Mitochondrial DNA Damage and Its Repair Mechanisms in Aging Oocytes


Abstract
1. Introduction
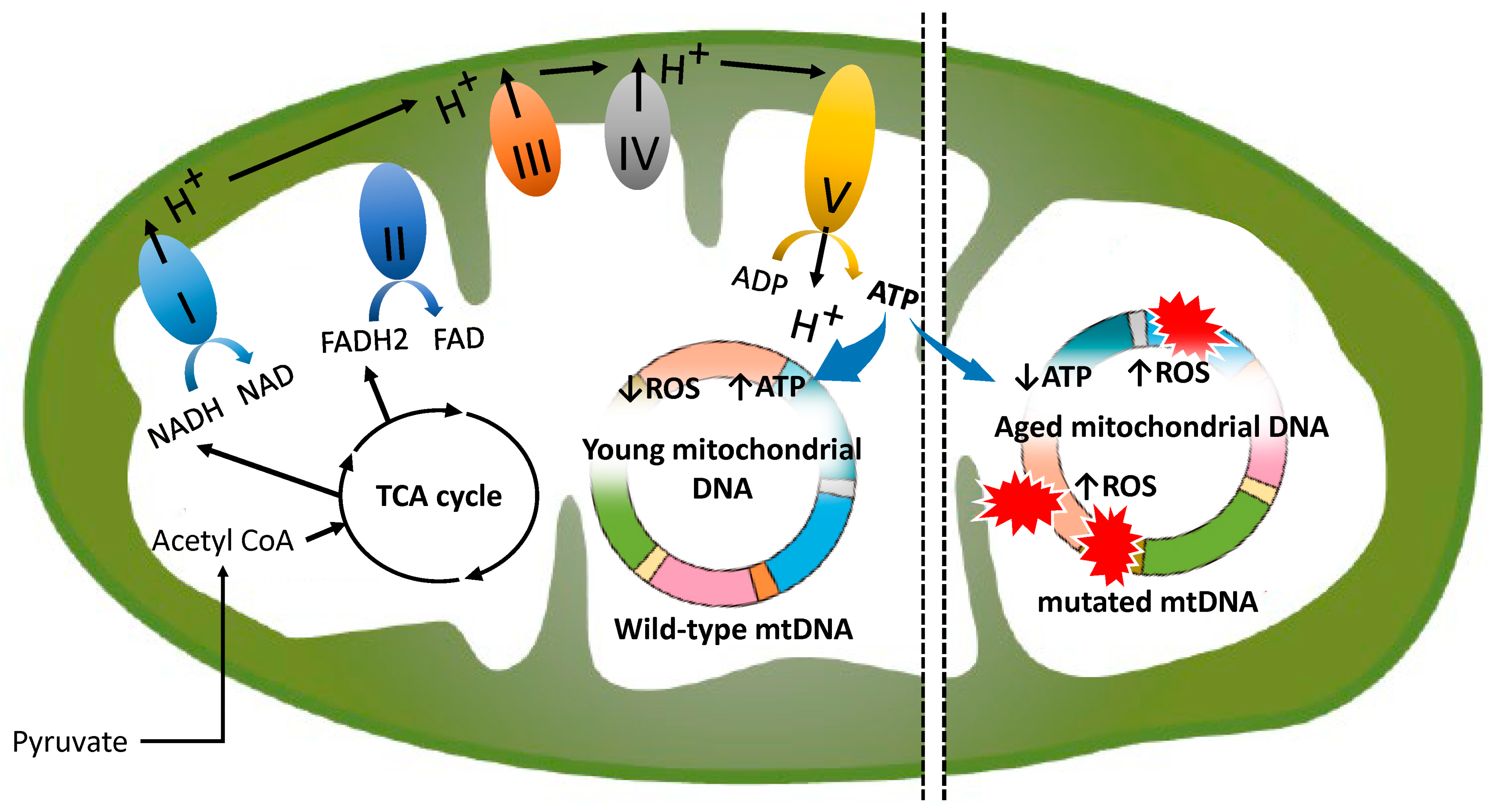
2. Normal Mitochondrial Function
3. Age-Related Changes in Mitochondria
4. Ovarian Ageing as the Free Radical Theory
5. Molecular Mechanism Underlying Oocyte Aging Caused by mtDNA Mutations
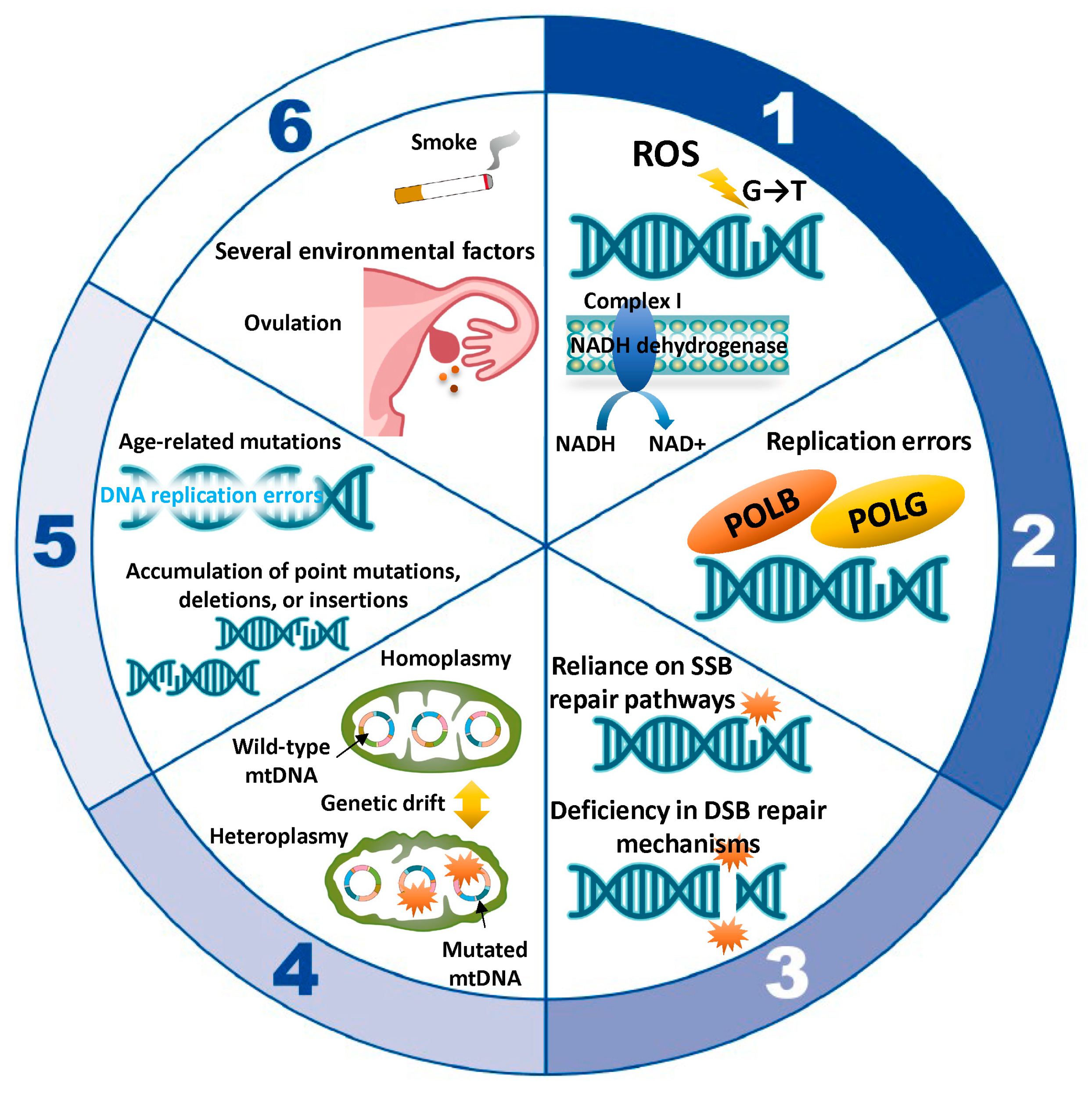
5.1. ROS-Induced Damage
5.2. Replication Errors
5.3. Mitochondrial DNA Repair Deficiencies
5.4. Replicative Segregation and Genetic Drift
5.5. Age-Related Accumulation of Mutations
5.6. Environmental Factors
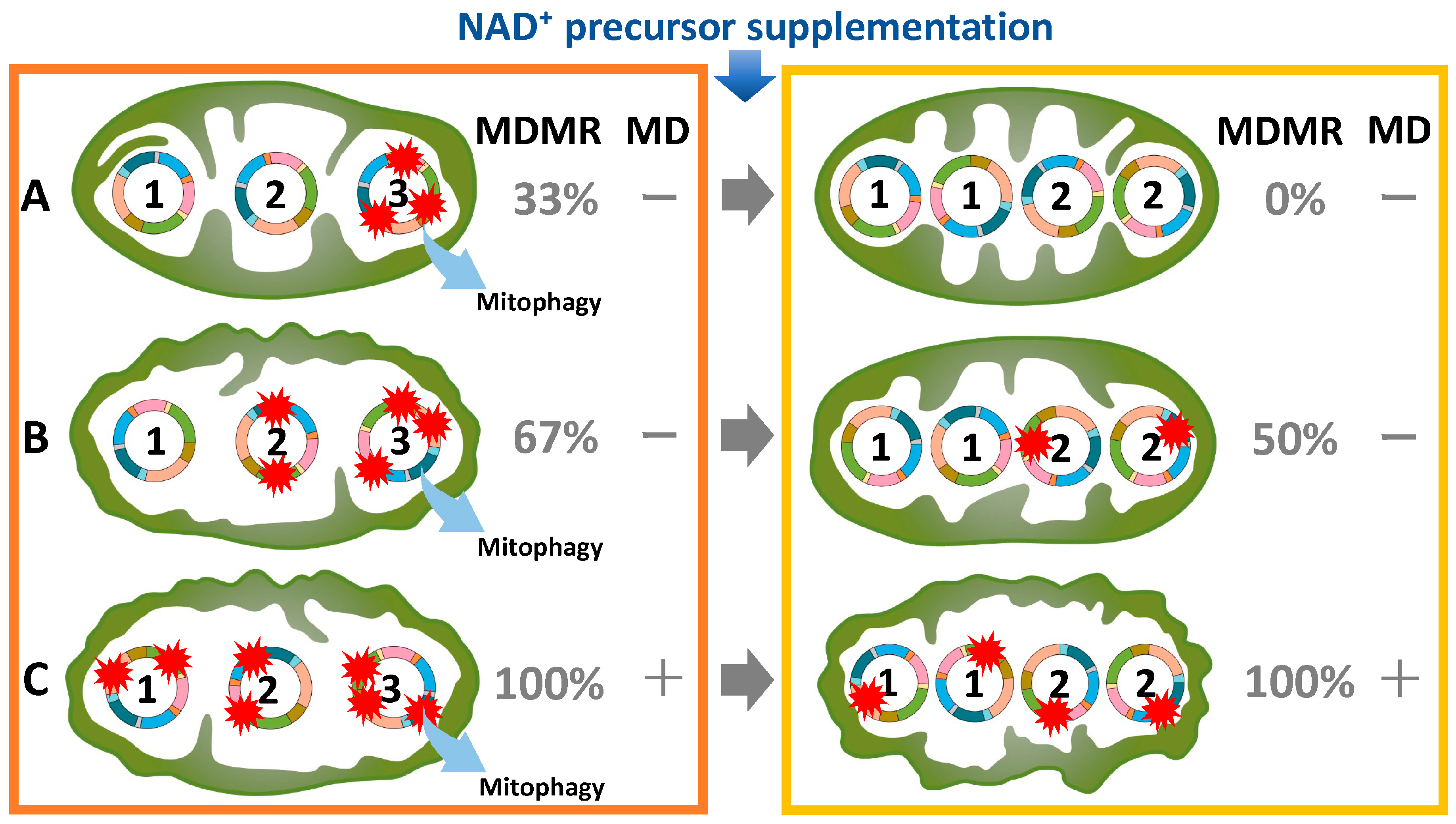
6. Heteroplasmy as a mtDNA Variant
7. Age-Related Alterations in mtDNA Mutations and Copy Number
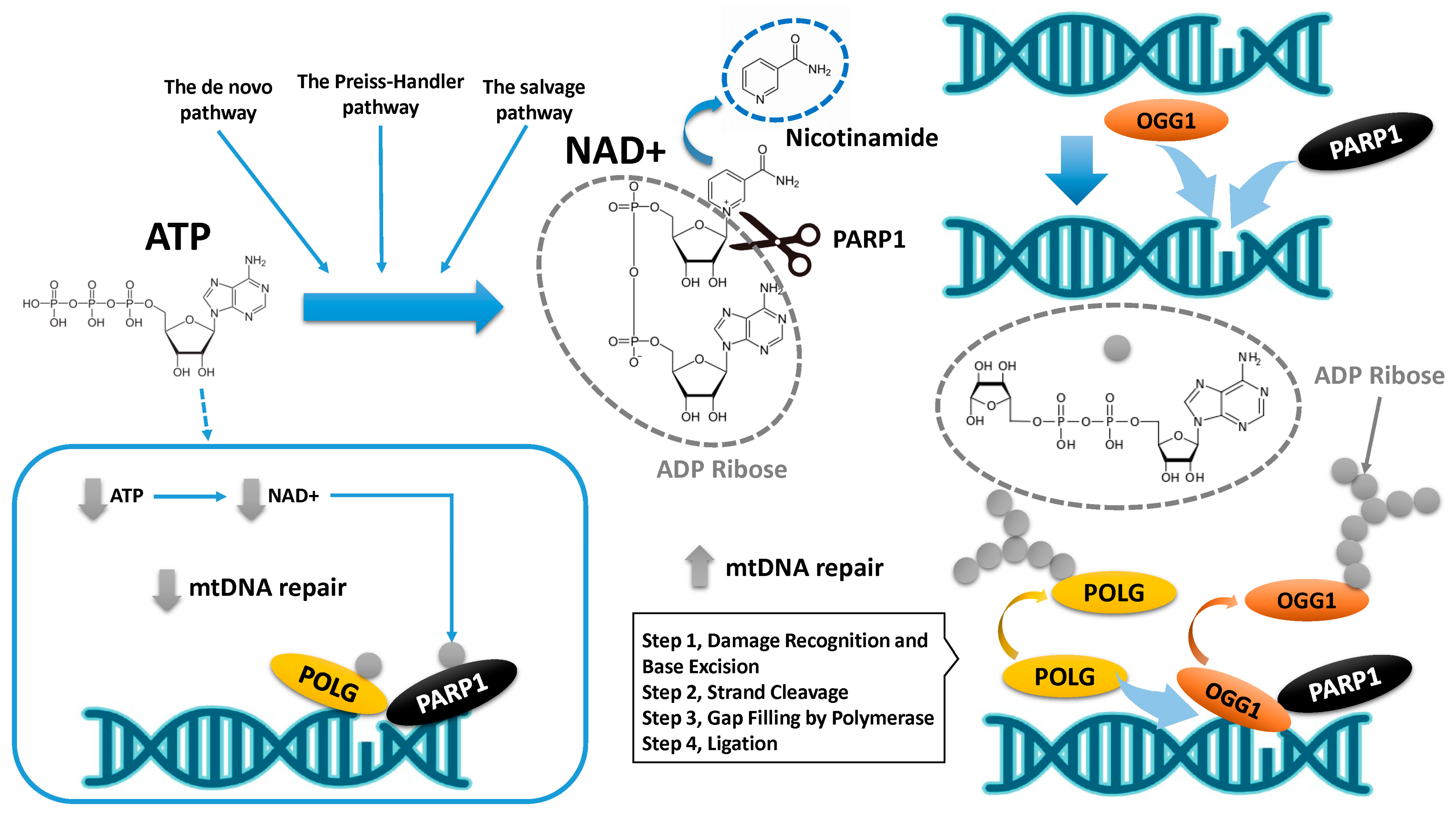
8. mtDNA Damage and Its Repair Mechanism
9. Conclusions
10. Future Direction
11. Materials and Methods
Search Strategy and Selection Criteria
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fathalla, M.F. Impact of reproductive evolutionary mismatch on women’s health and the need for action and research. Int. J. Gynaecol. Obstet. 2019, 144, 129–134. [Google Scholar] [CrossRef]
- O’Brien, Y.; Wingfield, M.B. Reproductive ageing-turning back the clock? Ir. J. Med. Sci. 2019, 188, 161–167. [Google Scholar] [CrossRef]
- Kisielewski, R.; Tołwińska, A.; Mazurek, A.; Laudański, P. Inflammation and ovarian cancer--current views. Ginekol. Pol. 2013, 84, 293–297. [Google Scholar] [CrossRef]
- Faddy, M.J. Follicle dynamics during ovarian ageing. Mol. Cell. Endocrinol. 2000, 163, 43–48. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists Committee on Gynecologic Practice and Practice Committee. Female age-related fertility decline. Fertil. Steril. 2014, 101, 633–634. [Google Scholar] [CrossRef]
- May-Panloup, P.; Boucret, L.; Chao de la Barca, J.M.; Desquiret-Dumas, V.; Ferré-L’Hotellier, V.; Morinière, C.; Descamps, P.; Procaccio, V.; Reynier, P. Ovarian ageing: The role of mitochondria in oocytes and follicles. Hum. Reprod. Update 2016, 22, 725–743. [Google Scholar] [CrossRef]
- Silber, S.J.; Kato, K.; Aoyama, N.; Yabuuchi, A.; Skaletsky, H.; Fan, Y.; Shinohara, K.; Yatabe, N.; Kobayashi, T. Intrinsic fertility of human oocytes. Fertil. Steril. 2017, 107, 1232–1237. [Google Scholar] [CrossRef]
- Steuerwald, N.M.; Bermúdez, M.G.; Wells, D.; Munné, S.; Cohen, J. Maternal age-related differential global expression profiles observed in human oocytes. Reprod. Biomed. Online 2007, 14, 700–708. [Google Scholar] [CrossRef]
- Battaglia, D.E.; Goodwin, P.; Klein, N.A.; Soules, M.R. Influence of maternal age on meiotic spindle assembly in oocytes from naturally cycling women. Hum. Reprod. 1996, 11, 2217–2222. [Google Scholar] [CrossRef]
- Chiang, T.; Schultz, R.M.; Lampson, M.A. Age-dependent susceptibility of chromosome cohesion to premature separase activation in mouse oocytes. Biol. Reprod. 2011, 85, 1279–1283. [Google Scholar] [CrossRef]
- Zhu, Z.; Xu, W.; Liu, L. Ovarian aging: Mechanisms and intervention strategies. Med. Rev. 2022, 2, 590–610. [Google Scholar] [CrossRef]
- Van der Reest, J.; Nardini Cecchino, G.; Haigis, M.C.; Kordowitzki, P. Mitochondria: Their relevance during oocyte ageing. Ageing Res. Rev. 2021, 70, 101378. [Google Scholar] [CrossRef]
- He, H.; Wang, J.; Mou, X.; Liu, X.; Li, Q.; Zhong, M.; Luo, B.; Yu, Z.; Zhang, J.; Xu, T.; et al. Selective autophagic degradation of ACLY (ATP citrate lyase) maintains citrate homeostasis and promotes oocyte maturation. Autophagy 2023, 19, 163–179. [Google Scholar] [CrossRef]
- Zhang, D.; Keilty, D.; Zhang, Z.F.; Chian, R.C. Mitochondria in oocyte aging: Current understanding. Facts Views Vis. ObGyn 2017, 9, 29–38. [Google Scholar]
- Warzych, E.; Lipinska, P. Energy metabolism of follicular environment during oocyte growth and maturation. J. Reprod. Dev. 2020, 66, 1–7. [Google Scholar] [CrossRef]
- Gutnisky, C.; Dalvit, G.C.; Thompson, J.G.; Cetica, P.D. Pentose phosphate pathway activity: Effect on in vitro maturation and oxidative status of bovine oocytes. Reprod. Fertil. Dev. 2014, 26, 931–942. [Google Scholar] [CrossRef]
- Dubeibe Marin, D.F.; da Costa, N.N.; di Paula Bessa Santana, P.; de Souza, E.B.; Ohashi, O.M. Importance of lipid metabolism on oocyte maturation and early embryo development: Can we apply what we know to buffalo? Anim. Reprod. Sci. 2019, 211, 106220. [Google Scholar] [CrossRef]
