

Comparative Transcriptome Analysis Revealing the Potential Salt Tolerance Mechanism of Exogenous Abscisic Acid Application in Melilotus albus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Physiological Responses of M. albus Under ABA Treatment
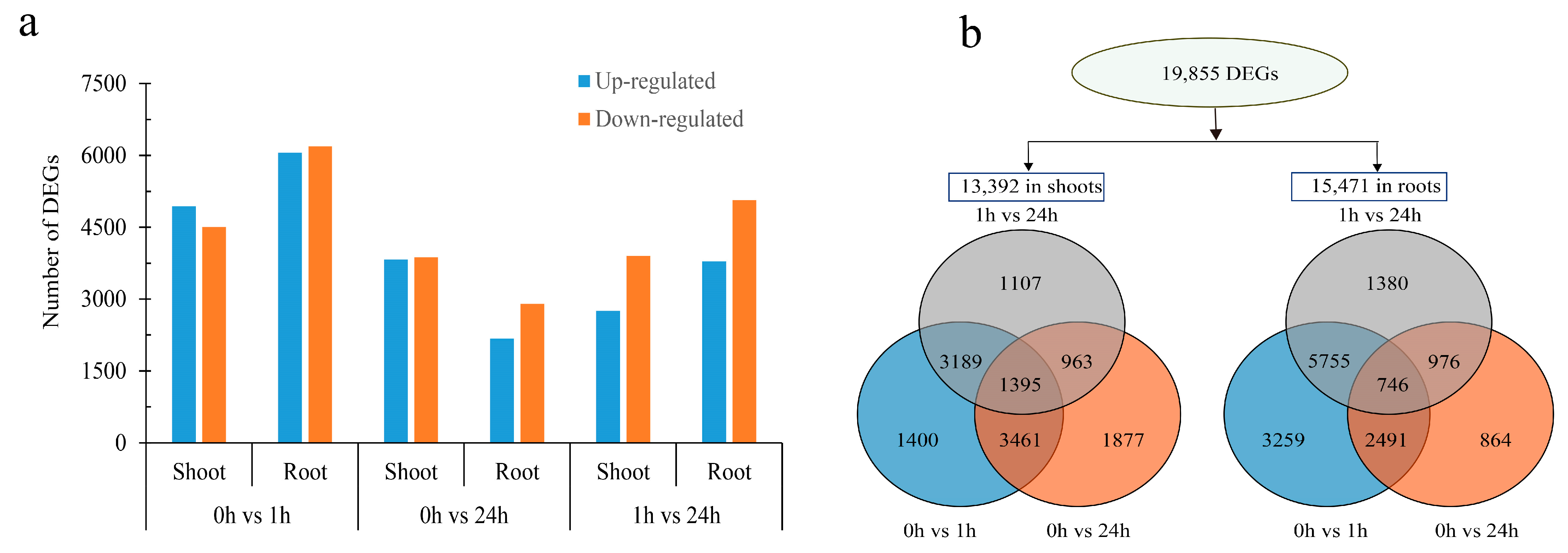
2.2. Comparative Transcriptome Analysis of M. albus to ABA Treatment
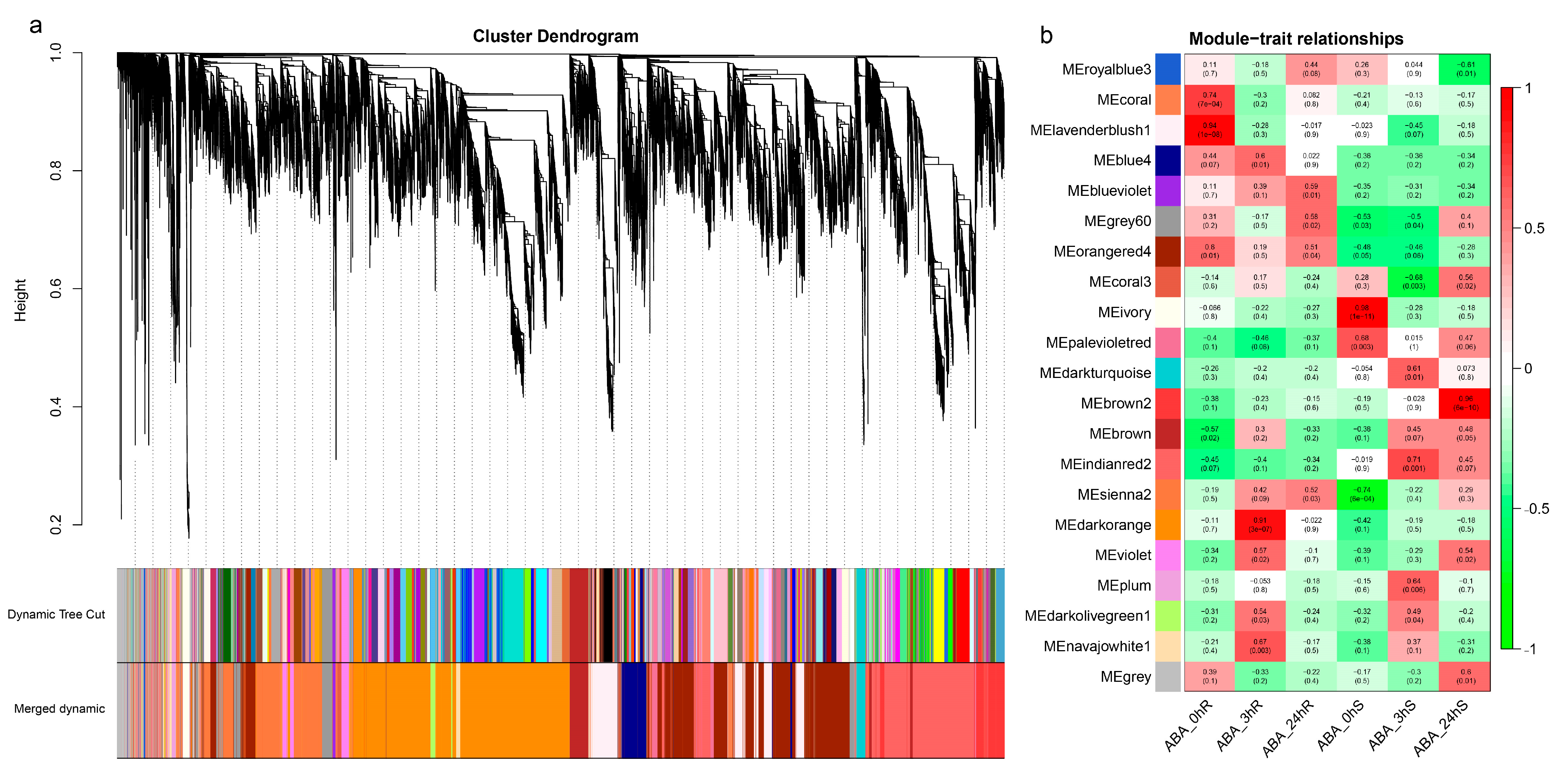
2.3. Weighted Correlation Network Analysis (WGCNA)
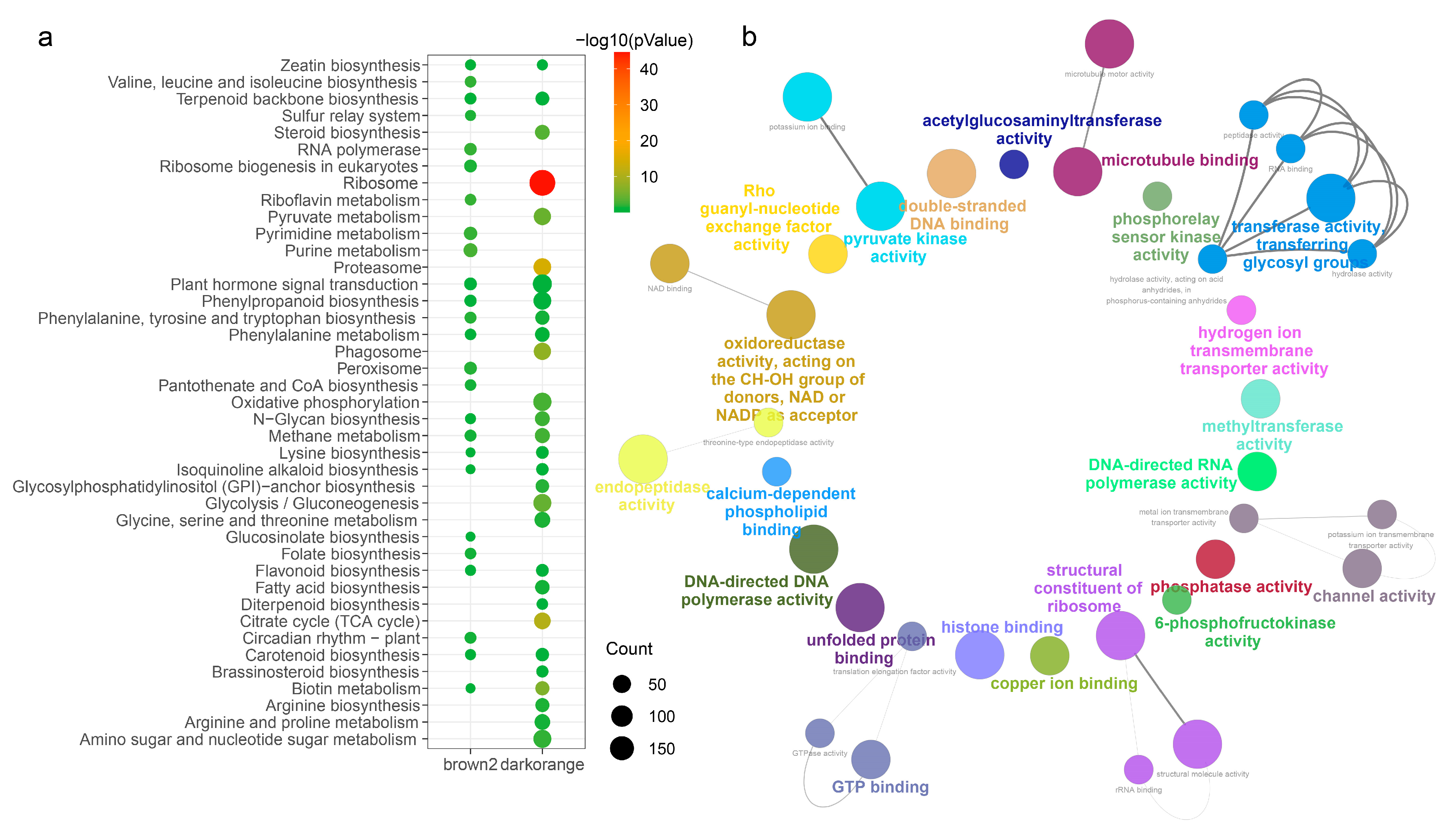
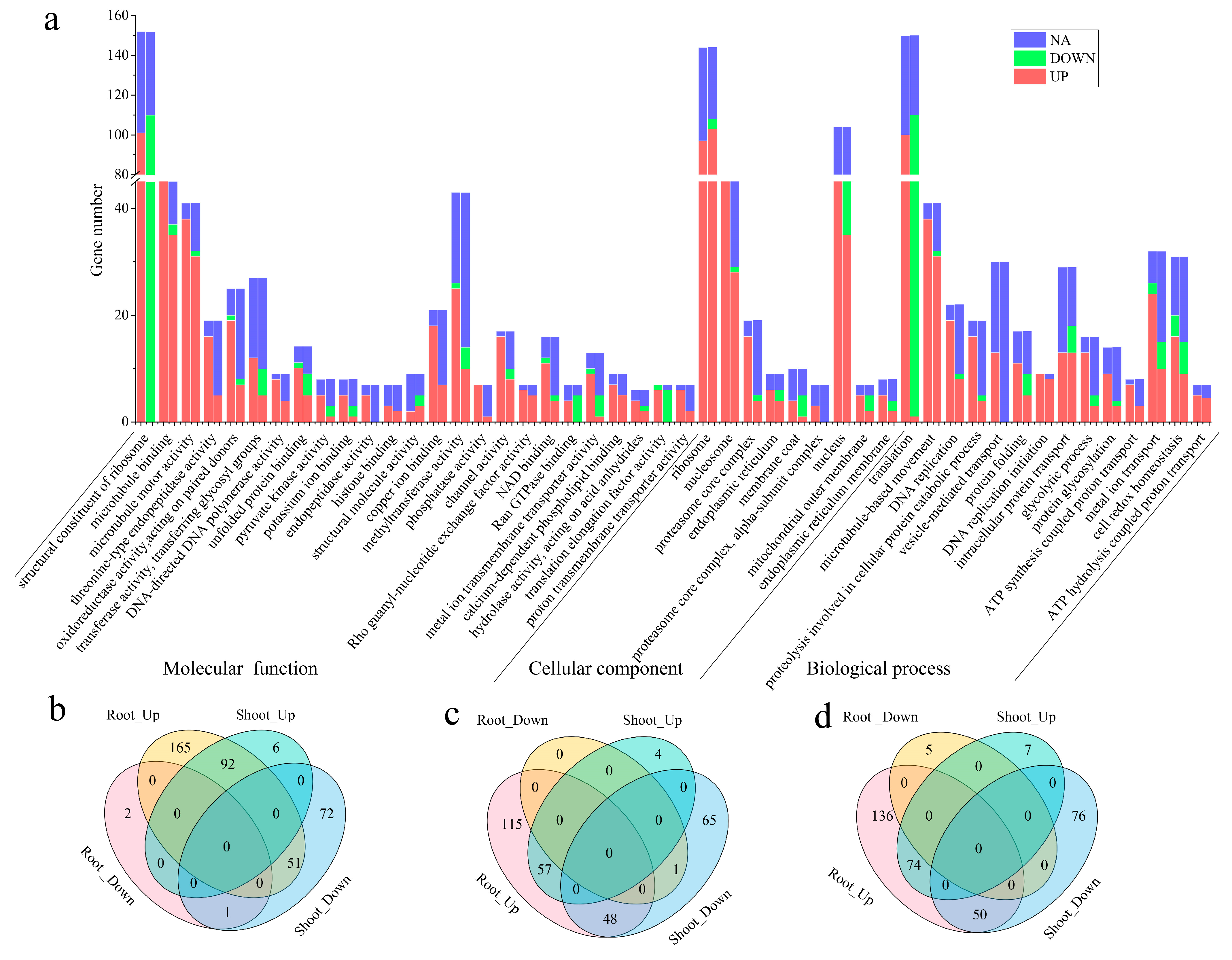
2.4. Functional Enrichment Analysis of Two Modules
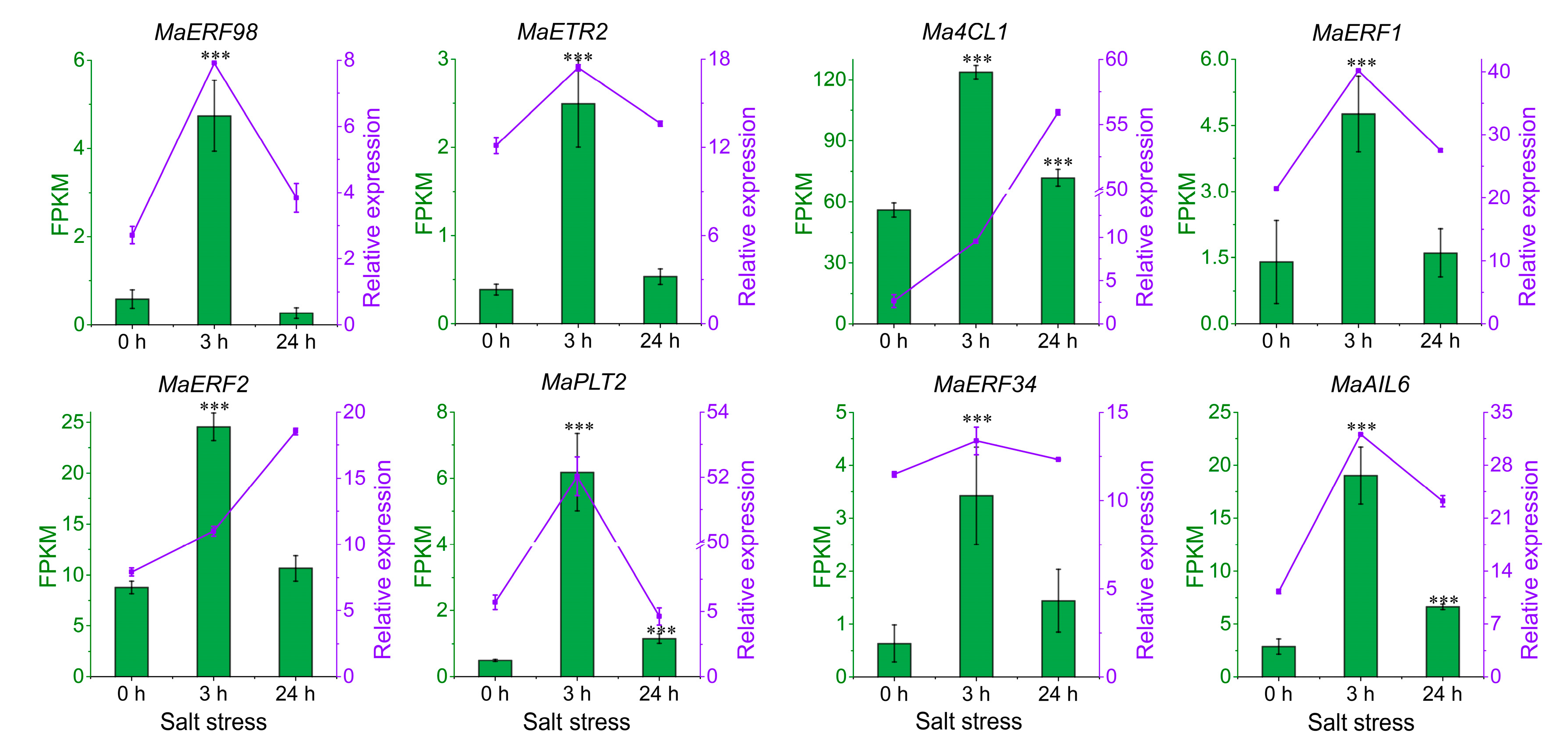
2.5. Gene Structures Analysis of Ma4CL Gene Family
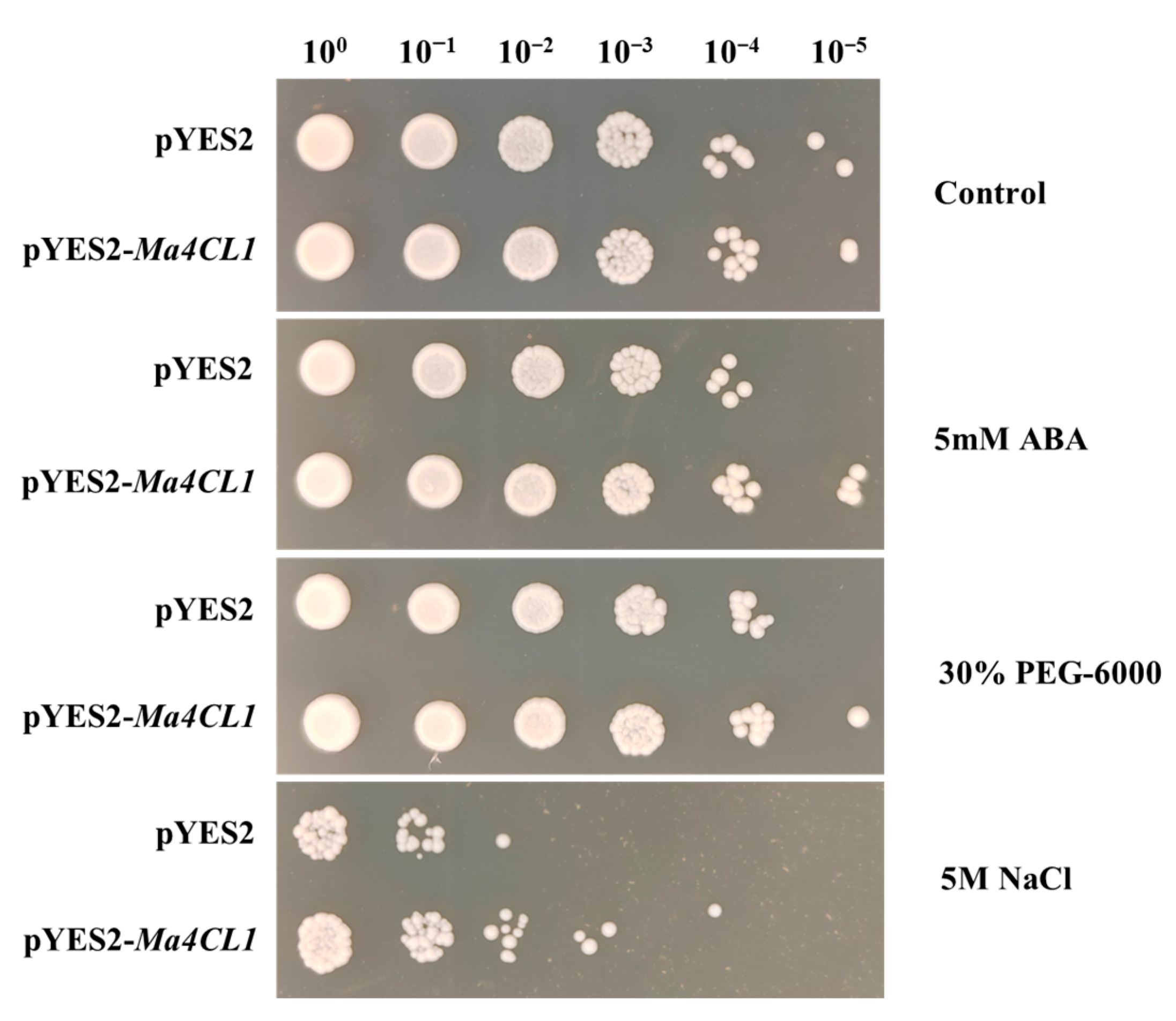
2.6. Ma4CL1 Enhanced Tolerance of Transgenic Yeast to ABA and Abiotic Stresses
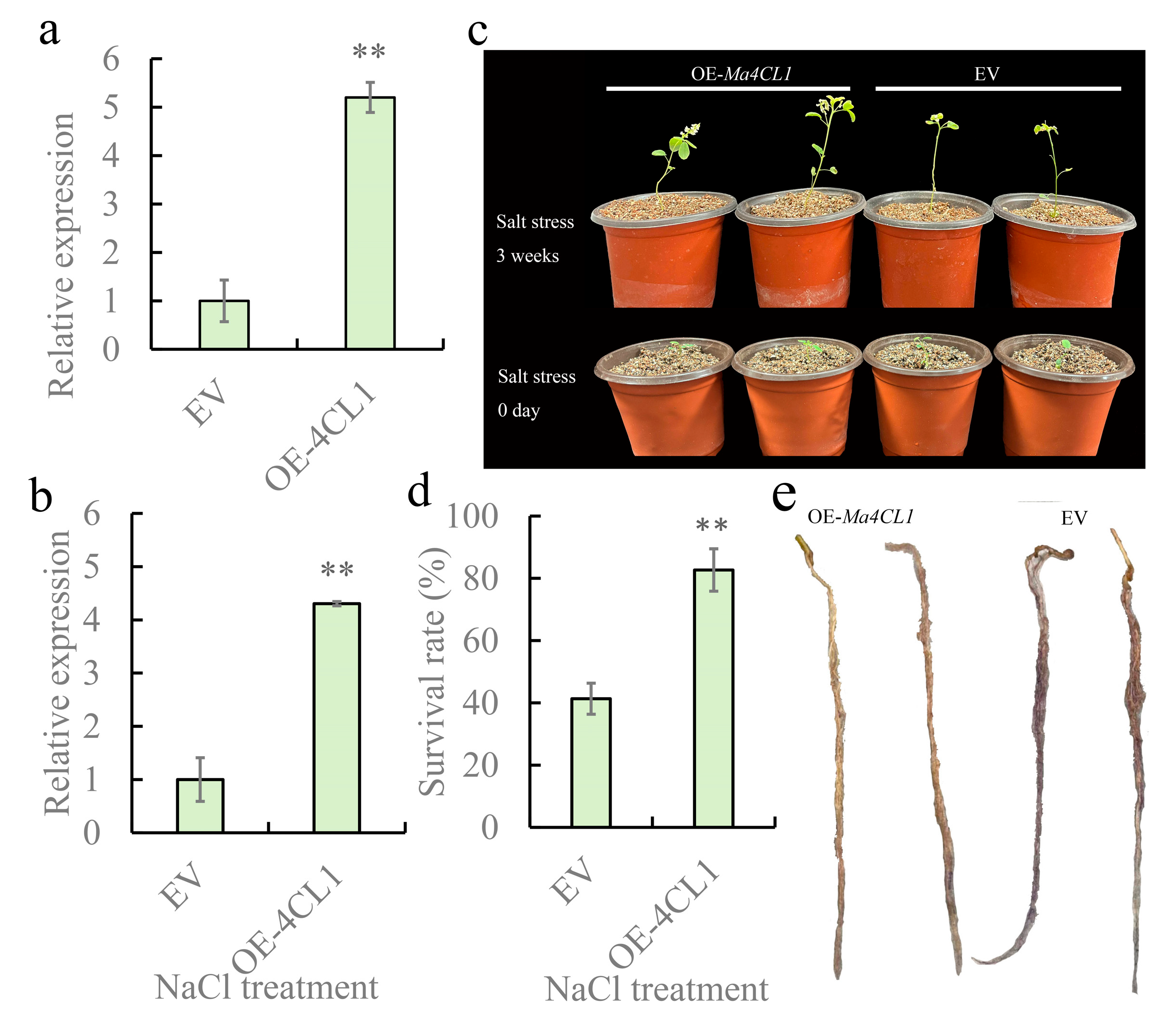
2.7. Overexpression of Ma4CL1 Enhanced the Salt Tolerance of M. albus
3. Materials and Methods
3.1. Plant Materials, ABA Treatments and Physiological Indicators Measurement
3.2. RNA Extraction, Library Construction and Sequencing
3.3. Functional Annotation and Enrichment Analysis
3.4. Gene Co-Expression Network Analysis
3.5. Quantitative Real-Time PCR Analysis (qPCR)
3.6. Heterologous Expression of Ma4CL in Yeast
3.7. Ma4CL1 Overexpression Vector Construction and Transformation into M. albus
3.8. Salt Stress and Histochemical Staining Measurement
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clark, A. Managing Cover Crops Profitably, 3rd ed.; Sustainable Agriculture Network: Beltsville, MD, USA, 2007. [Google Scholar]
- Nair, R.M.; Whittall, A.; Hughes, S.J.; Craig, A.D.; Revell, D.K.; Miller, S.M.; Powell, T.; Auricht, G.C. Variation in coumarin content of Melilotus species grown in South Australia. N. Z. J. Agric. Res. 2010, 53, 201–213. [Google Scholar] [CrossRef]
- Zhang, J.; Di, H.; Luo, K.; Jahufer, Z.; Wu, F.; Duan, Z.; Stewart, A.; Yan, Z.; Wang, Y. Coumarin content, morphological variation, and molecular phylogenetics of Melilotus. Molecules 2018, 23, E810. [Google Scholar] [CrossRef] [PubMed]
