
Targeting Ferroptosis in Parkinson’s Disease: Mechanisms and Emerging Therapeutic Strategies

 , ,
, , 
Abstract
1. Introduction
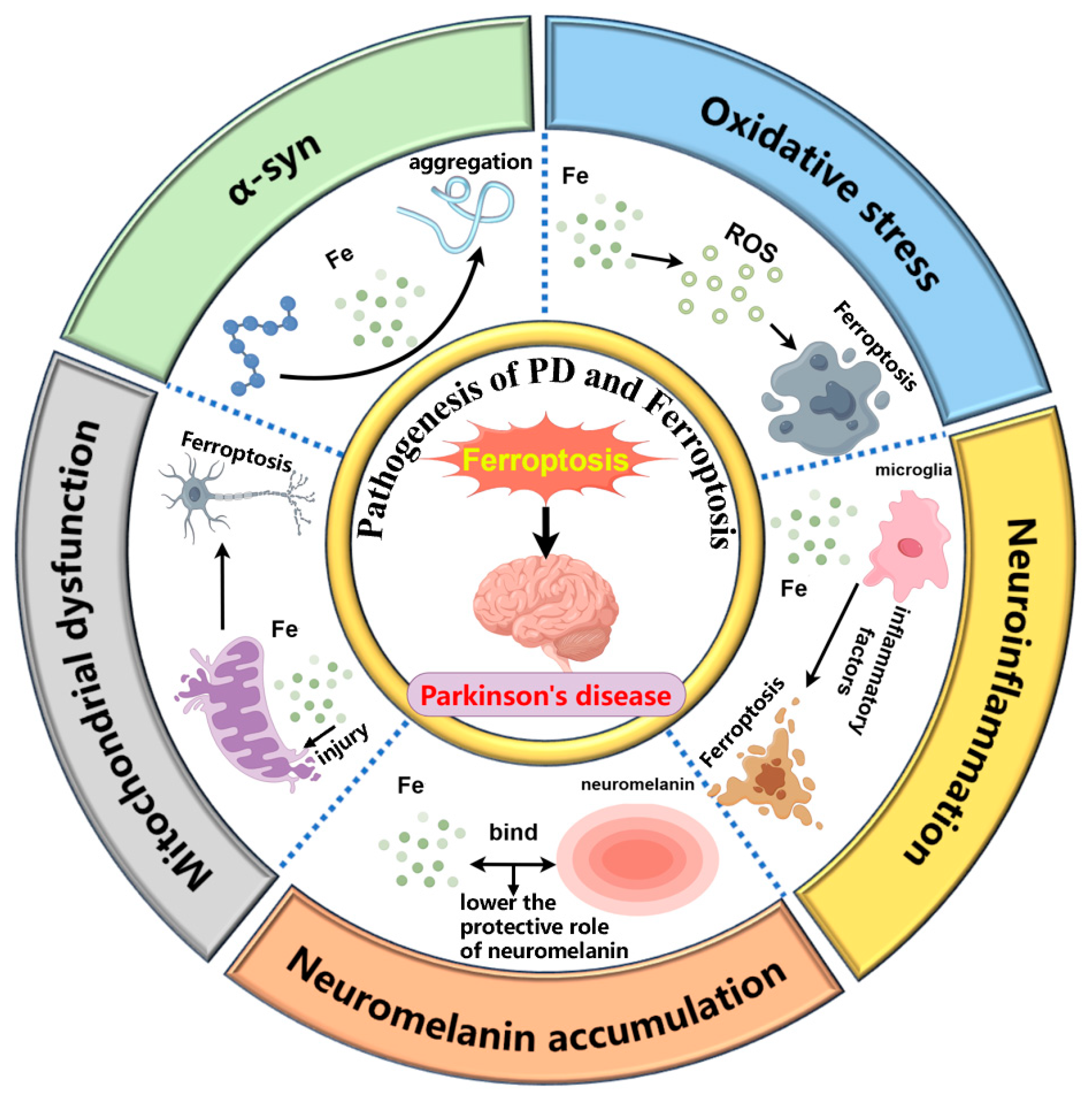
2. The Role of Ferroptosis in PD Pathogenesis
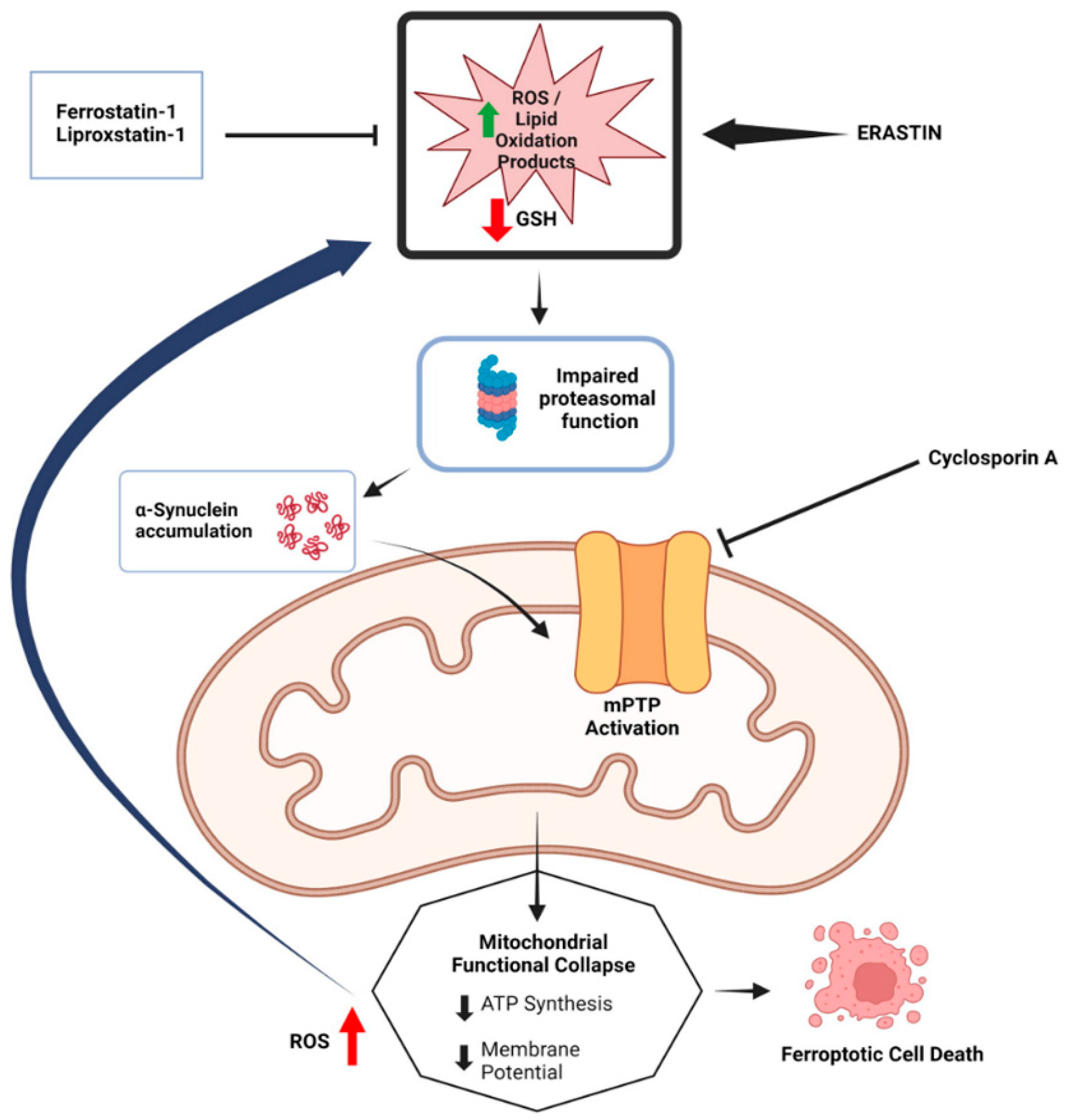
2.1. α-syn and Ferroptosis
2.2. Oxidative Stress and Ferroptosis
2.3. Mitochondrial Dysfunction and Ferroptosis
2.4. Microglia Associated Neuroinflammation and Ferroptosis
2.5. NM Accumulation and Ferroptosis
3. Therapeutic Approaches Targeting Ferroptosis in PD
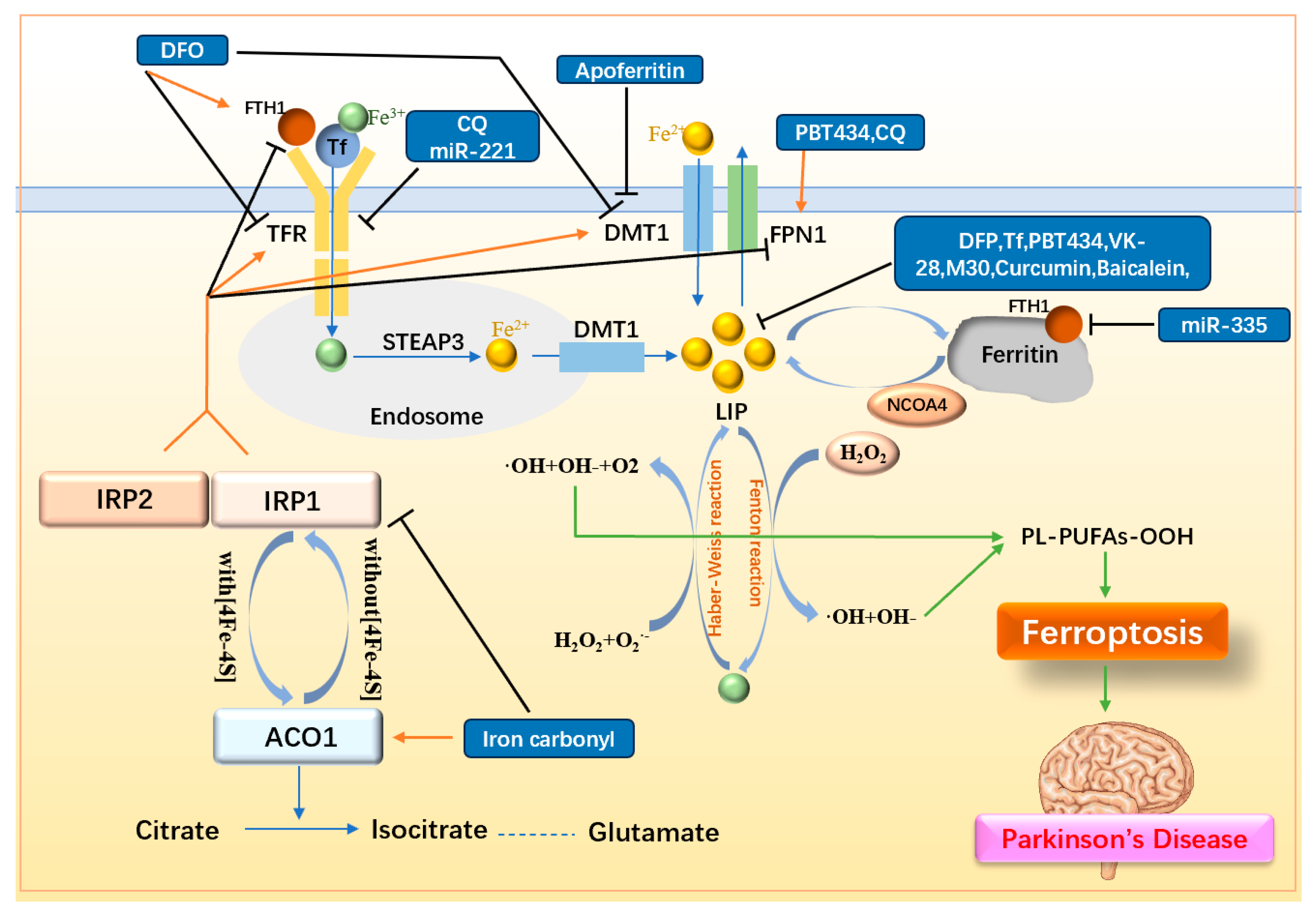
3.1. Decreasing Intracellular Iron Load
3.1.1. Simple Iron Chelators
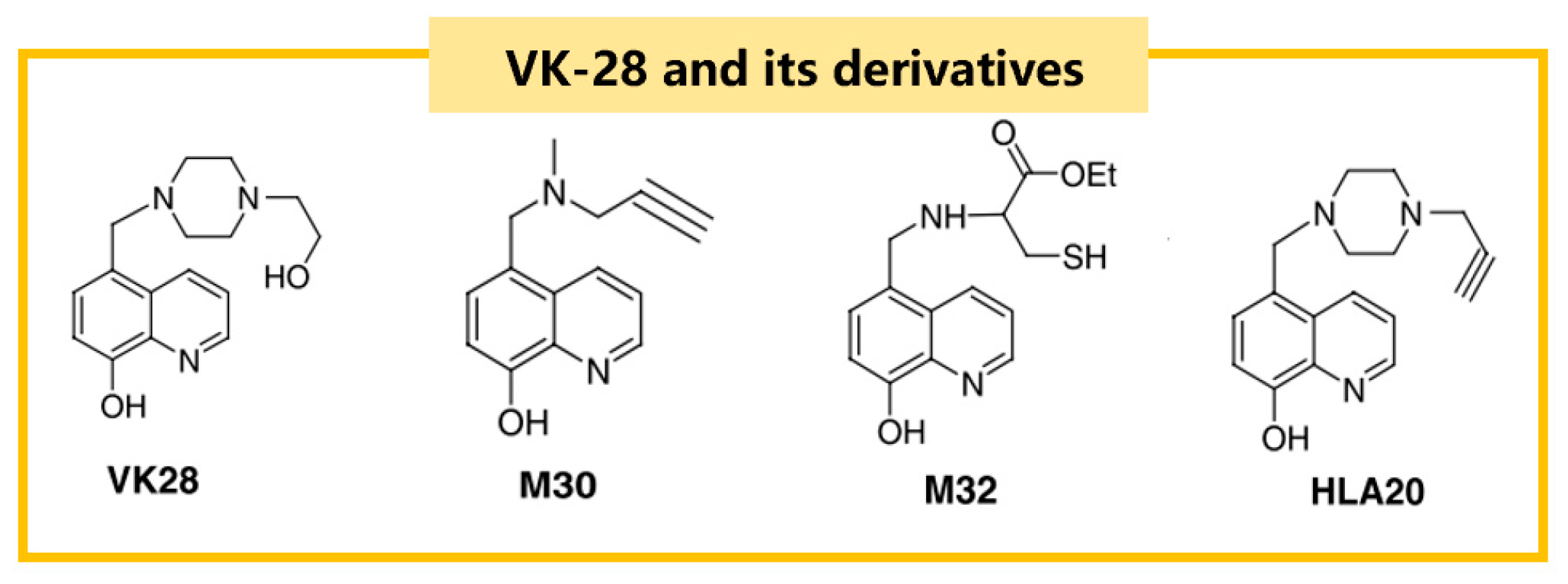
3.1.2. Iron Chelators with Multi-Dimensional Function
Hydroxyquinoline
Clioquinol
Flavonoids
3.1.3. Other Iron Metabolism-Related Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Inducer/Cell or Animal Models | Pharmacology | Mechanism | Reference |
|---|---|---|---|---|
| DFO | MPTP/mice | ↓motor deficiency ↑TH positive neurons ↓iron-positive cells in SN and striatum | iron chelation ↓α-syn, DMT1, TFR | [61,64] |
| DFP | MPTP/mice | ↑GSH ↓MDA ↓8-oxodeoxyguanosine ↑dopamine in striatum | iron chelation ↓unstable LIP in mitochondria | [64] |
| Transferrin | MPTP/mice | ↓motor deficiency | iron chelation | [66] |
| PBT434 | 6-OHDA/mice MPTP/mice Transgenic mice(hA35T α-syn) | ↓neurons loss in SN ↓oxidative damage markers | iron chelation ↓α-syn in SN ↑FPN1 ↑DJ-1 | [67] |
| VK-28 | 6-OHDA/rats | ↓neurotoxicity ↑dopaminergic neurons | iron chelation | [69] |
| M30 | Lactacystin/mice | ↓dopaminergic neurons loss | iron chelation ↓MAO-A, MAO-B non-selectively ↑bcl-2 levels ↓microglial activation ↓proteasome inhibition | [70] |
| HLA20 | 6-OHDA/P19 | neuroprotection | iron chelation ↓MAO-B selectively | [68] |
| CQ | MPTP/monkeys | ↓iron in SN ↓ROS ↓4-HNE ↑serum SOD ↑GSH↓MDA | Iron chelator ↑FPN1 ↓TFR2 ↑AKT/mTOR ↓p53-mediated cell death | [73] |
| Curcumin | 6-OHDA/rats | ↑dopamine in striatum ↑TH positive neurons in striatum ↓iron-stained cells | iron chelation | [74] |
| Baicalein | Vitamin K/SK-N-MC | ↓iron content ↓ROS ↓MDA | iron chelation ↓Bax ↓caspase-9 ↑bcl-2 | [75] |
| MPTP/mice | ↓motor deficiency ↑dopaminergic neurons | iron chelation ↓nuclear shift of NF-κB | [76] | |
| SN4741 cells | ↓α-syn transmission | covalently bound to α-syn | [77] | |
| Apoferritin | MPTP/mice | ↓LIP | ↓DMT1, ACSL4 ↑FSP1 | [78] |
| Iron carbonyl | Retenone/rat | Control iron utilization | ↓IRP1 ↑ACO1 | [79] |
| miR-335 | 6-OHDA/rats 6-OHDA/PC12 | ↑LIP | ↓FTH1 | [80] |
| miR-221 | MPP+/SH-SY5Y | ↓LIP | ↓TFR2 | [81] |
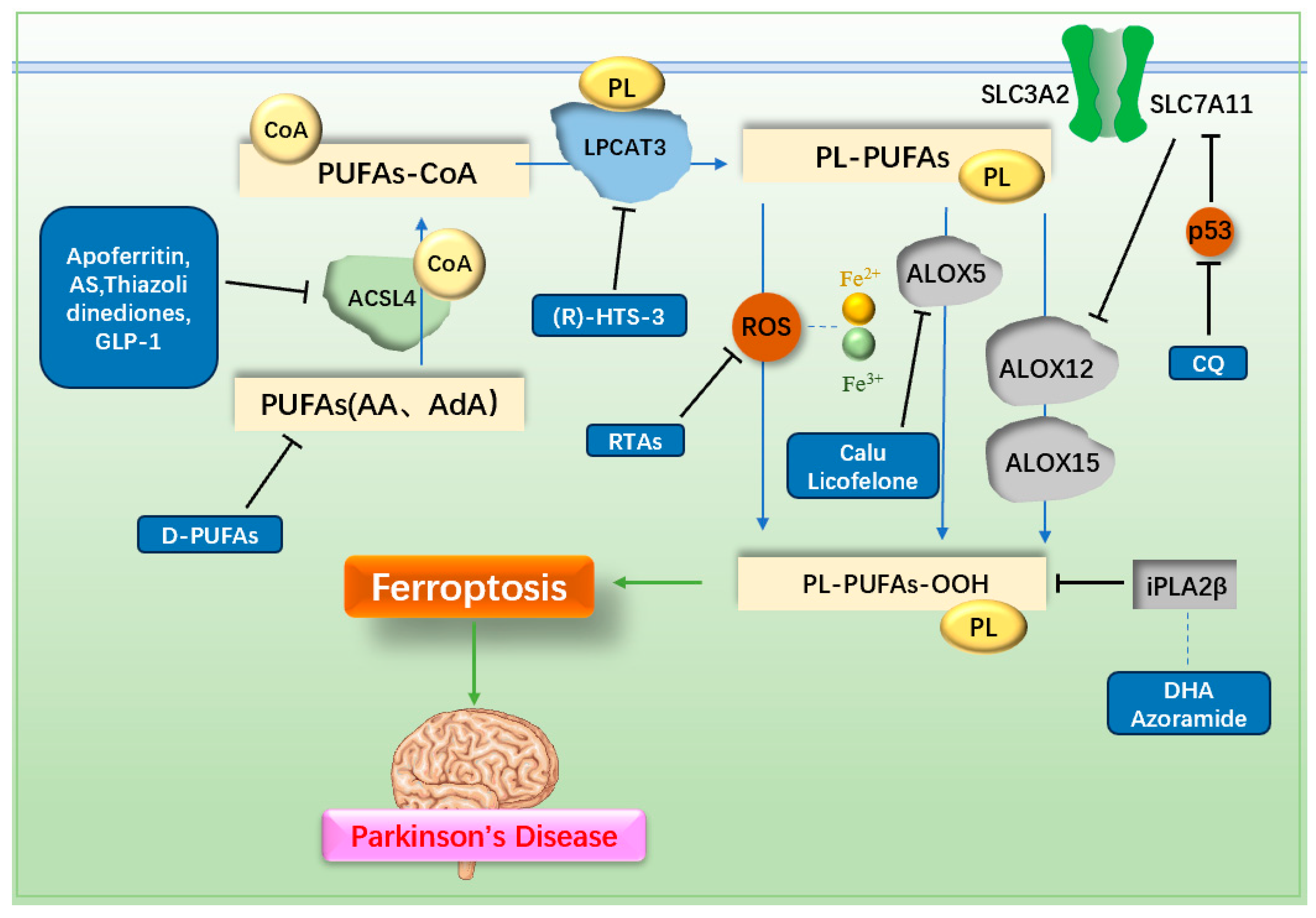
3.2. Decreasing LPO Generation
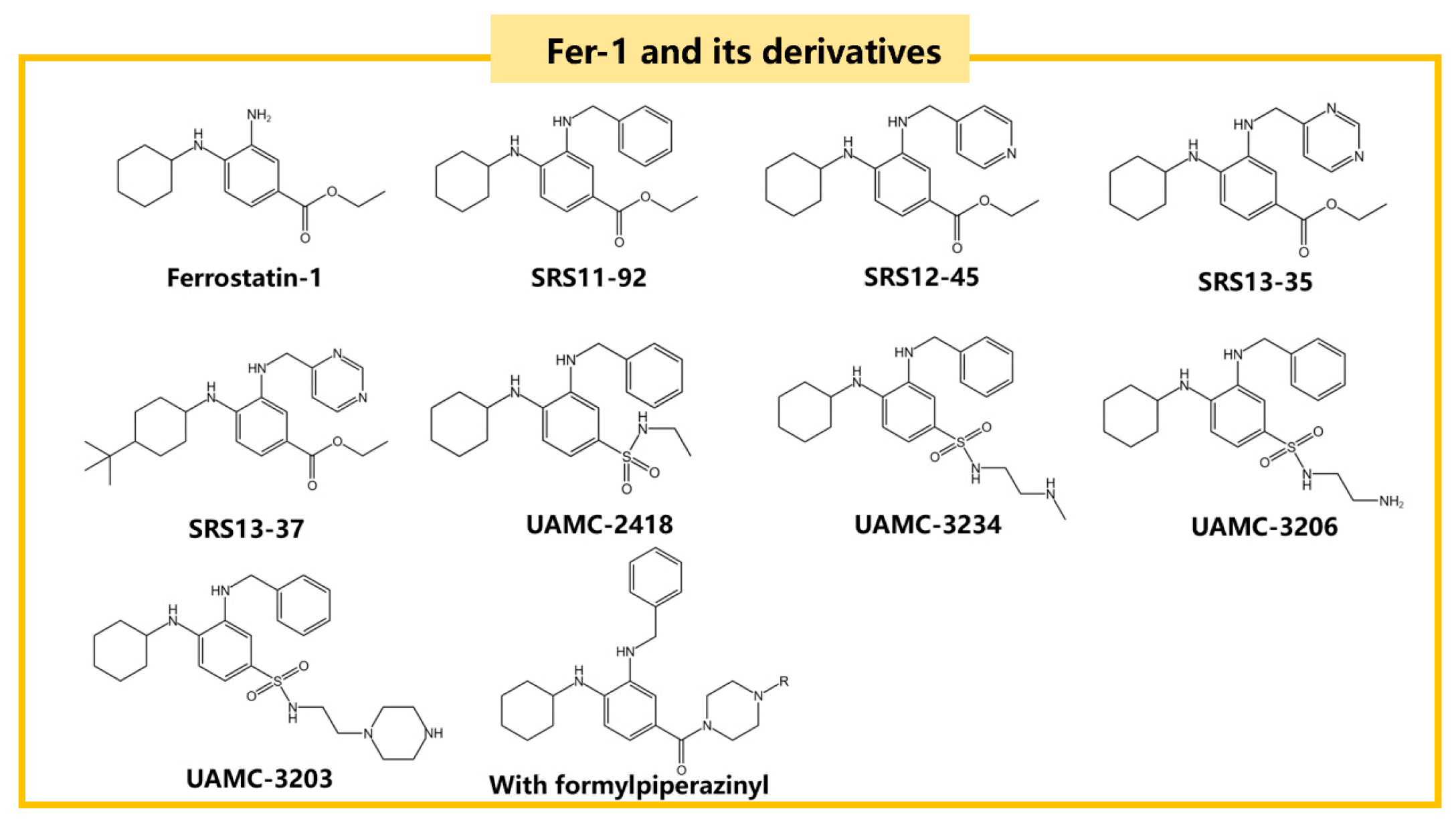
3.2.1. Radical Trapping Antioxidants (RTAs)
3.2.2. ACSL4 Inhibitor
3.2.3. LPCAT3 Inhibitor
3.2.4. ALOX5 Inhibitor
3.2.5. PUFAs Inhibitor
3.2.6. iPLA2β-Related Drugs
| Drugs | Inducer/Cell or Animal Models | Pharmacology | Mechanism | Reference |
|---|---|---|---|---|
| CuII(astm) | RSL3/mice cortical neurons | ↓LPO | RTA | [87] |
| Fer-1 | Glutamate/HT-22 | ↑GPX4 ↓MDA ↑SOD ↓ROS ↓LPO | RTA ↑Nrf2/GPX4 | [45] |
| TEMPO | Glutamate/mouse hippocampal cell lines | ↓LPO ↓cell death | RTA | [89] |