- Sugiura, K.; Pendola, F.L.; Eppig, J.J. Oocyte control of metabolic cooperativity between oocytes and companion granulosa cells: Energy metabolism. Dev. Biol. 2005, 279, 20–30. [Google Scholar] [CrossRef]
- Kobayashi, H.; Imanaka, S. Recent progress in metabolomics for analyzing common infertility conditions that affect ovarian function. Reprod. Med. Biol. 2024, 23, e12609. [Google Scholar] [CrossRef]
- May-Panloup, P.; Chretien, M.F.; Malthiery, Y.; Reynier, P. Mitochondrial DNA in the oocyte and the developing embryo. Curr. Top. Dev. Biol. 2007, 77, 51–83. [Google Scholar] [CrossRef]
- MacLennan, M.; Crichton, J.H.; Playfoot, C.J.; Adams, I.R. Oocyte development, meiosis and aneuploidy. Semin. Cell Dev. Biol. 2015, 45, 68–76. [Google Scholar] [CrossRef]
- Charalambous, C.; Webster, A.; Schuh, M. Aneuploidy in mammalian oocytes and the impact of maternal ageing. Nat. Rev. Mol. Cell Biol. 2023, 24, 27–44. [Google Scholar] [CrossRef]
- Sánchez-Baracaldo, P.; Cardona, T. On the origin of oxygenic photosynthesis and Cyanobacteria. New Phytol. 2020, 225, 1440–1446. [Google Scholar] [CrossRef]
- Baffy, G. Mitochondrial uncoupling in cancer cells: Liabilities and opportunities. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 655–664. [Google Scholar] [CrossRef]
- Boengler, K.; Kosiol, M.; Mayr, M.; Schulz, R.; Rohrbach, S. Mitochondria and ageing: Role in heart, skeletal muscle and adipose tissue. J. Cachexia Sarcopenia Muscle 2017, 8, 349–369. [Google Scholar] [CrossRef]
- Podolak, A.; Woclawek-Potocka, I.; Lukaszuk, K. The Role of Mitochondria in Human. Fertility and Early Embryo Development: What Can. We Learn. for Clinical Application of Assessing and Improving Mitochondrial DNA? Cells 2022, 11, 797. [Google Scholar] [CrossRef]
- Trushina, E.; Trushin, S.; Hasan, M.F. Mitochondrial complex I as a therapeutic target for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 483–495. [Google Scholar] [CrossRef]
- De Paula, W.B.; Lucas, C.H.; Agip, A.N.; Vizcay-Barrena, G.; Allen, J.F. Energy, ageing, fidelity and sex: Oocyte mitochondrial DNA as a protected genetic template. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20120263. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Wang, L.; Tang, J.; Wang, L.; Tan, F.; Song, H.; Zhou, J.; Li, F. Oxidative stress in oocyte aging and female reproduction. J. Cell. Physiol. 2021, 236, 7966–7983. [Google Scholar] [CrossRef]
- Dan Dunn, J.; Alvarez, L.A.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Bentov, Y.; Yavorska, T.; Esfandiari, N.; Jurisicova, A.; Casper, R.F. The contribution of mitochondrial function to reproductive aging. J. Assist. Reprod. Genet. 2011, 28, 773–783. [Google Scholar] [CrossRef]
- Mikwar, M.; MacFarlane, A.J.; Marchetti, F. Mechanisms of oocyte aneuploidy associated with advanced maternal age. Mutat. Res. Rev. Mutat. Res. 2020, 785, 108320. [Google Scholar] [CrossRef]
- Park, S.U.; Walsh, L.; Berkowitz, K.M. Mechanisms of ovarian aging. Reproduction 2021, 162, R19–R33. [Google Scholar] [CrossRef]
- Müller-Höcker, J.; Schäfer, S.; Weis, S.; Münscher, C.; Strowitzki, T. Morphological-cytochemical and molecular genetic analyses of mitochondria in isolated human oocytes in the reproductive age. Mol. Hum. Reprod. 1996, 2, 951–958. [Google Scholar] [CrossRef]
- Motta, P.M.; Nottola, S.A.; Makabe, S.; Heyn, R. Mitochondrial morphology in human fetal and adult female germ cells. Hum. Reprod. 2000, 15, 129–147. [Google Scholar] [CrossRef]
- Soares, M.; Sousa, A.P.; Fernandes, R.; Ferreira, A.F.; Almeida-Santos, T.; Ramalho-Santos, J. Aging-related mitochondrial alterations in bovine oocytes. Theriogenology 2020, 157, 218–225. [Google Scholar] [CrossRef]
- Liu, Y.; Han, M.; Li, X.; Wang, H.; Ma, M.; Zhang, S.; Guo, Y.; Wang, S.; Wang, Y.; Duan, N.; et al. Age-related changes in the mitochondria of human mural granulosa cells. Hum. Reprod. 2017, 32, 2465–2473. [Google Scholar] [CrossRef]
- Cox, R.T.; Poulton, J.; Williams, S.A. The role of mitophagy during oocyte aging in human, mouse, and Drosophila: Implications for oocyte quality and mitochondrial disease. Reprod. Fertil. 2021, 2, R113–R129. [Google Scholar] [CrossRef]