- Stickler, F.C.; Johnson, I.J. Dry matter and nitrogen production of legumes and legume associations in the fall of the seeding year 1. Agron. J. 1959, 51, 135–137. [Google Scholar] [CrossRef]
- Wang, Y. Study on Rapid and Accurate Dection Methods for Coumarin Content and Its Application in Melilotus spp.; Lanzhou University: Lanzhou, China, 2021. [Google Scholar]
- Wu, F.; Duan, Z.; Xu, P.; Yan, Q.; Meng, M.; Cao, M.; Jones, C.S.; Zong, X.; Zhou, P.; Wang, Y.; et al. Genome and systems biology of Melilotus albus provides insights into coumarins biosynthesis. Plant Biotechnol. J. 2022, 20, 592–609. [Google Scholar] [CrossRef] [PubMed]
- Zong, X.; Wang, S.; Han, Y.; Zhao, Q.; Xu, P.; Yan, Q.; Wu, F.; Zhang, J. Genome-wide profiling of the potential regulatory network of lncRNA and mRNA in Melilotus albus under salt stress. Environ. Exp. Bot. 2021, 189, 104548. [Google Scholar] [CrossRef]
- Raghavendra, A.S.; Gonugunta, V.K.; Christmann, A.; Grill, E. ABA perception and signalling. Trends Plant Sci. 2010, 15, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Parent, B.; Hachez, C.; Redondo, E.; Simonneau, T.; Chaumont, F.; Tardieu, F. Drought and abscisic acid effects on aquaporin content translate into changes in hydraulic conductivity and leaf growth rate: A trans-scale approach. Plant Physiol. 2009, 149, 2000–2012. [Google Scholar] [CrossRef]
- Fujita, Y.; Fujita, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 2011, 124, 509–525. [Google Scholar] [CrossRef]
- Wang, Y.; Tao, X.; Tang, X.M.; Xiao, L.; Sun, J.L.; Yan, X.F.; Li, D.; Deng, H.Y.; Ma, X.R. Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genom. 2013, 14, 841. [Google Scholar] [CrossRef]
- Singh, S.P.; Saini, M.K.; Singh, J.; Pongener, A.; Sidhu, G.S. Preharvest application of abscisic acid promotes anthocyanins accumulation in pericarp of litchi fruit without adversely affecting postharvest quality. Postharvest Biol. Tec. 2014, 96, 14–22. [Google Scholar] [CrossRef]
