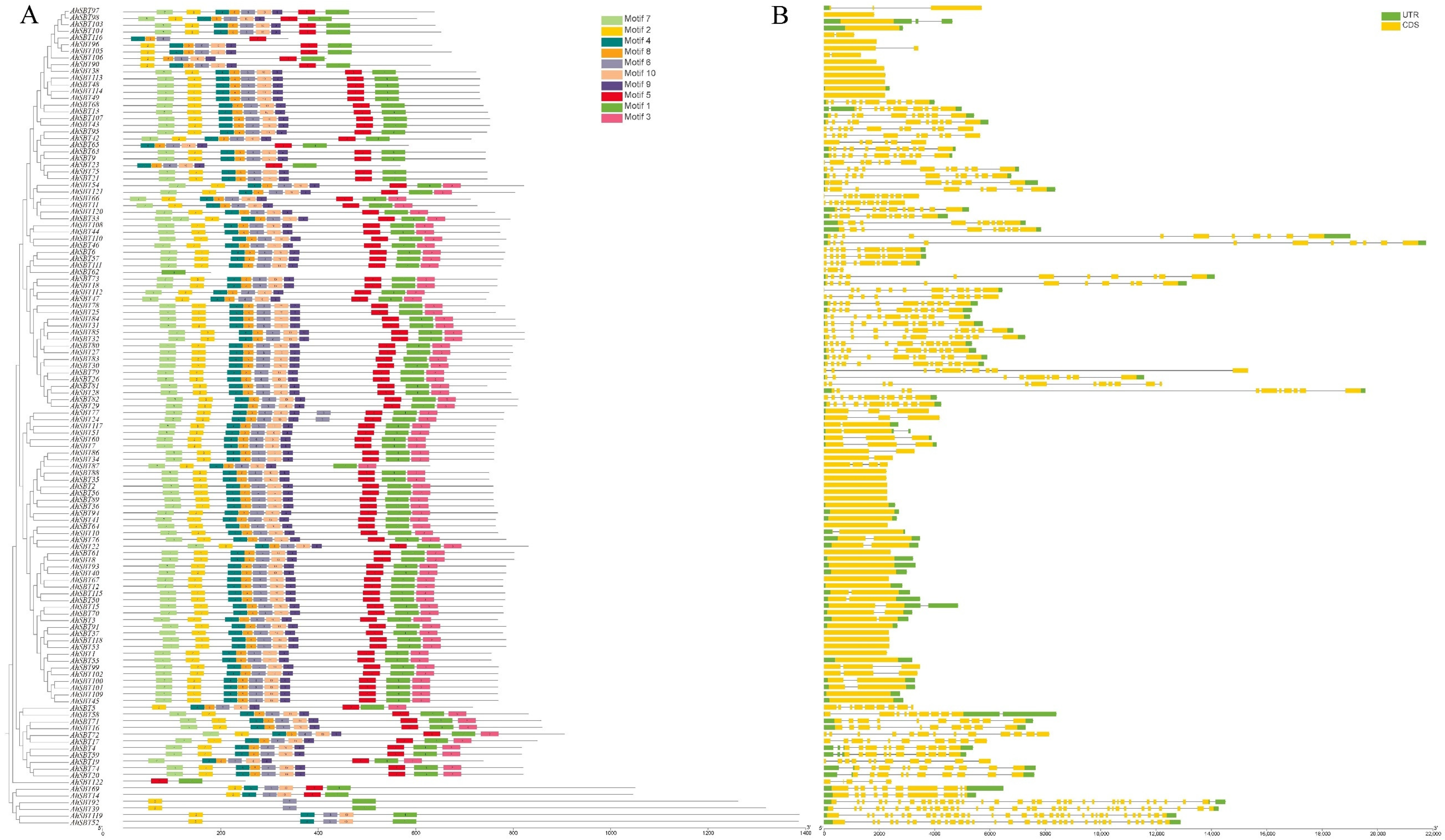
2.3. Analysis of AhSBTs Protein Conserved Domain and Structure
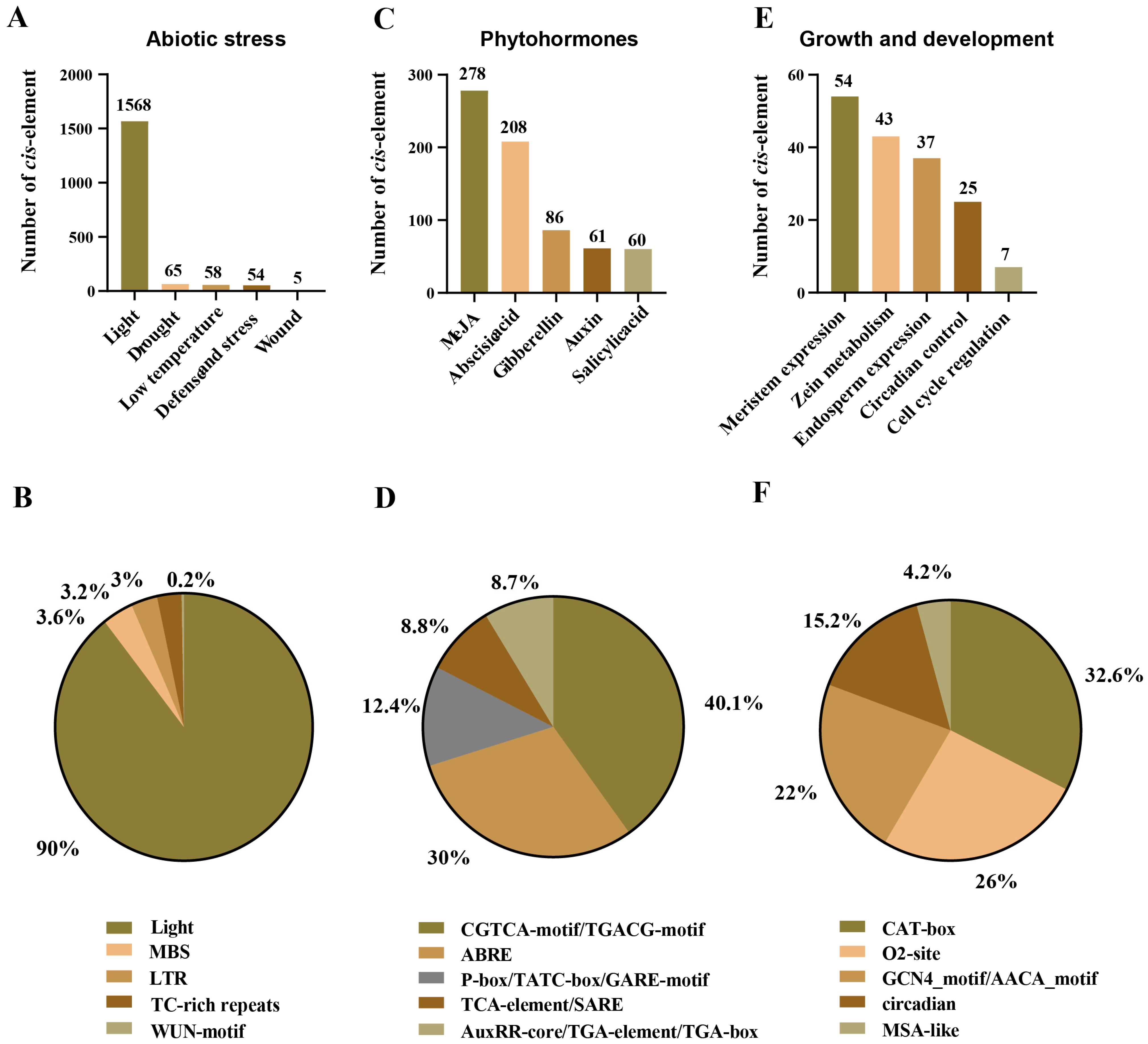
2.4. Cis-Elements: Crucial Components in the Promoter Regions of AhSBTs
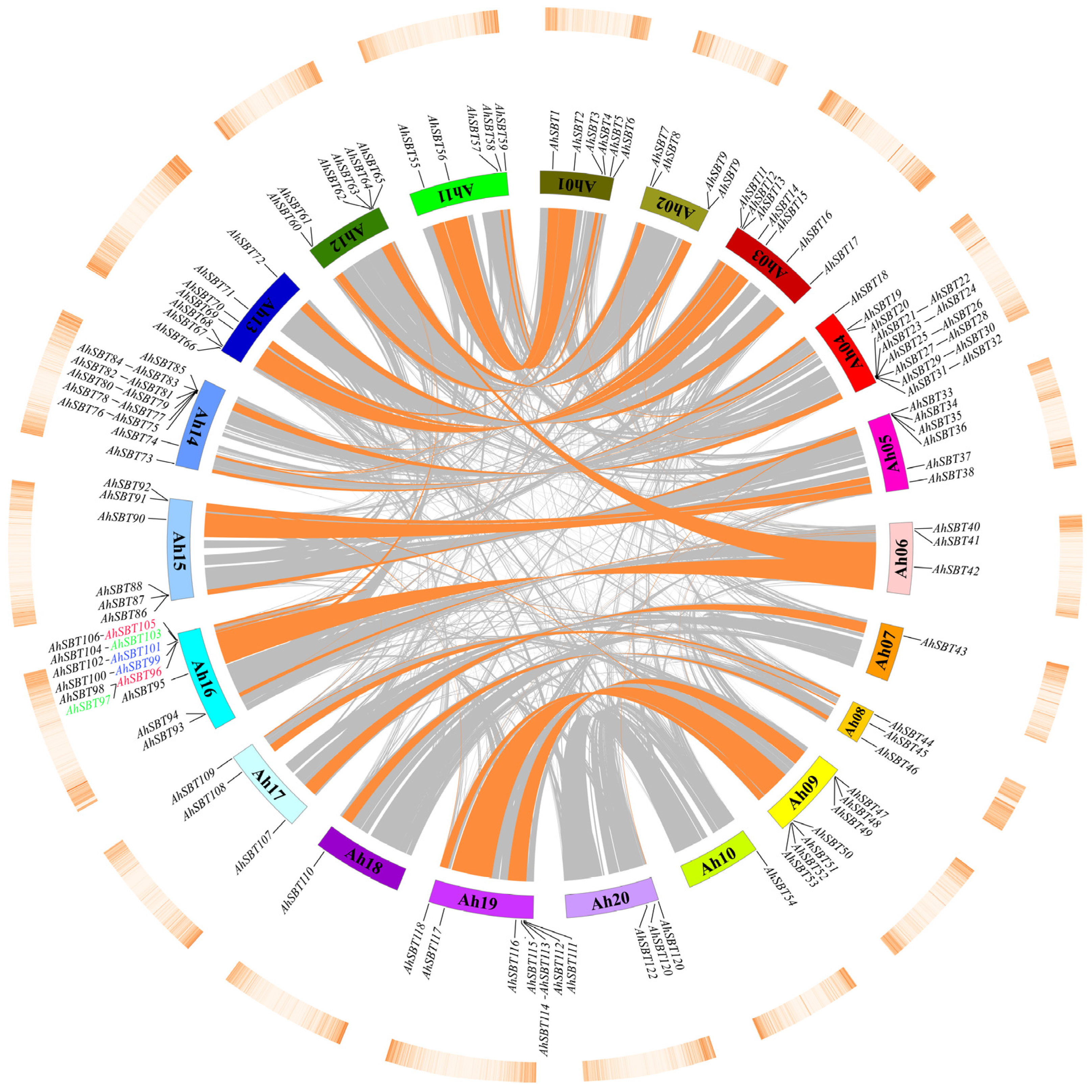
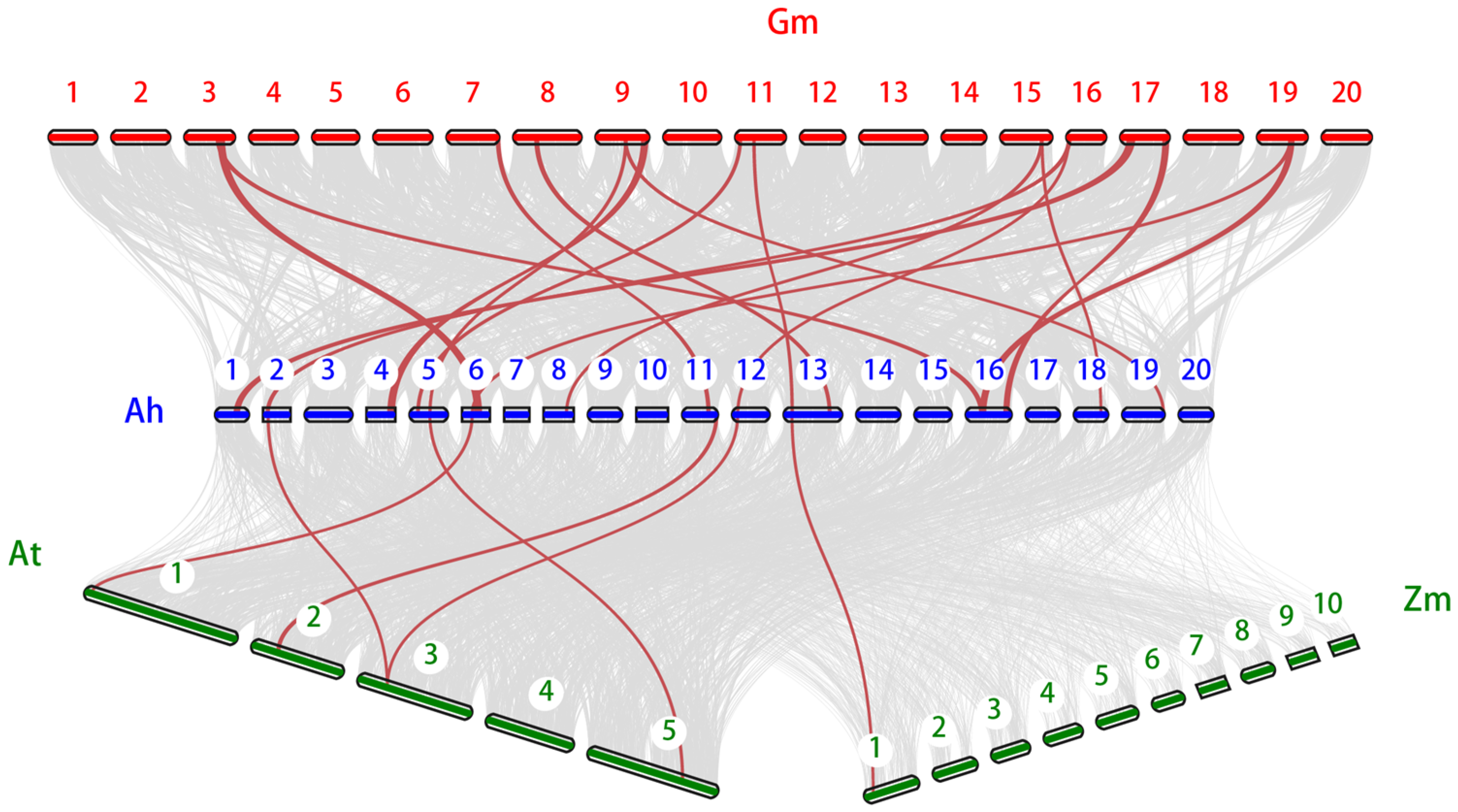
2.5. Tandem Duplication and Synteny Analyses of AhSBTs
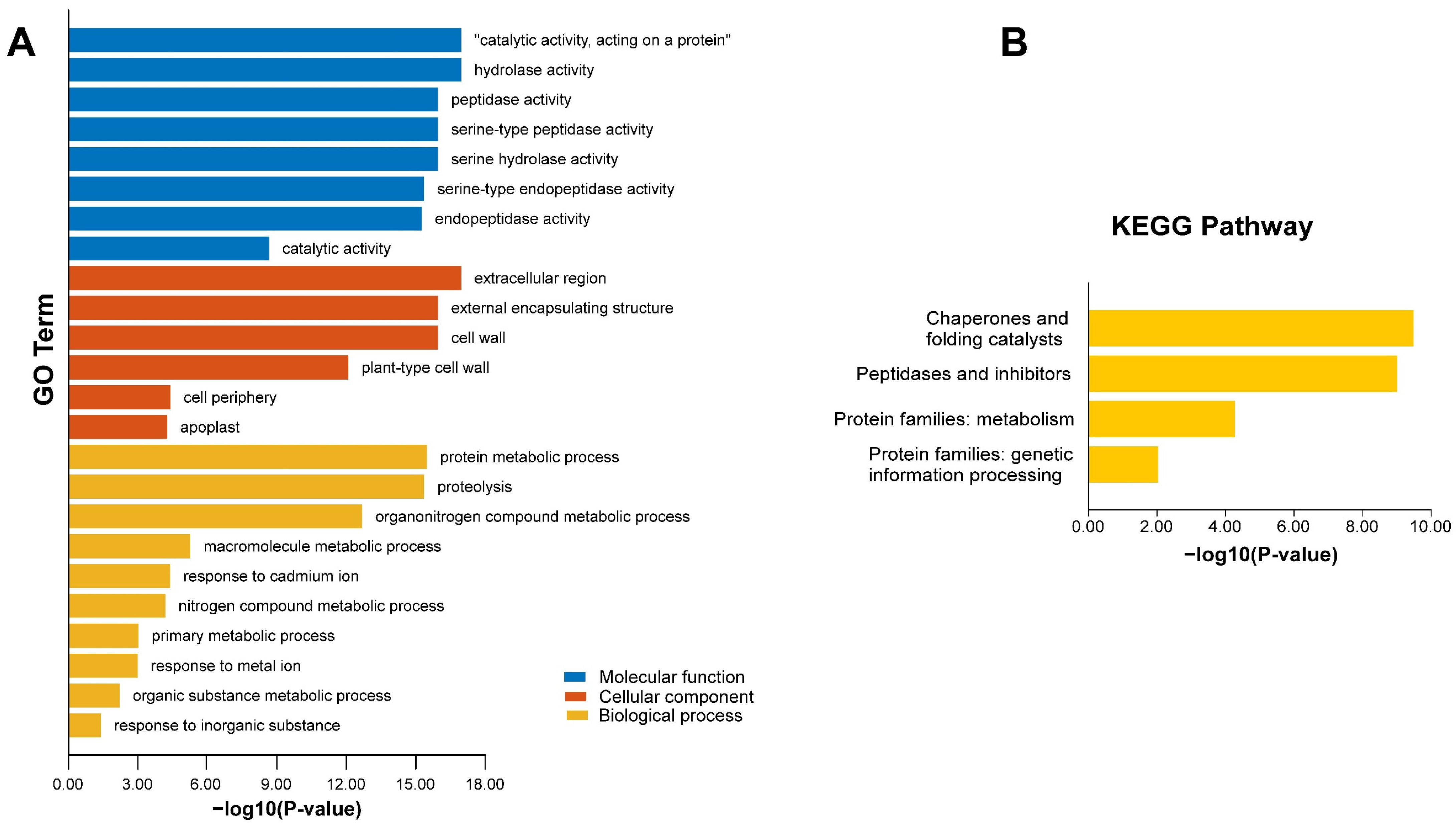
2.6. AhSBTs Enrichment Analysis Using Kyoto Encyclopedia of Genes and Genomics and Gene Ontology
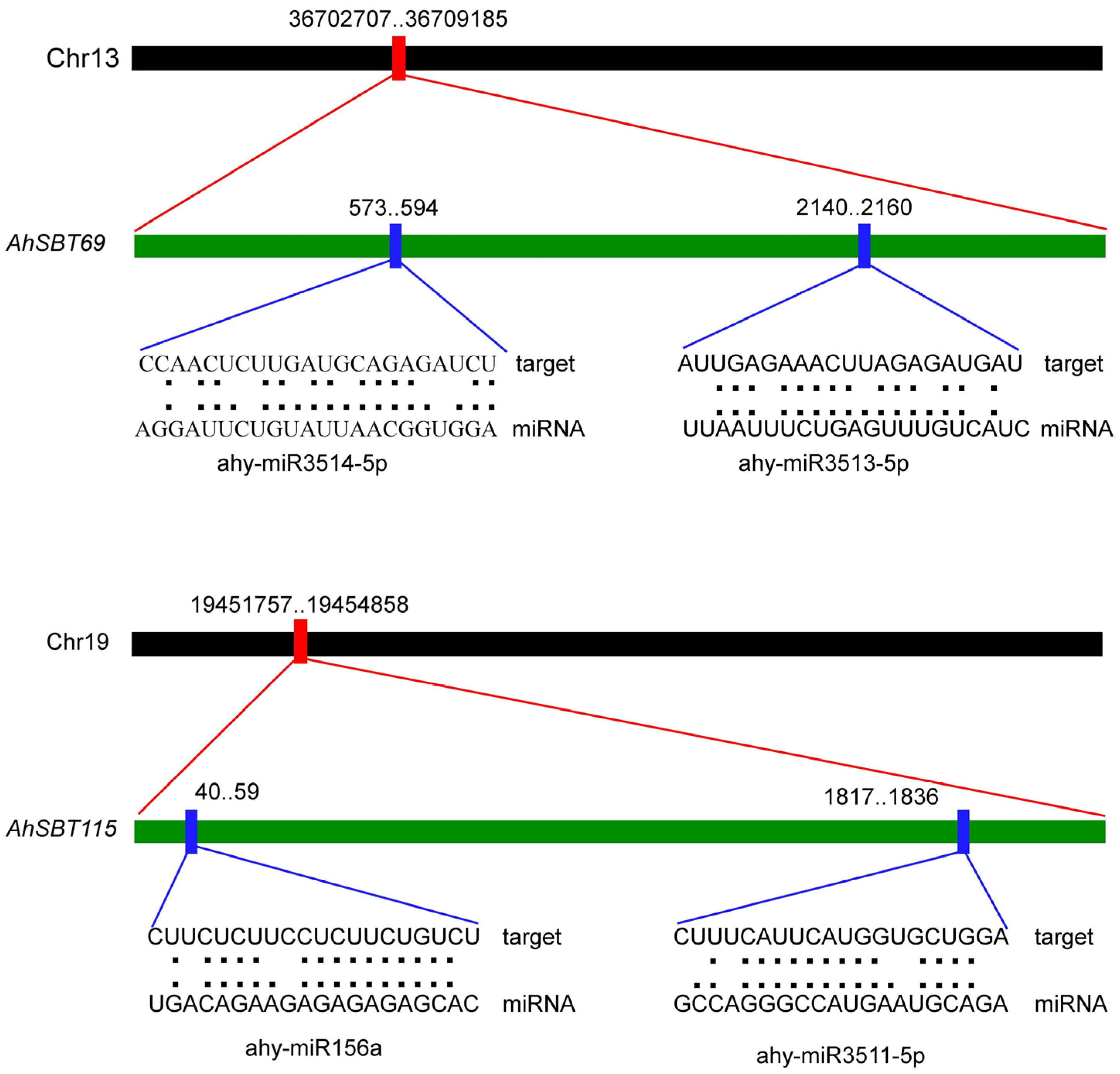
2.7. Exploration of miRNAs Targeting AhSBTs
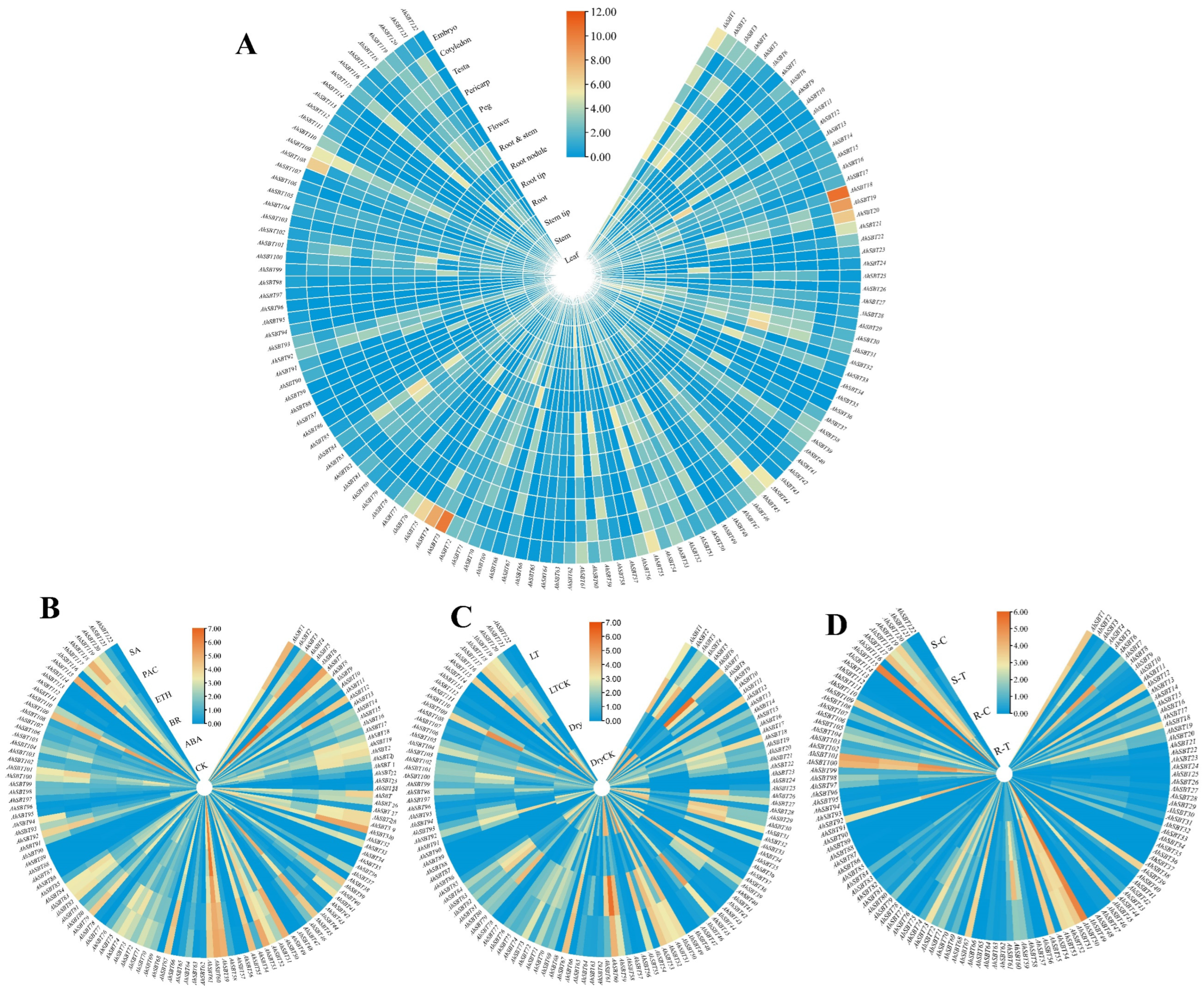
2.8. Profiling the Expression of AhSBTs in Various Developmental Tissues
2.9. Expression of AhSBTs Under Bacterial Wilt, Hormones, and Abiotic Treatments
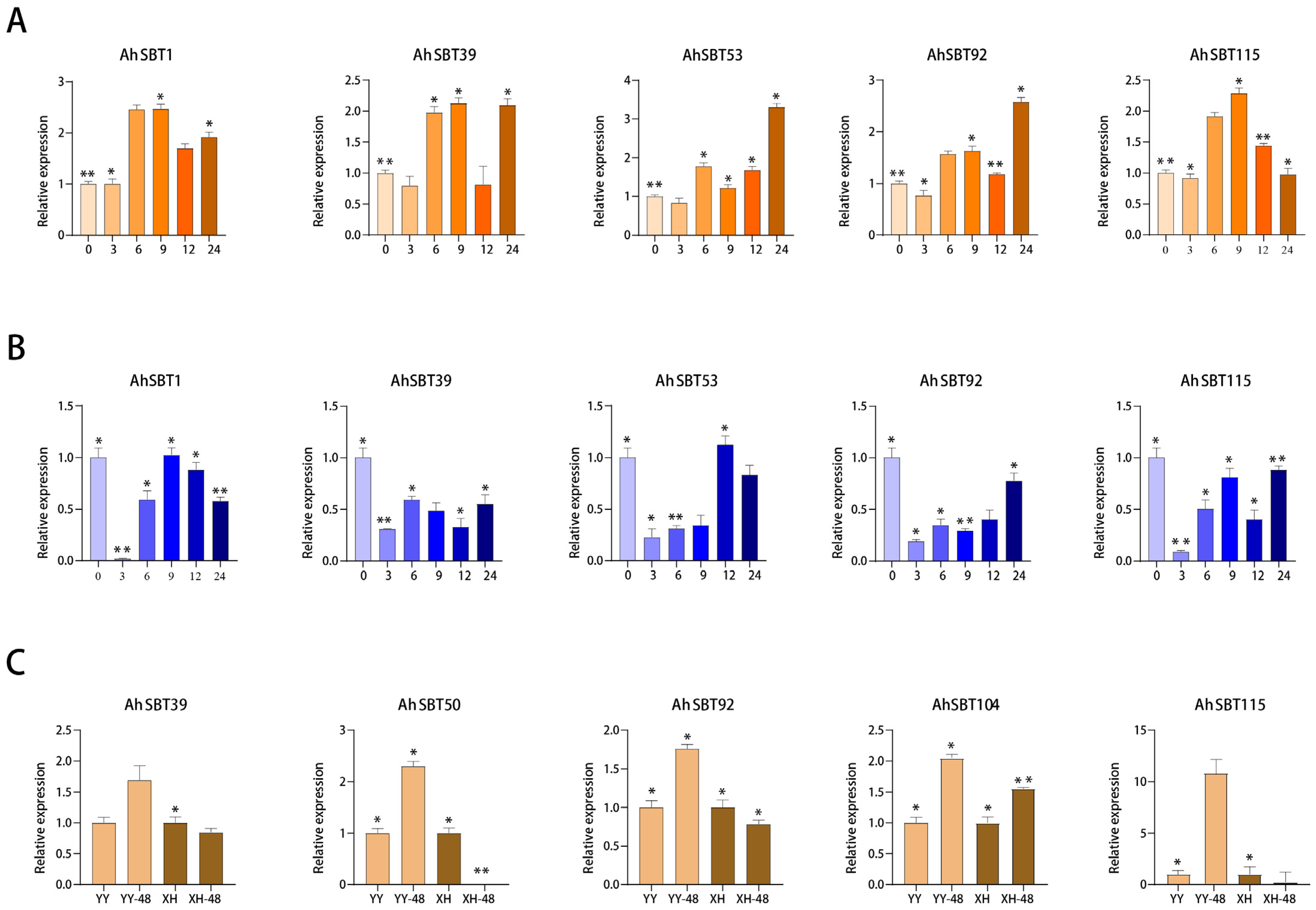
2.10. Profiling Expression of AhSBTs Under Bacterial Wilt, ABA and Cold Treatment Using qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Identification of AhSBTs Across the Genome
4.2. SBTs Distribution Across Chromosomes, Evolutionary Phylogenetic Analysis, and Covariance Study
4.3. Analysis of Gene Structure and Motifs
4.4. Promoter Analysis of AhSBTs
4.5. Prediction of Potential miRNAs Targeting AhSBTs and Assessment of Functional Annotation
4.6. Expression Patterns of the AhSBTs
4.7. Plant Material and Stress Conditions
4.8. RNA Extraction and qRT-PCR-Based Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AhSBT | Arachis hypogaea subtilases |
| Ah | Arachis hypogaea |
| ASP | Aspartic acid |
| At | Arabidopsis thaliana |
| Chr | Chromosome |
| FPKM | Fragments per kilobase of transcript per million mapped reads |