| AS | RSL3/HT22 | ↓LPO | ↓ACSL4 | [100] |
| Calusenamide | MPTP/mice | ↓LPO | ↓ALOX5 activation/nuclear translocation | [103] |
| Licofelone | MPTP/mice | ↑locomotor ability | ↓ALOX5 | [104] |
| D-PUFAs | Mixed cultures of cortical neurons of rats | ↓α-syn-induced LPO and α-syn-induced cell death | compete with PUFAs | [105,106] |
| DHA | PLA2G6D331Y/D331Y mice | Improve motor deficits ↓neuroinflammation | ↓LPO | [109] |
| Azoramide | PLA2G6D331Y dopaminergic neurons | Improve DA damage | ↑ER function ↑CREB signaling | [107] |
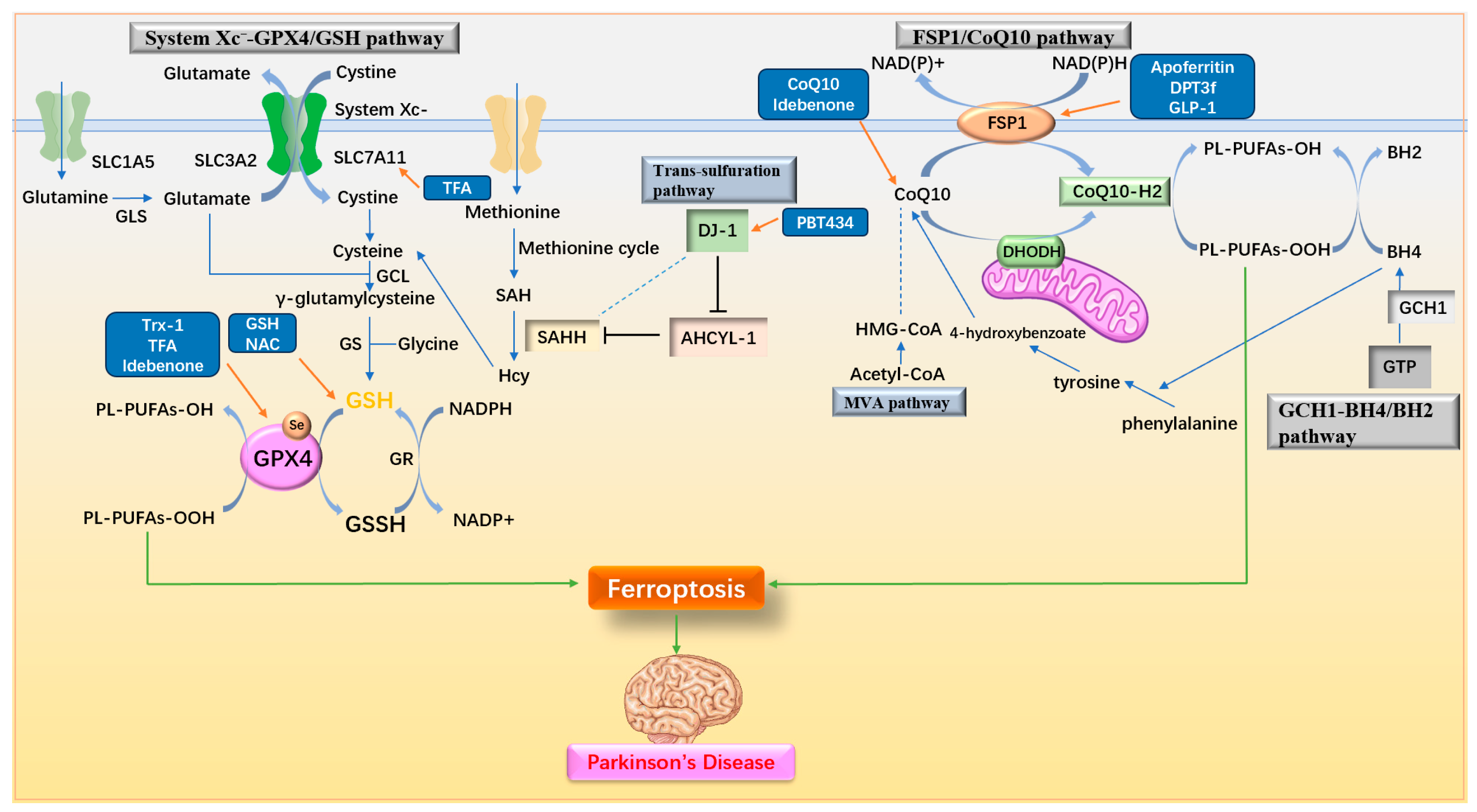
3.3. Regulating Antioxidant Pathway
3.3.1. Regulating System Xc−-GPX4/GSH Pathway
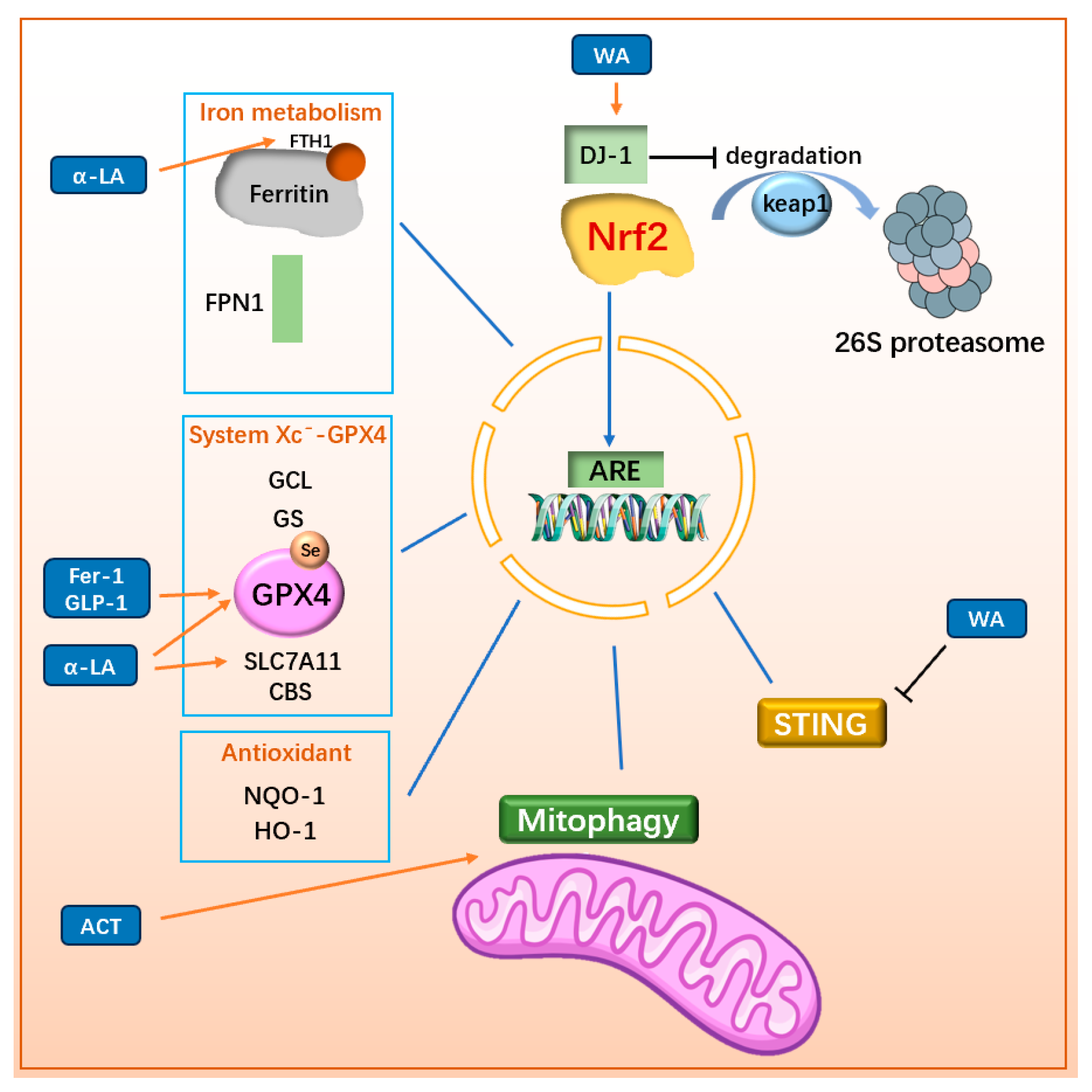
3.3.2. Regulating Nrf2-Related Pathway
3.3.3. Regulating FSP1/CoQ10 Pathway
3.3.4. Multifunctional Antioxidant Regulator
| Drugs | Inducer/Cell or Animal Models | Pharmacology | Mechanism | Reference |
|---|---|---|---|---|
| Trx-1 | MPP+/PC12 MPP+/SH-SY5Y MPTP/mice | ↑GSH | ↑GPX4 | [117] |
| TFA | MPTP/mice | ↑GSH | ↑SLC7A11/GPX4 | [118] |
| α-LA | MPP+/PC12 | ↓MDA, 4-HNE, iron, ROS | ↑PI3K/Akr/Nrf2/ SLC7A11/GPX4 | [129] |
| 6-OHDA/PC12 | ↓iron, LPO ↓mitochondrial damage | ↑SIRT1/Nrf2/ FTH1/GPX4 | [130] | |
| Fer-1 | Glutamate/HT-22 | ↓MDA ↑SOD ↓ROS ↓LPO | RTA ↑Nrf2/GPX4 | [45] |
| ACT | MPP+/SH-SY5Y MPTP/mice | ↑mitochondrial integrity in dopaminergic neurons ↓LPO | ↑Nrf2-mitophagy | [132] |
| WA | MPTP/mice | ↓loss of dopaminergic neurons ↓neuroinflammation ↓movement disorder | ↑DJ1/Nrf2 while ↓STING | [35,133] |
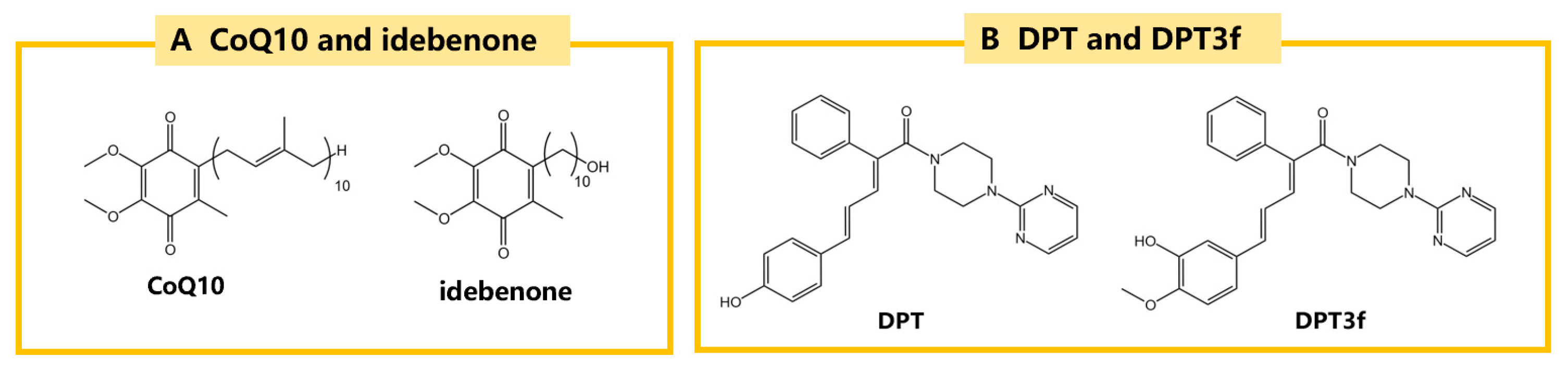
| CoQ10 | MPTP/mice | ↑survival of dopaminergic neurons and TH positive neurons ↓α-syn aggregation | ↑CoQ10 | [138] |
| Idebenone | rotenone/rats | ↓movement disorder ↑survival of TH positive neurons | ↑GPX4 ↑NAD(P)H dihydrogenase [quinone]-1 | [46] |
| MPTP/mice | improve PD symptoms | ↑Parkin/PINK1 mitophagy | [143] | |
| DPT3f | Ischemic stroke | ↑CoQ10 | ↑FSP1/CoQ10 | [144] |
| Novel Probiotic L. lactis MG1363-pMG36e-GLP-1 | MPTP/mice | Neurotrophy ↑CoQ10 ↓LPO ↓α-syn ↑TH positive nerve cells ↑BBB integrity ↑intestinal barrier reverse dysbacteriosis | ↑Keap1/Nrf2/GPX4 ↑FSP1/CoQ10 ↓ACSL4 | [101] |
4. Conclusions
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. Mr. Parkinson’s Letter on the Committee of Apothecaries. Med. Phys. J. 1813, 29, 132–133. [Google Scholar] [PubMed]
- Kalinderi, K.; Papaliagkas, V.; Fidani, L. Pharmacogenetics and levodopa induced motor complications. Int. J. Neurosci. 2019, 129, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Valldeoriola, F.; Torres, V. Predicting impulse control disorder in Parkinson’s Disease: Is there a formula? Eur. Neuropsychopharmacol. 2023, 72, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef]
- Yu, Y.; Yan, Y.; Niu, F.; Wang, Y.; Chen, X.; Su, G.; Liu, Y.; Zhao, X.; Qian, L.; Liu, P.; et al. Ferroptosis: A cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021, 7, 193. [Google Scholar] [CrossRef]
- Mahoney-Sánchez, L.; Bouchaoui, H.; Ayton, S.; Devos, D.; Duce, J.A.; Devedjian, J.C. Ferroptosis and its potential role in the physiopathology of Parkinson’s Disease. Progress. Neurobiol. 2021, 196, 101890. [Google Scholar] [CrossRef]