- Hu, B.; Li, J.; Wang, D.; Wang, H.; Qin, Y.; Hu, G.; Zhao, J. Transcriptome profiling of Litchi chinensis pericarp in response to exogenous cytokinins and abscisic acid. Plant Growth Regul. 2018, 84, 437–450. [Google Scholar] [CrossRef]
- Mou, W.; Li, D.; Luo, Z.; Mao, L.; Ying, T. Transcriptomic analysis reveals possible influences of ABA on secondary metabolism of pigments, flavonoids and antioxidants in tomato fruit during ripening. PLoS ONE 2015, 10, e0129598. [Google Scholar] [CrossRef] [PubMed]
- Murcia, G.; Fontana, A.; Pontin, M.; Baraldi, R.; Bertazza, G.; Piccoli, P.N. ABA and GA3 regulate the synthesis of primary and secondary metabolites related to alleviation from biotic and abiotic stresses in grapevine. Phytochemistry 2017, 135, 34–52. [Google Scholar] [CrossRef]
- Mareri, L.; Parrotta, L.; Cai, G. Environmental stress and plants. Int. J. Mol. Sci. 2022, 23, 5416. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, M.; Xie, B.; Jiang, X.; Gai, Y. Functional characteristics analysis of dehydrins in Larix kaempferi under osmotic stress. Int. J. Mol. Sci. 2021, 22, 1715. [Google Scholar] [CrossRef]
- Sharma, S.; Villamor, J.G.; Verslues, P.E. Essential role of tissue-specific proline synthesis and catabolism in growth and redox balance at low water potential. Plant Physiol. 2011, 157, 292–304. [Google Scholar] [CrossRef]
- Habibpourmehraban, F.; Masoomi-Aladizgeh, F.; Haynes, P.A. Effect of ABA pre-treatment on rice plant transcriptome response to multiple abiotic stress. Biomolecules 2023, 13, 1554. [Google Scholar] [CrossRef]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF transcription factor regulatory networks in hormone and abiotic stress responses in Arabidopsis. Front. Plant Sci. 2019, 10, 228. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Guo, J.; Wang, G. Effects of exogenous ABA on the synthesis of flavonoids in Ginkgo biloba leaves in vivo. J. Nanjing For. Univ. 2023, 47, 88. [Google Scholar]