- Wilding, M.; Dale, B.; Marino, M.; di Matteo, L.; Alviggi, C.; Pisaturo, M.L.; Lombardi, L.; De Placido, G. Mitochondrial aggregation patterns and activity in human oocytes and preimplantation embryos. Hum. Reprod. 2001, 16, 909–917. [Google Scholar] [CrossRef]
- Ben-Meir, A.; Burstein, E.; Borrego-Alvarez, A.; Chong, J.; Wong, E.; Yavorska, T.; Naranian, T.; Chi, M.; Wang, Y.; Bentov, Y.; et al. Coenzyme Q10 restores oocyte mitochondrial function and fertility during reproductive aging. Aging Cell 2015, 14, 887–895. [Google Scholar] [CrossRef]
- Li, C.J.; Lin, L.T.; Tsai, H.W.; Chern, C.U.; Wen, Z.H.; Wang, P.H.; Tsui, K.H. The Molecular Regulation in the Pathophysiology in Ovarian Aging. Aging Dis. 2021, 12, 934–949. [Google Scholar] [CrossRef]
- Van Blerkom, J. Mitochondrial function in the human oocyte and embryo and their role in developmental competence. Mitochondrion 2011, 11, 797–813. [Google Scholar] [CrossRef]
- Eichenlaub-Ritter, U. Oocyte ageing and its cellular basis. Int. J. Dev. Biol. 2012, 56, 841–852. [Google Scholar] [CrossRef]
- Schon, E.A.; Kim, S.H.; Ferreira, J.C.; Magalhaes, P.; Grace, M.; Warburton, D.; Gross, S.J. Chromosomal non-disjunction in human oocytes: Is there a mitochondrial connection. Hum. Reprod. 2000, 15 (Suppl. S2), 160–172. [Google Scholar] [CrossRef]
- Van Blerkom, J.; Davis, P.W.; Lee, J. ATP content of human oocytes and developmental potential and outcome after in-vitro fertilization and embryo transfer. Hum. Reprod. 1995, 10, 415–424. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, X.Q.; Lu, S.; Guo, Y.L.; Ma, X. Deficit of mitochondria-derived ATP during oxidative stress impairs mouse MII oocyte spindles. Cell Res. 2006, 16, 841–850. [Google Scholar] [CrossRef]
- Meldrum, D.R.; Casper, R.F.; Diez-Juan, A.; Simon, C.; Domar, A.D.; Frydman, R. Aging and the environment affect gamete and embryo potential: Can we intervene. Fertil. Steril. 2016, 105, 548–559. [Google Scholar] [CrossRef]
- Xing, X.; Liang, Y.; Li, Y.; Zhao, Y.; Zhang, Y.; Li, Z.; Li, Z.; Wu, Z. Fisetin Delays Postovulatory Oocyte Aging by Regulating Oxidative Stress and Mitochondrial Function through Sirt1 Pathway. Molecules 2023, 28, 5533. [Google Scholar] [CrossRef]
- Luo, D.; Zhang, J.B.; Li, S.P.; Liu, W.; Yao, X.R.; Guo, H.; Jin, Z.L.; Jin, Y.X.; Yuan, B.; Jiang, H.; et al. Imperatorin Ameliorates the Aging-Associated Porcine Oocyte Meiotic Spindle Defects by Reducing Oxidative Stress and Protecting Mitochondrial Function. Front. Cell Dev. Biol. 2020, 8, 592433. [Google Scholar] [CrossRef]
- Guillaumet-Adkins, A.; Yanez, Y.; Peris-Diaz, M.D.; Calabria, I.; Palanca-Ballester, C.; Sandoval, J. Epigenetics and oxidative stress in aging. Oxid. Med. Cell. Longev. 2017, 2017, 9175806. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I. Reactive oxygen species and the free radical theory of aging. Free Radic. Biol. Med. 2013, 60, 1–4. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef]
- Lin, X.; Dai, Y.; Tong, X.; Xu, W.; Huang, Q.; Jin, X.; Li, C.; Zhou, F.; Zhou, H.; Lin, X.; et al. Excessive oxidative stress in cumulus granulosa cells induced cell senescence contributes to endometriosis-associated infertility. Redox Biol. 2020, 30, 101431. [Google Scholar] [CrossRef]
- Long, S.; Zheng, Y.; Deng, X.; Guo, J.; Xu, Z.; Scharffetter-Kochanek, K.; Dou, Y.; Jiang, M. Maintaining mitochondrial DNA copy number mitigates ROS-induced oocyte decline and female reproductive aging. Commun. Biol. 2024, 7, 1229. [Google Scholar] [CrossRef]
- Kumar, M.; Pathak, D.; Kriplani, A.; Ammini, A.C.; Talwar, P.; Dada, R. Nucleotide variations in mitochondrial DNA and supra-physiological ROS levels in cytogenetically normal cases of premature ovarian insufficiency. Arch. Gynecol. Obstet. 2010, 282, 695–705. [Google Scholar] [CrossRef]
- Su, W.P.; Li, C.J.; Lin, L.T.; Lin, P.H.; Wen, Z.H.; Sheu, J.J.; Tsui, K.H. Boosting mitochondrial function and metabolism in aging female germ cells with dual ROCK/ROS inhibition. Biomed. Pharmacother. 2023, 163, 114888. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, E.Y.; Lee, K.A. GAS6 ameliorates advanced age-associated meiotic defects in mouse oocytes by modulating mitochondrial function. Aging 2021, 13, 18018–18032. [Google Scholar] [CrossRef]