- Nagatsu, T.; Sawada, M. Biochemistry of postmortem brains in Parkinson’s disease: Historical overview and future prospects. In Neuropsychiatric Disorders An Integrative Approach. Journal of Neural Transmission; Springer: Vienna, Austria, 2007; pp. 113–120. [Google Scholar]
- Jiang, H.; Wang, J.; Rogers, J.; Xie, J. Brain Iron Metabolism Dysfunction in Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 3078–3101. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carter, C.J.; Wells, F.R.; Javoy-Agid, F.; Agid, Y.; Lees, A.; Jenner, P.; Marsden, C.D. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J. Neurochem. 1989, 52, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Melki, R. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J. Park. Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef]
- Nikam, S.; Nikam, P.; Ahaley, S.K.; Sontakke, A.V. Oxidative stress in Parkinson’s disease. Indian J. Clin. Biochem. 2009, 24, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 216–231. [Google Scholar] [CrossRef] [PubMed]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef]
- Zhang, W.; Zecca, L.; Wilson, B.; Ren, H.W.; Wang, Y.J.; Wang, X.M.; Hong, J.S. Human neuromelanin: An endogenous microglial activator for dopaminergic neuron death. Front. Biosci. (Elite Ed) 2013, 5, 1–11. [Google Scholar] [CrossRef]
- Sulzer, D.; Edwards, R.H. The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef]
- Zhao, Q.; Tao, Y.; Zhao, K.; Ma, Y.; Xu, Q.; Liu, C.; Zhang, S.; Li, D. Structural Insights of Fe(3+) Induced α-synuclein Fibrillation in Parkinson’s Disease. J. Mol. Biol. 2023, 435, 167680. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef]
- Saito, Y.; Kawashima, A.; Ruberu, N.N.; Fujiwara, H.; Koyama, S.; Sawabe, M.; Arai, T.; Nagura, H.; Yamanouchi, H.; Hasegawa, M.; et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003, 62, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Wang, Q.; Xiong, X.; Chen, X.; Tu, Z.; Li, B.; Zhang, F.; Chen, C.; Pan, M.; Xu, T.; et al. Deficiency of parkin causes neurodegeneration and accumulation of pathological α-synuclein in monkey models. J. Clin. Investig. 2024, 134, e179633. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Y.; Qu, L.; Chen, B.; Jiang, H.; Song, N.; Xie, J. Iron-induced oxidative stress contributes to α-synuclein phosphorylation and up-regulation via polo-like kinase 2 and casein kinase 2. Neurochem. Int. 2019, 125, 127–135. [Google Scholar] [CrossRef]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Angelova, P.R.; Choi, M.L.; Berezhnov, A.V.; Horrocks, M.H.; Hughes, C.D.; De, S.; Rodrigues, M.; Yapom, R.; Little, D.; Dolt, K.S.; et al. Alpha synuclein aggregation drives ferroptosis: An interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020, 27, 2781–2796. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remião, F.; Silva, R. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef]
- Mahoney-Sanchez, L.; Bouchaoui, H.; Boussaad, I.; Jonneaux, A.; Timmerman, K.; Berdeaux, O.; Ayton, S.; Krüger, R.; Duce, J.A.; Devos, D.; et al. Alpha synuclein determines ferroptosis sensitivity in dopaminergic neurons via modulation of ether-phospholipid membrane composition. Cell Rep. 2022, 40, 111231. [Google Scholar] [CrossRef]