- Hamberger, B.; Hahlbrock, K. The 4-coumarate:CoA ligase gene family in Arabidopsis thaliana comprises one rare, sinapate-activating and three commonly occurring isoenzymes. Proc. Natl. Acad. Sci. USA 2004, 101, 2209–2214. [Google Scholar] [CrossRef]
- Robe, K.; Izquierdo, E.; Vignols, F.; Rouached, H.; Dubos, C. The Coumarins: Secondary metabolites playing a primary role in plant nutrition and health. Trends Plant Sci. 2021, 26, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kim, J.I.; Pysh, L.; Chapple, C. Four isoforms of Arabidopsis thaliana 4-coumarate: CoA ligase (4CL) have overlapping yet distinct roles in phenylpropanoid metabolism. Plant Physiol. 2015, 169, 2409–2421. [Google Scholar] [CrossRef]
- Nie, T.; Sun, X.; Wang, S.; Wang, D.; Ren, Y.; Chen, Q. Genome-wide identification and expression analysis of the 4-coumarate: CoA ligase gene family in Solanum tuberosum. Int. J. mol. Sci. 2023, 24, 1642. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.E.K.; Choi, B.; Cho, B.K.; Kim, J.B.; Bae, H. Regulation of 4CL, encoding 4-coumarate: Coenzyme a ligase, expression in kenaf under diverse stress conditions. Plant Omics 2013, 6, 254–262. [Google Scholar]
- Chen, X.; Wang, H.; Li, X.; Ma, K.; Zeng, F. Molecular cloning and functional analysis of 4-Coumarate:CoA ligase 4 (4CL-like 1) from Fraxinus mandshurica and its role in abiotic stress tolerance and cell wall synthesis. BMC Plant Biol. 2019, 19, 231. [Google Scholar] [CrossRef]
- Ma, J.; Zuo, D.; Zhang, X.; Li, H.; Ye, H.; Zhang, N.; Li, M.; Dang, M.; Geng, F.; Zhou, H.; et al. Genome-wide identification analysis of the 4-Coumarate: CoA ligase (4CL) gene family expression profiles in Juglans regia and its wild relatives J. Mandshurica resistance and salt stress. BMC Plant Biol. 2024, 24, 211. [Google Scholar] [CrossRef]