- Arbeithuber, B.; Cremona, M.A.; Hester, J.; Barrett, A.; Higgins, B.; Anthony, K.; Chiaromonte, F.; Diaz, F.J.; Makova, K.D. Advanced age increases frequencies of de novo mitochondrial mutations in macaque oocytes and somatic tissues. Proc. Natl. Acad. Sci. USA 2022, 119, e2118740119. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, A.M.; Beau-Faller, M.; Debieuvre, D.; Ouafik, L.; Westeel, V.; Rouquette, I.; Mazières, J.; Bringuier, P.P.; Monnet, I.; Escande, F.; et al. Outcomes of Patients with Advanced NSCLC From the Intergroupe Francophone de Cancérologie Thoracique Biomarkers France Study by KRAS Mutation Subtypes. JTO Clin. Res. Rep. 2020, 1, 100052. [Google Scholar] [CrossRef]
- Nishigori, C.; Hattori, Y.; Toyokuni, S. Role of reactive oxygen species in skin carcinogenesis. Antioxid. Redox Signal. 2004, 6, 561–570. [Google Scholar] [CrossRef]
- Matkarimov, B.T.; Saparbaev, M.K. DNA Repair and Mutagenesis in Vertebrate Mitochondria: Evidence for Asymmetric DNA Strand Inheritance. Adv. Exp. Med. Biol. 2020, 1241, 77–100. [Google Scholar] [CrossRef]
- Kaufman, B.A.; Van Houten, B. POLB: A new role of DNA polymerase beta in mitochondrial base excision repair. DNA Repair 2017, 60, A1–A5. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Niki, E. Lipid peroxidation products as oxidative stress biomarkers. Biofactors 2008, 34, 171–180. [Google Scholar] [CrossRef]
- Samuel, P.; Tsapekos, M.; de Pedro, N.; Liu, A.G.; Casey Lippmeier, J.; Chen, S. Ergothioneine Mitigates Telomere Shortening under Oxidative Stress Conditions. J. Diet. Suppl. 2022, 19, 212–225. [Google Scholar] [CrossRef]
- Sasaki, H.; Hamatani, T.; Kamijo, S.; Iwai, M.; Kobanawa, M.; Ogawa, S.; Miyado, K.; Tanaka, M. Impact of Oxidative Stress on Age-Associated Decline in Oocyte Developmental Competence. Front. Endocrinol. 2019, 10, 811. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Geng, X.; Zheng, W.; Tang, J.; Xu, B.; Shi, Q. Current understanding of ovarian aging. Sci. China Life Sci. 2012, 55, 659–669. [Google Scholar] [CrossRef]
- Koyama, K.; Kang, S.S.; Huang, W.; Yanagawa, Y.; Takahashi, Y.; Nagano, M. Aging-related changes in in vitro-matured bovine oocytes: Oxidative stress, mitochondrial activity and ATP content after nuclear maturation. J. Reprod. Dev. 2014, 60, 136–142. [Google Scholar] [CrossRef]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.; Kumar, M.; Kalmbach, K. Oocyte competency is the key to embryo potential. Fertil. Steril. 2015, 103, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Cree, L.M.; Samuels, D.C.; de Sousa Lopes, S.C.; Rajasimha, H.K.; Wonnapinij, P.; Mann, J.R.; Dahl, H.H.; Chinnery, P.F. A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 2008, 40, 249–254. [Google Scholar] [CrossRef]
- John, J.S. The control of mtDNA replication during differentiation and development. Biochim. Biophys. Acta 2014, 1840, 1345–1354. [Google Scholar] [CrossRef]
- Cotterill, M.; Harris, S.E.; Collado Fernandez, E.; Lu, J.; Huntriss, J.D.; Campbell, B.K.; Picton, H.M. The activity and copy number of mitochondrial DNA in ovine oocytes throughout oogenesis in vivo and during oocyte maturation in vitro. Mol. Hum. Reprod. 2013, 19, 444–450. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef]
- Wai, T.; Teoli, D.; Shoubridge, E.A. The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat. Genet. 2008, 40, 1484–1488. [Google Scholar] [CrossRef]
- May-Panloup, P.; Chretien, M.F.; Savagner, F.; Vasseur, C.; Jean, M.; Malthiery, Y.; Reynier, P. Increased sperm mitochondrial DNA content in male infertility. Hum. Reprod. 2003, 18, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Ao, A.; Zhang, X.; Cyr, D.; Dufort, D.; Shoubridge, E.A. The role of mitochondrial DNA copy number in mammalian fertility. Biol. Reprod. 2010, 83, 52–62. [Google Scholar] [CrossRef]
- Chiaratti, M.R.; Garcia, B.M.; Carvalho, K.F.; Machado, T.S.; Ribeiro, F.K.D.S.; Macabelli, C.H. The role of mitochondria in the female germline: Implications to fertility and inheritance of mitochondrial diseases. Cell Biol. Int. 2018, 42, 711–724. [Google Scholar] [CrossRef]