- Shamoto-Nagai, M.; Hisaka, S.; Naoi, M.; Maruyama, W. Modification of α-synuclein by lipid peroxidation products derived from polyunsaturated fatty acids promotes toxic oligomerization: Its relevance to Parkinson disease. J. Clin. Biochem. Nutr. 2018, 62, 207–212. [Google Scholar] [CrossRef]
- Ganguly, U.; Singh, S.; Bir, A.; Ghosh, A.; Chakrabarti, S.S.; Saini, R.V.; Saso, L.; Bisaglia, M.; Chakrabarti, S. Alpha-synuclein interaction with mitochondria is the final mechanism of ferroptotic death induced by erastin in SH-SY5Y cells. Free Radic. Res. 2024, 58, 217–228. [Google Scholar] [CrossRef]
- Wu, L.; Liu, M.; Liang, J.; Li, N.; Yang, D.; Cai, J.; Zhang, Y.; He, Y.; Chen, Z.; Ma, T. Ferroptosis as a New Mechanism in Parkinson’s Disease Therapy Using Traditional Chinese Medicine. Front. Pharmacol. 2021, 12, 659584. [Google Scholar] [CrossRef]
- Vallerga, C.L.; Zhang, F.; Fowdar, J.; McRae, A.F.; Qi, T.; Nabais, M.F.; Zhang, Q.; Kassam, I.; Henders, A.K.; Wallace, L.; et al. Analysis of DNA methylation associates the cystine-glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 2020, 11, 1238. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Chen, X.; Jiang, L.; Lu, B.; Yuan, M.; Zhu, D.; Zhu, H.; He, Q.; Yang, B.; Ying, M. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 2020, 11, 1251. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef]
- Jian, X.; Zhao, G.; Chen, H.; Wang, Y.; Li, J.; Xie, L.; Li, B. Revealing a novel contributing landscape of ferroptosis-related genes in Parkinson’s disease. Comput. Struct. Biotechnol. J. 2022, 20, 5218–5225. [Google Scholar] [CrossRef]
- Chen, Y.P.; Yu, S.H.; Zhang, G.H.; Hou, Y.B.; Gu, X.J.; Ou, R.W.; Shen, Y.; Song, W.; Chen, X.P.; Zhao, B.; et al. The mutation spectrum of Parkinson-disease-related genes in early-onset Parkinson’s disease in ethnic Chinese. Eur. J. Neurol. 2022, 29, 3218–3228. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Wu, H.; Wang, F.; Ta, N.; Zhang, T.; Gao, W. The Multifaceted Regulation of Mitochondria in Ferroptosis. Life 2021, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gelé, P.; Pétrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Avcı, B.; Günaydın, C.; Güvenç, T.; Yavuz, C.K.; Kuruca, N.; Bilge, S.S. Idebenone Ameliorates Rotenone-Induced Parkinson’s Disease in Rats Through Decreasing Lipid Peroxidation. Neurochem. Res. 2021, 46, 513–522. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, M.; Queralt-Martín, M.; Gurnev, P.A.; Rosencrans, W.M.; Rovini, A.; Jacobs, D.; Abrantes, K.; Hoogerheide, D.P.; Bezrukov, S.M.; Rostovtseva, T.K. Restricting α-synuclein transport into mitochondria by inhibition of α-synuclein-VDAC complexation as a potential therapeutic target for Parkinson’s disease treatment. Cell. Mol. Life Sci. 2022, 79, 368. [Google Scholar] [CrossRef]
- Scheiblich, H.; Eikens, F.; Wischhof, L.; Opitz, S.; Jüngling, K.; Cserép, C.; Schmidt, S.V.; Lambertz, J.; Bellande, T.; Pósfai, B.; et al. Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Neuron 2024, 112, 3106–3125.e3108. [Google Scholar] [CrossRef] [PubMed]