- Müller, M.; Munné-Bosch, S. Ethylene response factors: A key regulatory hub in hormone and stress signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.G.; Li, H.; Liu, J.Y. Molecular characterization of three ethylene responsive element binding factor genes from cotton. J. Integr. Plant Biol. 2010, 52, 485–495. [Google Scholar] [CrossRef]
- Yang, H.; Yu, C.; Yan, J.; Wang, X.; Chen, F.; Zhao, Y.; Wei, W. Overexpression of the Jatropha curcas JcERF1 gene coding an AP2/ERF-type transcription factor increases tolerance to salt in transgenic tobacco. Biochemistry 2014, 79, 1226–1236. [Google Scholar] [CrossRef]
- Cheng, M.C.; Liao, P.M.; Kuo, W.W.; Lin, T.P. The Arabidopsis ETHYLENE RESPONSE FACTOR1 regulates abiotic stress-responsive gene expression by binding to different cis-acting elements in response to different stress signals. Plant Physiol. 2013, 162, 1566–1582. [Google Scholar] [CrossRef]
- Bahieldin, A.; Atef, A.; Edris, S.; Gadalla, N.O.; Ali, H.M.; Hassan, S.M.; Al-Kordy, M.A.; Ramadan, A.M.; Makki, R.M.; Al-Hajar, A.S.; et al. Ethylene responsive transcription factor ERF109 retards PCD and improves salt tolerance in plant. BMC Plant Biol. 2016, 16, 216. [Google Scholar] [CrossRef]
- Park, S.J.; Park, S.; Kim, Y.; Hyeon, D.Y.; Park, H.; Jeong, J.; Jeong, U.; Yoon, Y.S.; You, D.; Kwak, J.; et al. Ethylene responsive factor34 mediates stress—Induced leaf senescence by regulating salt stress-responsive genes. Plant Cell Environ. 2022, 45, 1719–1733. [Google Scholar] [CrossRef]
- Duan, Z.; Yan, Q.; Wu, F.; Wang, Y.; Wang, S.; Zong, X.; Zhou, P.; Zhang, J. Genome-Wide analysis of the UDP-glycosyltransferase family reveals its roles in coumarin biosynthesis and abiotic stress in Melilotus albus. Int. J. Mol. Sci. 2021, 22, 10826. [Google Scholar] [CrossRef]
- Duan, Z. Study on MaUGT79 Regulated by MaMYB4 Involving in Scopolin Biosynthesis and Drought Stress in Melilotus albus; Lanzhou University: Lanzhou, China, 2023. [Google Scholar]