| Gm | Glycin max |
| GO | Gene ontology |
| HIS | Histidine |
| HMM | Hidden Markov model |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| miRNAs | Micro RNAs |
| Mya | Million years ago |
| qRT-PCR | Quantitative real-time PCR |
| SER | Serine |
| STQ | Peanut cultivar Shitouqi |
| Zm | Zea mays |
References
- Schaller, A. A cut above the rest: The regulatory function of plant proteases. Planta 2004, 220, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Siezen, R.J.; Leunissen, J.A. Subtilases: The superfamily of subtilisin-like serine proteases. Protein Sci. 1997, 6, 501–523. [Google Scholar] [CrossRef] [PubMed]
- Truskina, J.; Brück, S.; Stintzi, A.; Boeuf, S.; Doll, N.M.; Fujita, S.; Geldner, N.; Schaller, A.; Ingram, G.C. A peptide-mediated, multilateral molecular dialogue for the coordination of pollen wall formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2201446119. [Google Scholar] [CrossRef] [PubMed]
- Barrett, A.J.; Rawlings, N.D. Families and clans of serine peptidases. Arch. Biochem. Biophys. 1995, 318, 247–250. [Google Scholar] [CrossRef]
- Schaller, A.; Stintzi, A.; Graff, L. Subtilases—Versatile tools for protein turnover, plant development, and interactions with the environment. Physiol. Plant. 2012, 145, 52–66. [Google Scholar] [CrossRef]
- Rautengarten, C.; Steinhauser, D.; Büssis, D.; Stintzi, A.; Schaller, A.; Kopka, J.; Altmann, T. Inferring hypotheses on functional relationships of genes: Analysis of the Arabidopsis thaliana subtilase gene family. PLoS Comput. Biol. 2005, 1, e40. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.N.; Veliz, C.G.; Criado, M.V.; Signorini, A.; Simonetti, E.; Caputo, C. Identification and expression analysis of 11 subtilase genes during natural and induced senescence of barley plants. J. Plant Physiol. 2017, 211, 70–80. [Google Scholar] [CrossRef]
- Tripathi, L.P.; Sowdhamini, R. Cross genome comparisons of serine proteases in Arabidopsis and rice. BMC Genom. 2006, 7, 200. [Google Scholar] [CrossRef]
- Dai, M.; Zhou, N.; Zhang, Y.; Zhang, Y.; Ni, K.; Wu, Z.; Liu, L.; Wang, X.; Chen, Q. Genome-wide analysis of the SBT gene family involved in drought tolerance in cotton. Front. Plant Sci. 2022, 13, 1097732. [Google Scholar] [CrossRef]
- Cao, J.; Han, X.; Zhang, T.; Yang, Y.; Huang, J.; Hu, X. Genome-wide and molecular evolution analysis of the subtilase gene family in Vitis vinifera. BMC Genom. 2014, 15, 1116. [Google Scholar] [CrossRef]
- Reichardt, S.; Repper, D.; Tuzhikov, A.I.; Galiullina, R.A.; Planas-Marquès, M.; Chichkova, N.V.; Vartapetian, A.B.; Stintzi, A.; Schaller, A. The tomato subtilase family includes several cell death-related proteinases with caspase specificity. Sci. Rep. 2018, 8, 10531. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Zhou, G.; Ruan, H.; Zhao, J.; Hasi, A.; Zong, N. Genome-Wide Identification and Analysis of the Maize Serine Peptidase S8 Family Genes in Response to Drought at Seedling Stage. Plants 2023, 12, 369. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, V.; López, A.; Mauch-Mani, B.; Gil, M.J.; Vera, P. An extracellular subtilase switch for immune priming in Arabidopsis. PLoS Pathog. 2013, 9, e1003445. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Zhang, Z.; Wang, J.; Zuo, K. Characterization of a novel cotton subtilase gene GbSBT1 in response to extracellular stimulations and its role in Verticillium resistance. PLoS ONE 2016, 11, e0153988. [Google Scholar] [CrossRef]
- Ekchaweng, K.; Khunjan, U.; Churngchow, N. Molecular cloning and characterization of three novel subtilisin-like serine protease genes from Hevea brasiliensis. Physiol. Mol. Plant Pathol. 2017, 97, 79–95. [Google Scholar] [CrossRef]
- Budič, M.; Sabotič, J.; Meglič, V.; Kos, J.; Kidrič, M. Characterization of two novel subtilases from common bean (Phaseolus vulgaris L.) and their responses to drought. Plant Physiol. Biochem. 2013, 62, 79–87. [Google Scholar] [CrossRef]
- Galotta, M.F.; Pugliese, P.; Gutiérrez-Boem, F.H.; Veliz, C.G.; Criado, M.V.; Caputo, C.; Echeverria, M.; Roberts, I.N. Subtilase activity and gene expression during germination and seedling growth in barley. Plant Physiol. Biochem. 2019, 139, 197–206. [Google Scholar] [CrossRef]
- Zhuang, W.; Chen, H.; Yang, M.; Wang, J.; Pandey, M.K.; Zhang, C.; Chang, W.C.; Zhang, L.; Zhang, X.; Tang, R.; et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat. Genet. 2019, 51, 865–876. [Google Scholar] [CrossRef]
- Xue, T.; Liu, L.; Zhang, X.; Li, Z.; Sheng, M.; Ge, X.; Xu, W.; Su, Z. Genome-Wide Investigation and Co-Expression Network Analysis of SBT Family Gene in Gossypium. Int. J. Mol. Sci. 2023, 24, 5760. [Google Scholar] [CrossRef]
- Zhao, X.; Goher, F.; Chen, L.; Song, J.; Zhao, J. Genome-Wide Identification, Phylogeny and Expression Analysis of Subtilisin (SBT) Gene Family under Wheat Biotic and Abiotic Stress. Plants 2023, 12, 3065. [Google Scholar] [CrossRef]