- Joers, V.; Tansey, M.G.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2017, 155, 57–75. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; González-Billault, C.; Núñez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.K.; Zelic, M.; Han, Y.; Teeple, E.; Chen, L.; Sadeghi, M.; Shankara, S.; Guo, L.; Li, C.; Pontarelli, F.; et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 2023, 26, 12–26. [Google Scholar] [CrossRef]
- Zecca, L.; Casella, L.; Albertini, A.; Bellei, C.; Zucca, F.A.; Engelen, M.; Zadlo, A.; Szewczyk, G.; Zareba, M.; Sarna, T. Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson’s disease. J. Neurochem. 2008, 106, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, J.; Zhao, P.; Li, S.; Zhang, R.; Zhang, X.; Liu, D.; Zhang, B. Neuromelanin enhances the toxicity of α-synuclein in SK-N-SH cells. J. Neural Transm. 2012, 119, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebrón, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef]
- Tian, Y.; Lu, J.; Hao, X.; Li, H.; Zhang, G.; Liu, X.; Li, X.; Zhao, C.; Kuang, W.; Chen, D.; et al. FTH1 Inhibits Ferroptosis Through Ferritinophagy in the 6-OHDA Model of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1796–1812. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, T.; Bai, R.; Xie, Y. Hydroxypyridinone-Based Iron Chelators with Broad-Ranging Biological Activities. J. Med. Chem. 2020, 63, 14470–14501. [Google Scholar] [CrossRef]
- Yanatori, I.; Kishi, F. DMT1 and iron transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef]
- Bórquez, D.A.; Urrutia, P.J. Iron regulatory protein 1: The deadly switch of ferroptosis. Neural Regen. Res. 2024, 19, 477–478. [Google Scholar] [CrossRef]
- Su, Y.; Jiao, Y.; Cai, S.; Xu, Y.; Wang, Q.; Chen, X. The molecular mechanism of ferroptosis and its relationship with Parkinson’s disease. Brain Res. Bull. 2024, 213, 110991. [Google Scholar] [CrossRef]
- Zeng, X.; An, H.; Yu, F.; Wang, K.; Zheng, L.; Zhou, W.; Bao, Y.; Yang, J.; Shen, N.; Huang, D. Benefits of Iron Chelators in the Treatment of Parkinson’s Disease. Neurochem. Res. 2021, 46, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; McLean, C.; Bush, A.I.; Finkelstein, D.I. Transferrin protects against Parkinsonian neurotoxicity and is deficient in Parkinson’s substantia nigra. Signal Transduct. Target. Ther. 2016, 1, 16015. [Google Scholar] [CrossRef]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef]
- Zheng, H.; Gal, S.; Weiner, L.M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M.B. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: In vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem. 2005, 95, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Shachar, D.B.; Kahana, N.; Kampel, V.; Warshawsky, A.; Youdim, M.B. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacology 2004, 46, 254–263. [Google Scholar] [CrossRef]
- Zhu, W.; Xie, W.; Pan, T.; Xu, P.; Fridkin, M.; Zheng, H.; Jankovic, J.; Youdim, M.B.; Le, W. Prevention and restoration of lactacystin-induced nigrostriatal dopamine neuron degeneration by novel brain-permeable iron chelators. FASEB J. 2007, 21, 3835–3844. [Google Scholar] [CrossRef]
- Zheng, H.; Weiner, L.M.; Bar-Am, O.; Epsztejn, S.; Cabantchik, Z.I.; Warshawsky, A.; Youdim, M.B.; Fridkin, M. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorganic Med. Chem. 2005, 13, 773–783. [Google Scholar] [CrossRef]