- Daehwan, K.; Ben, L.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Mihaela, P.; Pertea, G.M.; Antonescu, C.M.; Tsung-Cheng, C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef] [PubMed]
- Beissbarth, T.; Speed, T.P. GOstat Find statistically over-represented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 2008, 9, 559. [Google Scholar] [CrossRef]
- Wang, S.; Duan, Z.; Zhang, J. Establishment of hairy root transformation system of Melilotus albus induced by Agrobacterium rhizogenes. Acta Agrestia Sin. 2021, 29, 2591. [Google Scholar]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef]
- Ko, D.K.; Brandizzi, F. Network--based approaches for understanding gene regulation and function in plants. Plant J. 2020, 104, 302–317. [Google Scholar] [CrossRef]
- Kosová, K.; Vítámvás, P.; Prášil, I.T.; Renaut, J. Plant proteome changes under abiotic stress-contribution of proteomics studies to understanding plant stress response. J. Proteom. 2011, 74, 1301–1322. [Google Scholar] [CrossRef] [PubMed]
- Kosová, K.; Vítámvás, P.; Prášil, I.T.; Klíma, M.; Renaut, J. Plant proteoforms under environmental stress: Functional proteins arising from a single gene. Front. Plant Sci. 2021, 12, 793113. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Fu, X.; Chu, Y.; Wu, P.; Liu, Y.; Ma, L.; Tian, H.; Zhu, B. Biosynthesis and the roles of plant sterols in development and stress responses. Int. J. Mol. Sci. 2022, 23, 2332. [Google Scholar] [CrossRef] [PubMed]
- Rogowska, A.; Szakiel, A. The role of sterols in plant response to abiotic stress. Phytochem. Rev. 2020, 19, 1525–1538. [Google Scholar] [CrossRef]
- Kumar, M.S.; Mawlong, I.; Ali, K.; Tyagi, A. Regulation of phytosterol biosynthetic pathway during drought stress in rice. Plant Physiol. Biochem. 2018, 129, 11–20. [Google Scholar] [CrossRef]