- Tani, M.; Shinmura, K.; Kohno, T.; Shiroishi, T.; Wakana, S.; Kim, S.R.; Nohmi, T.; Kasai, H.; Takenoshita, S.; Nagamachi, Y.; et al. Genomic structure and chromosomal localization of the mouse Ogg1 gene that is involved in the repair of 8-hydroxyguanine in DNA damage. Mamm. Genome 1998, 9, 32–37. [Google Scholar] [CrossRef]
- Yang, L.; Lin, X.; Tang, H.; Fan, Y.; Zeng, S.; Jia, L.; Li, Y.; Shi, Y.; He, S.; Wang, H.; et al. Mitochondrial DNA mutation exacerbates female reproductive aging via impairment of the NADH/NAD(+) redox. Aging Cell 2020, 19, e13206. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef]
- Spikings, E.C.; Alderson, J.; John, J.C.S. Regulated mitochondrial DNA replication during oocyte maturation is essential for successful porcine embryonic development. Biol. Reprod. 2007, 76, 327–335. [Google Scholar] [CrossRef]
- Dahal, S.; Raghavan, S.C. Mitochondrial genome stability in human: Understanding the role of DNA repair pathways. Biochem. J. 2021, 478, 1179–1197. [Google Scholar] [CrossRef]
- Spikings, E.C.; Alderson, J.; John, J.C.S. Transmission of mitochondrial DNA following assisted reproduction and nuclear transfer. Hum. Reprod. Update 2006, 12, 401–415. [Google Scholar] [CrossRef]
- Rebolledo-Jaramillo, B.; Su, M.S.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef]
- Aretz, I.; Jakubke, C.; Osman, C. Power to the daughters—Mitochondrial and mtDNA transmission during cell division. Biol. Chem. 2020, 401, 533–546. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef]
- Prá, D.; Franke, S.I.; Henriques, J.A.; Fenech, M. Iron and genome stability: An update. Mutat. Res. /Fundam. Mol. Mech. Mutagen. 2012, 733, 92–99. [Google Scholar] [CrossRef]
- Lagouge, M.; Larsson, N.G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef]
- Lakshmanan, L.N.; Yee, Z.; Ng, L.F.; Gunawan, R.; Halliwell, B.; Gruber, J. Clonal expansion of mitochondrial DNA deletions is a private mechanism of aging in long-lived animals. Aging Cell 2018, 17, e12814. [Google Scholar] [CrossRef]
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and ageing. Biochim. Biophys. Acta 2010, 1797, 961–967. [Google Scholar] [CrossRef]
- Hammond, E.R.; Green, M.P.; Shelling, A.N.; Berg, M.C.; Peek, J.C.; Cree, L.M. Oocyte mitochondrial deletions and heteroplasmy in a bovine model of ageing and ovarian stimulation. Mol. Hum. Reprod. 2016, 22, 261–271. [Google Scholar] [CrossRef]
- Ferreira, A.F.; Soares, M.; Almeida-Santos, T.; Ramalho-Santos, J.; Sousa, A.P. Aging and oocyte competence: A molecular cell perspective. WIREs Mech. Dis. 2023, 15, e1613. [Google Scholar] [CrossRef]
- Sun, Q.Y.; Miao, Y.L.; Schatten, H. Towards a new understanding on the regulation of mammalian oocyte meiosis resumption. Cell Cycle 2009, 8, 2741–2747. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Fan, L.H.; Jing, Y.; Li, J.; Ouyang, Y.C.; Wang, Z.B.; Hou, Y.; Sun, Q.Y. N-acetyl-L-cysteine (NAC) delays post-ovulatory oocyte aging in mouse. Aging 2019, 11, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Wilson, I.J.; Yu-Wai-Man, P.; Coxhead, J.; Deehan, D.; Horvath, R.; Taylor, R.W.; Samuels, D.C.; Santibanez-Koref, M.; Chinnery, P.F. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 2013, 22, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Lu, J.; Ma, F.; Keinan, A.; Gu, Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 10654–10659. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Larsson, N.G. Keeping mtDNA in shape between generations. PLoS Genet. 2014, 10, e1004670. [Google Scholar] [CrossRef]
- Jansen, R.P.; de Boer, K. The bottleneck: Mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol. Cell. Endocrinol. 1998, 145, 81–88. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Sen, A.; Boix, J.; Pla-Martín, D. Endosomal-dependent mitophagy coordinates mitochondrial nucleoid and mtDNA elimination. Autophagy 2023, 19, 2609–2610. [Google Scholar] [CrossRef]
- Ross, J.M.; Stewart, J.B.; Hagström, E.; Brené, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013, 501, 412–415. [Google Scholar] [CrossRef]
- Chatzovoulou, K.; Mayeur, A.; Gigarel, N.; Jabot-Hanin, F.; Hesters, L.; Munnich, A.; Frydman, N.; Bonnefont, J.P.; Steffann, J. Mitochondrial DNA mutations do not impact early human embryonic development. Mitochondrion 2021, 58, 59–63. [Google Scholar] [CrossRef]