- Asao, M. Clioquinol and S.M.O.N.: Reanalysis of original data. Lancet 1979, 1, 446. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, W.; Zeng, W.; Cheng, A.; Shi, R.; Zhengli, C. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging 2020, 12, 9515–9533. [Google Scholar] [CrossRef] [PubMed]
- Du, X.-X.; Xu, H.-M.; Jiang, H.; Song, N.; Wang, J.; Xie, J.-X. Curcumin protects nigral dopaminergic neurons by iron-chelation in the 6-hydroxydopamine rat model of Parkinson’s disease. Neurosci. Bull. 2012, 28, 253–258. [Google Scholar] [CrossRef]
- Yazdanparast, M.M.a.R. Protective Effects of Flavonoid Baicalein against Menadione-Induced Damage in SK-N-MC Cells. CellBio 2013, 2, 35–44. [Google Scholar]
- Lee, E.; Park, H.R.; Ji, S.T.; Lee, Y.; Lee, J. Baicalein attenuates astroglial activation in the 1-methyl-4-phenyl-1,2,3,4-tetrahydropyridine-induced Parkinson’s disease model by downregulating the activations of nuclear factor-κB, ERK, and JNK. J. Neurosci. Res. 2014, 92, 130–139. [Google Scholar] [CrossRef]
- Li, X.; Zhang, G.; Nie, Q.; Wu, T.; Jiao, L.; Zheng, M.; Wan, X.; Li, Y.; Wu, S.; Jiang, B.; et al. Baicalein blocks α-synuclein secretion from SN4741 cells and facilitates α-synuclein polymerization to big complex. Neurosci. Lett. 2017, 655, 109–114. [Google Scholar] [CrossRef]
- Song, L.M.; Xiao, Z.X.; Zhang, N.; Yu, X.Q.; Cui, W.; Xie, J.X.; Xu, H.M. Apoferritin improves motor deficits in MPTP-treated mice by regulating brain iron metabolism and ferroptosis. iScience 2021, 24, 102431. [Google Scholar] [CrossRef]
- Berry, T.M.; Moustafa, A.A. A novel treatment strategy to prevent Parkinson’s disease: Focus on iron regulatory protein 1 (IRP1). Int. J. Neurosci. 2023, 133, 67–76. [Google Scholar] [CrossRef]
- Li, X.; Si, W.; Li, Z.; Tian, Y.; Liu, X.; Ye, S.; Huang, Z.; Ji, Y.; Zhao, C.; Hao, X.; et al. miR-335 promotes ferroptosis by targeting ferritin heavy chain 1 in in vivo and in vitro models of Parkinson’s disease. Int. J. Mol. Med. 2021, 47, 61. [Google Scholar] [CrossRef]
- Asci, R.; Vallefuoco, F.; Andolfo, I.; Bruno, M.; De Falco, L.; Iolascon, A. Trasferrin receptor 2 gene regulation by microRNA 221 in SH-SY5Y cells treated with MPP⁺ as Parkinson’s disease cellular model. Neurosci. Res. 2013, 77, 121–127. [Google Scholar] [CrossRef]
- Iqbal, S.; Jabeen, F.; Kahwa, I.; Omara, T. Suberosin Alleviates Thiazolidinedione-Induced Cardiomyopathy in Diabetic Rats by Inhibiting Ferroptosis via Modulation of ACSL4-LPCAT3 and PI3K-AKT Signaling Pathways. Cardiovasc. Toxicol. 2023, 23, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Chou, V.P.; Ko, N.; Holman, T.R.; Manning-Boğ, A.B. Gene-environment interaction models to unmask susceptibility mechanisms in Parkinson’s disease. J. Vis. Exp. 2014, 83, e50960. [Google Scholar] [CrossRef]
- Koppula, P.; Zhuang, L.; Gan, B. Cytochrome P450 reductase (POR) as a ferroptosis fuel. Protein Cell 2021, 12, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, M.; Tamtaji, O.R.; Dadgostar, E.; Daneshvar Kakhaki, R.; Bahmani, F.; Abolhassani, J.; Aarabi, M.H.; Kouchaki, E.; Memarzadeh, M.R.; Asemi, Z. The effects of omega-3 fatty acids and vitamin E co-supplementation on clinical and metabolic status in patients with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Neurochem. Int. 2017, 108, 183–189. [Google Scholar] [CrossRef]