- Hou, Q.; Wang, L.; Qi, Y.; Yan, T.; Zhang, F.; Zhao, W.; Wan, X. A systematic analysis of the subtilase gene family and expression and subcellular localization investigation of anther-specific members in maize. Plant Physiol. Biochem. 2023, 203, 108041. [Google Scholar] [CrossRef] [PubMed]
- Bertioli, D.J.; Seijo, G.; Freitas, F.O.; Valls, J.F.; Leal-Bertioli, S.C.; Moretzsohn, M.C.J.P.G.R. An overview of peanut and its wild relatives. Plant Genet. Resour. 2011, 9, 134–149. [Google Scholar] [CrossRef]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938–950. [Google Scholar] [CrossRef]
- Ren, R.; Wang, H.; Guo, C.; Zhang, N.; Zeng, L.; Chen, Y.; Ma, H.; Qi, J. Widespread whole genome duplications contribute to genome complexity and species diversity in angiosperms. Mol. Plant 2018, 11, 414–428. [Google Scholar] [CrossRef]
- Babenko, V.N.; Rogozin, I.B.; Mekhedov, S.L.; Koonin, E.V. Prevalence of intron gain over intron loss in the evolution of paralogous gene families. Nucleic Acids Res. 2004, 32, 3724–3733. [Google Scholar] [CrossRef]
- Rose, R.; Schaller, A.; Ottmann, C. Structural features of plant subtilases. Plant Signal. Behav. 2010, 5, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Tan-Wilson, A.; Bandak, B.; Prabu-Jeyabalan, M. The PA domain is crucial for determining optimum substrate length for soybean protease C1: Structure and kinetics correlate with molecular function. Plant Physiol. Biochem. 2012, 53, 27–32. [Google Scholar] [CrossRef]