- Steffann, J.; Monnot, S.; Bonnefont, J.P. mtDNA mutations variously impact mtDNA maintenance throughout the human embryofetal development. Clin. Genet. 2015, 88, 416–424. [Google Scholar] [CrossRef]
- Schon, E.A.; DiMauro, S.; Hirano, M.; Gilkerson, R.W. Therapeutic prospects for mitochondrial disease. Trends Mol. Med. 2010, 16, 268–276. [Google Scholar] [CrossRef]
- Boucret, L.; Bris, C.; Seegers, V.; Goudenège, D.; Desquiret-Dumas, V.; Domin-Bernhard, M.; Ferré-L’Hotellier, V.; Bouet, P.E.; Descamps, P.; Reynier, P.; et al. Deep sequencing shows that oocytes are not prone to accumulate mtDNA heteroplasmic mutations during ovarian ageing. Hum. Reprod. 2017, 32, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Hartley, F.; Alageel, A.; Appeltant, R.; Gray, N.; Repapi, E.; Wells, D.; Williams, S.A.; Poulton, J. No evidence for age-related differences in mitochondrial RNA quality in the female germline. Reprod. Fertil. 2022, 3, 198–206. [Google Scholar] [CrossRef] [PubMed]
- De Paula, W.B.; Agip, A.N.; Missirlis, F.; Ashworth, R.; Vizcay-Barrena, G.; Lucas, C.H.; Allen, J.F. Female and male gamete mitochondria are distinct and complementary in transcription, structure, and genome function. Genome Biol. Evol. 2013, 5, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta 2013, 1833, 1979–1984. [Google Scholar] [CrossRef]
- Barritt, J.A.; Cohen, J.; Brenner, C.A. Mitochondrial DNA point mutation in human oocytes is associated with maternal age. Reprod. Biomed. Online 2000, 1, 96–100. [Google Scholar] [CrossRef]
- Chan, C.C.; Liu, V.W.; Lau, E.Y.; Yeung, W.S.; Ng, E.H.; Ho, P.C. Mitochondrial DNA content and 4977 bp deletion in unfertilized oocytes. Mol. Hum. Reprod. 2005, 11, 843–846. [Google Scholar] [CrossRef]
- Morimoto, N.; Hashimoto, S.; Yamanaka, M.; Nakano, T.; Satoh, M.; Nakaoka, Y.; Iwata, H.; Fukui, A.; Morimoto, Y.; Shibahara, H. Mitochondrial oxygen consumption rate of human embryos declines with maternal age. J. Assist. Reprod. Genet. 2020, 37, 1815–1821. [Google Scholar] [CrossRef]
- Barritt, J.A.; Brenner, C.A.; Cohen, J.; Matt, D.W. Mitochondrial DNA rearrangements in human oocytes and embryos. Mol. Hum. Reprod. 1999, 5, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.L.; Niven-Fairchild, T.; Powell, S.; Buradagunta, S. Mitochondrial deoxyribonucleic acid deletions in oocytes and reproductive aging in women. Fertil. Steril. 1995, 64, 577–583. [Google Scholar] [CrossRef]
- Brenner, C.A.; Wolny, Y.M.; Barritt, J.A.; Matt, D.W.; Munné, S.; Cohen, J. Mitochondrial DNA deletion in human oocytes and embryos. Mol. Hum. Reprod. 1998, 4, 887–892. [Google Scholar] [CrossRef]
- May-Panloup, P.; Chretien, M.F.; Jacques, C.; Vasseur, C.; Malthiery, Y.; Reynier, P. Low oocyte mitochondrial DNA content in ovarian insufficiency. Hum. Reprod. 2005, 20, 593–597. [Google Scholar] [CrossRef] [PubMed]
- May-Panloup, P.; Vignon, X.; Chretien, M.F.; Heyman, Y.; Tamassia, M.; Malthiery, Y.; Reynier, P. Increase of mitochondrial DNA content and transcripts in early bovine embryogenesis associated with upregulation of mtTFA and NRF1 transcription factors. Reprod. Biol. Endocrinol. 2005, 3, 65. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, M.; Alfarawati, S.; Hurd, D.; Paolucci, M.; Shovelton, J.; Fragouli, E.; Wells, D. Simultaneous assessment of aneuploidy, polymorphisms, and mitochondrial DNA content in human polar bodies and embryos with the use of a novel microarray platform. Fertil. Steril. 2014, 102, 1385–1392. [Google Scholar] [CrossRef]
- Murakoshi, Y.; Sueoka, K.; Takahashi, K.; Sato, S.; Sakurai, T.; Tajima, H.; Yoshimura, Y. Embryo developmental capability and pregnancy outcome are related to the mitochondrial DNA copy number and ooplasmic volume. J. Assist. Reprod. Genet. 2013, 30, 1367–1375. [Google Scholar] [CrossRef]
- Iwata, H.; Goto, H.; Tanaka, H.; Sakaguchi, Y.; Kimura, K.; Kuwayama, T.; Monji, Y. Effect of maternal age on mitochondrial DNA copy number, ATP content and IVF outcome of bovine oocytes. Reprod. Fertil. Dev. 2011, 23, 424–432. [Google Scholar] [CrossRef]
- Fragouli, E.; Spath, K.; Alfarawati, S.; Kaper, F.; Craig, A.; Michel, C.E.; Kokocinski, F.; Cohen, J.; Munne, S.; Wells, D. Altered levels of mitochondrial DNA are associated with female age, aneuploidy, and provide an independent measure of embryonic implantation potential. PLoS Genet. 2015, 11, e1005241. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, M.; Jiang, Z.; Seli, E. Mitochondrial dysfunction and ovarian aging. Am. J. Reprod. Immunol. 2017, 77, e12651. [Google Scholar] [CrossRef] [PubMed]