- Southon, A.; Szostak, K.; Acevedo, K.M.; Dent, K.A.; Volitakis, I.; Belaidi, A.A.; Barnham, K.J.; Crouch, P.J.; Ayton, S.; Donnelly, P.S.; et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. Br. J. Pharmacol. 2020, 177, 656–667. [Google Scholar] [CrossRef]
- Zilka, O.; Poon, J.F.; Pratt, D.A. Radical-Trapping Antioxidant Activity of Copper and Nickel Bis(Thiosemicarbazone) Complexes Underlies Their Potency as Inhibitors of Ferroptotic Cell Death. J. Am. Chem. Soc. 2021, 143, 19043–19057. [Google Scholar] [CrossRef]
- Jiang, X.; Wu, K.; Ye, X.Y.; Xie, T.; Zhang, P.; Blass, B.E.; Bai, R. Novel druggable mechanism of Parkinson’s disease: Potential therapeutics and underlying pathogenesis based on ferroptosis. Med. Res. Rev. 2023, 43, 872–896. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Devisscher, L.; Van Coillie, S.; Hofmans, S.; Van Rompaey, D.; Goossens, K.; Meul, E.; Maes, L.; De Winter, H.; Van Der Veken, P.; Vandenabeele, P.; et al. Discovery of Novel, Drug-Like Ferroptosis Inhibitors with in Vivo Efficacy. J. Med. Chem. 2018, 61, 10126–10140. [Google Scholar] [CrossRef]
- Ji, H.L.; Zhang, Y.F.; Zhang, N.Y.; Wang, K.M.; Meng, N.; Zhang, J.; Jiang, C.S. Design, synthesis, and evaluation of formylpiperazine analogs of Ferrostatin-1 as novel improved ferroptosis inhibitors. Bioorganic Med. Chem. 2024, 105, 117716. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.P.; Mazrad, Z.A.I.; Warne, N.M.; Ayton, S.; Bush, A.I.; Kempe, K. Schiff-Base Cross-Linked Poly(2-oxazoline) Micelle Drug Conjugates Possess Antiferroptosis Activity in Numerous In Vitro Cell Models. Biomacromolecules 2024, 25, 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- Kou, D.; Gao, Y.; Li, C.; Zhou, D.; Lu, K.; Wang, N.; Zhang, R.; Yang, Z.; Zhou, Y.; Chen, L.; et al. Intranasal Pathway for Nanoparticles to Enter the Central Nervous System. Nano Lett. 2023, 23, 5381–5390. [Google Scholar] [CrossRef]
- Goertsen, D.; Flytzanis, N.C.; Goeden, N.; Chuapoco, M.R.; Cummins, A.; Chen, Y.; Fan, Y.; Zhang, Q.; Sharma, J.; Duan, Y.; et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2022, 25, 106–115. [Google Scholar] [CrossRef]
- Yin, W.; Ma, H.; Qu, Y.; Ren, J.; Sun, Y.; Guo, Z.N.; Yang, Y. Exosomes: The next-generation therapeutic platform for ischemic stroke. Neural Regen. Res. 2025, 20, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Huang, Q.; Ru, Y.; Luo, Y.; Luo, X.; Liu, D.; Ma, Y.; Zhou, X.; Linghu, M.; Xu, W.; Gao, F.; et al. Identification of a targeted ACSL4 inhibitor to treat ferroptosis-related diseases. Sci. Adv. 2024, 10, eadk1200. [Google Scholar] [CrossRef]
- Yue, M.; Wei, J.; Chen, W.; Hong, D.; Chen, T.; Fang, X. Neurotrophic Role of the Next-Generation Probiotic Strain L. lactis MG1363-pMG36e-GLP-1 on Parkinson’s Disease via Inhibiting Ferroptosis. Nutrients 2022, 14, 4886. [Google Scholar] [CrossRef]