- Ottmann, C.; Rose, R.; Huttenlocher, F.; Cedzich, A.; Hauske, P.; Kaiser, M.; Huber, R.; Schaller, A. Structural basis for Ca2+-independence and activation by homodimerization of tomato subtilase 3. Proc. Natl. Acad. Sci. USA 2009, 106, 17223–17228. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Li, L.; Sahu, S.K.; Petersen, M.; Liu, X.; Melkonian, M.; Zhang, G.; Liu, H. Molecular evidence for origin, diversification and ancient gene duplication of plant subtilases (SBTs). Sci. Rep. 2019, 9, 12485. [Google Scholar] [CrossRef]
- Wang, G.; Ellendorff, U.; Kemp, B.; Mansfield, J.W.; Forsyth, A.; Mitchell, K.; Bastas, K.; Liu, C.-M.; Woods-Tör, A.; Zipfel, C.; et al. A Genome-Wide Functional Investigation into the Roles of Receptor-Like Proteins in Arabidopsis. Plant Physiol. 2008, 147, 503–517. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, F.; Zhou, T.; Fang, A.; Yu, Y.; Bi, C.; Xiao, S. In Silico Identification of the Full Complement of Subtilase-Encoding Genes and Characterization of the Role of TaSBT1.7 in Resistance Against Stripe Rust in Wheat. Phytopathology 2021, 111, 398–407. [Google Scholar] [CrossRef] [PubMed]
- D’Erfurth, I.; Le Signor, C.; Aubert, G.; Sanchez, M.; Vernoud, V.; Darchy, B.; Lherminier, J.; Bourion, V.; Bouteiller, N.; Bendahmane, A.; et al. A role for an endosperm-localized subtilase in the control of seed size in legumes. New Phytol. 2012, 196, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Liu, Y.; Hou, Z.; Zhang, Y.; Fang, Y.; Huang, Y.; Cai, H.; Qin, Y.; Cheng, Y. Genome-Wide Investigation of SBT Family Genes in Pineapple and Functional Analysis of AcoSBT1.12 in Floral Transition. Front. Genet. 2021, 12, 730821. [Google Scholar] [CrossRef]