- Seli, E. Mitochondrial DNA as a biomarker for in-vitro fertilization outcome. Curr. Opin. Obstet. Gynecol. 2016, 28, 158–163. [Google Scholar] [CrossRef]
- Ravichandran, K.; McCaffrey, C.; Grifo, J.; Morales, A.; Perloe, M.; Munne, S.; Wells, D.; Fragouli, E. Mitochondrial DNA quantification as a tool for embryo viability assessment: Retrospective analysis of data from single euploid blastocyst transfers. Hum. Reprod. 2017, 32, 1282–1292. [Google Scholar] [CrossRef]
- Fragouli, E.; McCaffrey, C.; Ravichandran, K.; Spath, K.; Grifo, J.A.; Munne, S.; Wells, D. Clinical implications of mitochondrial DNA quantification on pregnancy outcomes: A blinded prospective non-selection study. Hum. Reprod. 2017, 32, 2340–2347. [Google Scholar] [CrossRef]
- Lledo, B.; Ortiz, J.A.; Morales, R.; García-Hernández, E.; Ten, J.; Bernabeu, A.; Llácer, J.; Bernabeu, R. Comprehensive mitochondrial DNA analysis and IVF outcome. Hum. Reprod. Open 2018, 2018, hoy023. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Su, J.; Taylor, D.; Scott, R.T. Telomere DNA deficiency is associated with development of human embryonic aneuploidy. PLoS Genet. 2011, 7, e1002161. [Google Scholar] [CrossRef]
- Kim, J.; Seli, E. Mitochondria as a biomarker for IVF outcome. Reproduction 2019, 157, R235–R242. [Google Scholar] [CrossRef]
- Akbari, M.; Nilsen, H.L.; Montaldo, N.P. Dynamic features of human mitochondrial DNA maintenance and transcription. Front. Cell Dev. Biol. 2022, 10, 984245. [Google Scholar] [CrossRef]
- Herrmann, G.K.; Yin, Y.W. The Role of Poly(ADP-ribose) Polymerase 1 in Nuclear and Mitochondrial Base Excision Repair. Biomolecules 2023, 13, 1195. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, G.K.; Russell, W.K.; Garg, N.J.; Yin, Y.W. Poly(ADP-ribose) polymerase 1 regulates mitochondrial DNA repair in an NAD-dependent manner. J. Biol. Chem. 2021, 296, 100309. [Google Scholar] [CrossRef]
- Ross, J.M.; Coppotelli, G.; Hoffer, B.J.; Olson, L. Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Sci. Rep. 2014, 4, 6569. [Google Scholar] [CrossRef]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabo, C.; Clark, R.S. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003, 278, 18426–18433. [Google Scholar] [CrossRef]
- Bertoldo, M.J.; Listijono, D.R.; Ho, W.J.; Riepsamen, A.H.; Goss, D.M.; Richani, D.; Jin, X.L.; Mahbub, S.; Campbell, J.M.; Habibalahi, A.; et al. NAD(+) Repletion Rescues Female Fertility during Reproductive Aging. Cell Rep. 2020, 30, 1670–1681.e7. [Google Scholar] [CrossRef] [PubMed]
- Di Emidio, G.; Falone, S.; Artini, P.G.; Amicarelli, F.; D’Alessandro, A.M.; Tatone, C. Mitochondrial Sirtuins in Reproduction. Antioxidants 2021, 10, 1047. [Google Scholar] [CrossRef]
- Tatone, C.; Di Emidio, G.; Barbonetti, A.; Carta, G.; Luciano, A.M.; Falone, S.; Amicarelli, F. Sirtuins in gamete biology and reproductive physiology: Emerging roles and therapeutic potential in female and male infertility. Hum. Reprod. Update 2018, 24, 267–289. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef]
- Wan, W.; Hua, F.; Fang, P.; Li, C.; Deng, F.; Chen, S.; Ying, J.; Wang, X. Regulation of Mitophagy by Sirtuin Family Proteins: A Vital Role in Aging and Age-Related Diseases. Front. Aging Neurosci. 2022, 14, 845330. [Google Scholar] [CrossRef]
- Liang, J.; Huang, F.; Song, Z.; Tang, R.; Zhang, P.; Chen, R. Impact of NAD+ metabolism on ovarian aging. Immun. Ageing 2023, 20, 70. [Google Scholar] [CrossRef]
- Zhan, T.; Xiong, H.; Pang, J.; Zhang, W.; Ye, Y.; Liang, Z.; Huang, X.; He, F.; Jian, B.; He, W.; et al. Modulation of NAD+ biosynthesis activates SIRT1 and resists cisplatin-induced ototoxicity. Toxicol. Lett. 2021, 349, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.L.; Gibb, Z.; Swegen, A.; Grupen, C.G. NAD(+), Sirtuins and PARPs: Enhancing oocyte developmental competence. J. Reprod. Dev. 2022, 68, 345–354. [Google Scholar] [CrossRef]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Homer, H.A. Understanding oocyte ageing: Can. we influence the process as clinicians? Curr. Opin. Obstet. Gynecol. 2021, 33, 218–224. [Google Scholar] [CrossRef]