- Hong, Y.; Wang, Z.; Shi, H.; Yao, J.; Liu, X.; Wang, F.; Zeng, L.; Xie, Z.; Zhu, J.K. Reciprocal regulation between nicotinamide adenine dinucleotide metabolism and abscisic acid and stress response pathways in Arabidopsis. PLoS Genet. 2020, 16, e1008892. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, P.; Li, B.; Wang, W.; Zhu, B. Phosphoribosyltransferases and their roles in plant development and abiotic stress response. Int. J. Mol. Sci. 2023, 24, 11828. [Google Scholar] [CrossRef]
- Sinha, S.C.; Smith, J.L. The PRT protein family. Curr. Opin. Struct. Biol. 2001, 11, 733–739. [Google Scholar] [CrossRef]
- Singh, A.; Prasad, S.S.; Das, U.; Ingle, K.P.; Patil, K.; Shukla, P.K.; Ramteke, P.W.; Rajeshwar, J.R.; Gayathri, M.; Senthilmurugan, P. Regulation of nucleotide metabolism in response to salt stress. In Genetics of Salt Tolerance in Plants: A Central Dogma Perspective and Strategies for Enhancement; CABI GB: São Paulo, Brazil, 2024; pp. 9–23. [Google Scholar]
- Miao, C.; Xiao, L.; Hua, K.; Zou, C.; Zhao, Y.; Bressan, R.A.; Zhu, J.K. Mutations in a subfamily of abscisic acid receptor genes promote rice growth and productivity. Proc. Natl. Acad. Sci. USA. 2018, 115, 6058–6063. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, M.; Li, L.; Xu, Z.; Chen, X.; Guo, J.; Ma, Y. Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. J. Exp. Bot. 2009, 60, 3781–3796. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.; Zhang, J.; Chen, J.; Wu, T.; Zhu, S.; Yan, S.; Zhao, X.; Zhong, G. Expressing a Citrus ortholog of Arabidopsis ERF1 enhanced cold-tolerance in tobacco. Sci. Hortic. 2014, 174, 65–76. [Google Scholar] [CrossRef]
- Wu, L.; Chen, X.; Ren, H.; Zhang, Z.; Zhang, H.; Wang, J.; Wang, X.C.; Huang, R. ERF protein JERF1 that transcriptionally modulates the expression of abscisic acid biosynthesis-related gene enhances the tolerance under salinity and cold in tobacco. Planta 2007, 226, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ji, J.; Wang, G.; Guan, C.; Jin, C. LchERF, a novel