- Jamla, M.; Patil, S.; Joshi, S.; Khare, T.; Kumar, V. MicroRNAs and their exploration for developing heavy metal-tolerant plants. J. Plant Growth Regul. 2021, 41, 2579–2595. [Google Scholar] [CrossRef]
- Zhao, C.; Xia, H.; Cao, T.; Yang, Y.; Zhao, S.; Hou, L.; Zhang, Y.; Li, C.; Zhang, X.; Wang, X. Small RNA and degradome deep sequencing reveals peanut microRNA roles in response to pathogen infection. Plant Mol. Biol. Rep. 2015, 33, 1013–1029. [Google Scholar] [CrossRef]
- Yao, X.; Chen, J.; Zhou, J.; Yu, H.; Ge, C.; Zhang, M.; Gao, X.; Dai, X.; Yang, Z.-N.; Zhao, Y. An essential role for miRNA167 in maternal control of embryonic and seed development. Plant Physiol. 2019, 180, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Waadt, R.; Seller, C.A.; Hsu, P.-K.; Takahashi, Y.; Munemasa, S.; Schroeder, J.I. Plant hormone regulation of abiotic stress responses. Nat. Rev. Mol. Cell Biol. 2022, 23, 680–694. [Google Scholar] [CrossRef]
- Li, Y.; Luo, W.; Sun, Y.; Chang, H.; Ma, K.; Zhao, Z.; Lu, L. Identification and expression analysis of miR160 and their target genes in cucumber. Biochem. Genet. 2022, 60, 127–152. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Stajich, J.E. An Introduction to BioPerl. Methods Mol. Biol. 2007, 406, 535–548. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Zhang, Z. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences. Genom. Proteom. Bioinform. 2022, 20, 536–540. [Google Scholar] [CrossRef]