- Liang, J.; Huang, F.; Hao, X.; Zhang, P.; Chen, R. Nicotinamide mononucleotide supplementation rescues mitochondrial and energy metabolism functions and ameliorates inflammatory states in the ovaries of aging mice. MedComm 2024, 5, e727. [Google Scholar] [CrossRef]
- Yang, Q.; Li, H.; Wang, H.; Chen, W.; Zeng, X.; Luo, X.; Xu, J.; Sun, Y. Deletion of enzymes for de novo NAD(+) biosynthesis accelerated ovarian aging. Aging Cell 2023, 22, e13904. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Cui, Z.; Gao, Q.; Rui, R.; Xiong, B. Nicotinamide Mononucleotide Supplementation Reverses the Declining Quality of Maternally Aged Oocytes. Cell Rep. 2020, 32, 107987. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zhou, Y.; Tang, W.; Ren, C.; Jiang, A.; Wang, X.; Qian, X.; Zhou, Z.; Gong, A. Long-term treatment of Nicotinamide mononucleotide improved age-related diminished ovary reserve through enhancing the mitophagy level of granulosa cells in mice. J. Nutr. Biochem. 2022, 101, 108911. [Google Scholar] [CrossRef] [PubMed]
- Dalton, C.M.; Szabadkai, G.; Carroll, J. Measurement of ATP in single oocytes: Impact of maturation and cumulus cells on levels and consumption. J. Cell. Physiol. 2014, 229, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Smits, M.A.J.; Schomakers, B.V.; van Weeghel, M.; Wever, E.J.M.; Wüst, R.C.I.; Dijk, F.; Janssens, G.E.; Goddijn, M.; Mastenbroek, S.; Houtkooper, R.H.; et al. Human ovarian aging is characterized by oxidative damage and mitochondrial dysfunction. Hum. Reprod. 2023, 38, 2208–2220. [Google Scholar] [CrossRef]
- Pinto, M.; Moraes, C.T. Mechanisms linking mtDNA damage and aging. Free Radic. Biol. Med. 2015, 85, 250–258. [Google Scholar] [CrossRef]
- Ribeiro, S.; Sousa, M. In Vitro Fertilisation and Intracytoplasmic Sperm Injection predictive factors: A review of the effect of female age, ovarian reserve, male age, and male factor on IVF/ICSI treatment outcomes. JBRA Assist. Reprod. 2023, 27, 97–111. [Google Scholar] [CrossRef]
- Kobayashi, H.; Yoshimoto, C.; Matsubara, S.; Shigetomi, H.; Imanaka, S. Altered Energy Metabolism, Mitochondrial Dysfunction, and Redox Imbalance Influencing Reproductive Performance in Granulosa Cells and Oocyte During Aging. Reprod. Sci. 2024, 31, 906–916. [Google Scholar] [CrossRef]
- Kobayashi, H.; Imanaka, S. Exploring potential pathways from oxidative stress to ovarian aging. J. Obstet. Gynaecol. Res. 2024; online ahead of print. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Search Mode | The Keyword and Search Term Combinations |
|---|---|
| Search term 1 | mitochondria |
| Search term 2 | aging |
| Search term 3 | oocytes OR granulosa cells OR cumulus cells |
| Search term 4 | mtDNA mutations OR mtDNA repair |
| Search term 5 | oxidative stress OR reactive oxygen species OR ROS |
| Search term 6 | nicotinamide adenine dinucleotide OR NAD OR NADH OR NADPH |
| Search term 7 | poly ADP-ribose polymerase OR PARP1 |
| Search | Search term 1 AND Search term 2 AND Search term 3 |
| Search term 1 AND Search term 2 AND Search term 3 AND Search term 4 | |
| Search term 1 AND Search term 2 AND Search term 3 AND Search term 5 | |
| Search term 1 AND Search term 2 AND Search term 3 AND Search term 6 | |
| Search term 1 AND Search term 2 AND Search term 3 AND Search term 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobayashi, H.; Imanaka, S. Mitochondrial DNA Damage and Its Repair Mechanisms in Aging Oocytes. Int. J. Mol. Sci. 2024, 25, 13144. https://doi.org/10.3390/ijms252313144
Kobayashi H, Imanaka S. Mitochondrial DNA Damage and Its Repair Mechanisms in Aging Oocytes. International Journal of Molecular Sciences. 2024; 25(23):13144. https://doi.org/10.3390/ijms252313144
Chicago/Turabian StyleKobayashi, Hiroshi, and Shogo Imanaka. 2024. "Mitochondrial DNA Damage and Its Repair Mechanisms in Aging Oocytes" International Journal of Molecular Sciences 25, no. 23: 13144. https://doi.org/10.3390/ijms252313144
APA StyleKobayashi, H., & Imanaka, S. (2024). Mitochondrial DNA Damage and Its Repair Mechanisms in Aging Oocytes. International Journal of Molecular Sciences, 25(23), 13144. https://doi.org/10.3390/ijms252313144