ethylene-responsive transcription factor from Lycium chinense, confers salt tolerance in transgenic tobacco. Plant Cell Rep. 2014, 33, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z. The relationship between lignification and endogenous hormone content of excised bamboo shoots. Sci. Agric. Sin. 2006, 39, 792–797. [Google Scholar]
- He, X.; Xue, F.; Zhang, L.; Guo, H.; Ma, L.; Yang, M. Overexpressing fusion proteins of 4-coumaroyl-CoA ligase (4CL) and stilbene synthase (STS) in tobacco plants leading to resveratrol accumulation and improved stress tolerance. Plant Biotechnol. Rep. 2018, 12, 295–302. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Wu, F.; Duan, Z.; Wang, S.; Qu, Y.; Ao, B.; Sun, X.; Zhang, J. Comparative Transcriptome Analysis Revealing the Potential Salt Tolerance Mechanism of Exogenous Abscisic Acid Application in Melilotus albus. Int. J. Mol. Sci. 2024, 25, 13261. https://doi.org/10.3390/ijms252413261
Chen L, Wu F, Duan Z, Wang S, Qu Y, Ao B, Sun X, Zhang J. Comparative Transcriptome Analysis Revealing the Potential Salt Tolerance Mechanism of Exogenous Abscisic Acid Application in Melilotus albus. International Journal of Molecular Sciences. 2024; 25(24):13261. https://doi.org/10.3390/ijms252413261
Chicago/Turabian StyleChen, Lijun, Fan Wu, Zhen Duan, Shengsheng Wang, Yuncan Qu, Bao Ao, Xiaojuan Sun, and Jiyu Zhang. 2024. "Comparative Transcriptome Analysis Revealing the Potential Salt Tolerance Mechanism of Exogenous Abscisic Acid Application in Melilotus albus" International Journal of Molecular Sciences 25, no. 24: 13261. https://doi.org/10.3390/ijms252413261
APA StyleChen, L., Wu, F., Duan, Z., Wang, S., Qu, Y., Ao, B., Sun, X., & Zhang, J. (2024). Comparative Transcriptome Analysis Revealing the Potential Salt Tolerance Mechanism of Exogenous Abscisic Acid Application in Melilotus albus. International Journal of Molecular Sciences, 25(24), 13261. https://doi.org/10.3390/ijms252413261