- Bertioli, D.J.; Cannon, S.B.; Froenicke, L.; Huang, G.; Farmer, A.D.; Cannon, E.K.; Liu, X.; Gao, D.; Clevenger, J.; Dash, S. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat. Genet. 2016, 48, 438–446. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server (2017 release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Fu, H.; Sharif, Y.; Abdullaziz, S.; Wang, L.; Zhang, Y.; Zhuang, Y. Genome-Wide Identification, Functional Characterization, and Stress-Responsive Expression Profiling of Subtilase (SBT) Gene Family in Peanut (Arachis hypogaea L.). Int. J. Mol. Sci. 2024, 25, 13361. https://doi.org/10.3390/ijms252413361
Li S, Fu H, Sharif Y, Abdullaziz S, Wang L, Zhang Y, Zhuang Y. Genome-Wide Identification, Functional Characterization, and Stress-Responsive Expression Profiling of Subtilase (SBT) Gene Family in Peanut (Arachis hypogaea L.). International Journal of Molecular Sciences. 2024; 25(24):13361. https://doi.org/10.3390/ijms252413361
Chicago/Turabian StyleLi, Shipeng, Huiwen Fu, Yasir Sharif, Sheidu Abdullaziz, Lihui Wang, Yongli Zhang, and Yuhui Zhuang. 2024. "Genome-Wide Identification, Functional Characterization, and Stress-Responsive Expression Profiling of Subtilase (SBT) Gene Family in Peanut (Arachis hypogaea L.)" International Journal of Molecular Sciences 25, no. 24: 13361. https://doi.org/10.3390/ijms252413361
APA StyleLi, S., Fu, H., Sharif, Y., Abdullaziz, S., Wang, L., Zhang, Y., & Zhuang, Y. (2024). Genome-Wide Identification, Functional Characterization, and Stress-Responsive Expression Profiling of Subtilase (SBT) Gene Family in Peanut (Arachis hypogaea L.). International Journal of Molecular Sciences, 25(24), 13361. https://doi.org/10.3390/ijms252413361