- Reed, A.; Ichu, T.A.; Milosevich, N.; Melillo, B.; Schafroth, M.A.; Otsuka, Y.; Scampavia, L.; Spicer, T.P.; Cravatt, B.F. LPCAT3 Inhibitors Remodel the Polyunsaturated Phospholipid Content of Human Cells and Protect from Ferroptosis. ACS Chem. Biol. 2022, 17, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Wang, M.; Huang, Z.H.; Wang, M.; Sun, W.Y.; Kurihara, H.; Huang, R.T.; Wang, R.; Huang, F.; Liang, L.; et al. ALOX5 inhibition protects against dopaminergic neurons undergoing ferroptosis. Pharmacol. Res. 2023, 193, 106779. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.; Khayatan, D.; Arab, Z.N.; Momtaz, S.; Zare, K.; Jafari, R.M.; Dehpour, A.R.; Abdolghaffari, A.H. Licofelone, a potent COX/5-LOX inhibitor and a novel option for treatment of neurological disorders. Prostaglandins Other Lipid Mediat. 2021, 157, 106587. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Abramov, A.Y.; Shchepinov, M.S. Lipid peroxidation is essential for α-synuclein-induced cell death. J. Neurochem. 2015, 133, 582–589. [Google Scholar] [CrossRef]
- Ke, M.; Chong, C.M.; Zeng, H.; Huang, M.; Huang, Z.; Zhang, K.; Cen, X.; Lu, J.H.; Yao, X.; Qin, D.; et al. Azoramide protects iPSC-derived dopaminergic neurons with PLA2G6 D331Y mutation through restoring ER function and CREB signaling. Cell Death Dis. 2020, 11, 130. [Google Scholar] [CrossRef]
- Chen, D.; Chu, B.; Yang, X.; Liu, Z.; Jin, Y.; Kon, N.; Rabadan, R.; Jiang, X.; Stockwell, B.R.; Gu, W. iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 2021, 12, 3644. [Google Scholar] [CrossRef]
- Yeh, T.H.; Liu, H.F.; Chiu, C.C.; Cheng, M.L.; Huang, G.J.; Huang, Y.C.; Liu, Y.C.; Huang, Y.Z.; Lu, C.S.; Chen, Y.C.; et al. PLA2G6 mutations cause motor dysfunction phenotypes of young-onset dystonia-parkinsonism type 14 and can be relieved by DHA treatment in animal models. Exp. Neurol. 2021, 346, 113863. [Google Scholar] [CrossRef]
- Liu, J.; Tan, J.; Tang, B.; Guo, J. Unveiling the role of iPLA(2)β in neurodegeneration: From molecular mechanisms to advanced therapies. Pharmacol. Res. 2024, 202, 107114. [Google Scholar] [CrossRef]
- Mischley, L.K.; Lau, R.C.; Shankland, E.G.; Wilbur, T.K.; Padowski, J.M. Phase IIb Study of Intranasal Glutathione in Parkinson’s Disease. J. Park. Dis. 2017, 7, 289–299. [Google Scholar] [CrossRef]
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Coles, L.D.; Tuite, P.J.; Öz, G.; Mishra, U.R.; Kartha, R.V.; Sullivan, K.M.; Cloyd, J.C.; Terpstra, M. Repeated-Dose Oral N-Acetylcysteine in Parkinson’s Disease: Pharmacokinetics and Effect on Brain Glutathione and Oxidative Stress. J. Clin. Pharmacol. 2018, 58, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Holmay, M.J.; Terpstra, M.; Coles, L.D.; Mishra, U.; Ahlskog, M.; Öz, G.; Cloyd, J.C.; Tuite, P.J. N-Acetylcysteine boosts brain and blood glutathione in Gaucher and Parkinson diseases. Clin. Neuropharmacol. 2013, 36, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e421. [Google Scholar] [CrossRef] [PubMed]
- Mischley, L.K.; Allen, J.; Bradley, R. Coenzyme Q10 deficiency in patients with Parkinson’s disease. J. Neurol. Sci. 2012, 318, 72–75. [Google Scholar] [CrossRef]
- Bai, L.; Yan, F.; Deng, R.; Gu, R.; Zhang, X.; Bai, J. Thioredoxin-1 Rescues MPP(+)/MPTP-Induced Ferroptosis by Increasing Glutathione Peroxidase 4. Mol. Neurobiol. 2021, 58, 3187–3197. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, G.; Chen, Y.; Yuan, W.; Cai, J.; Feng, A.; Fang, J.; Xu, Q.; Wu, X. Total flavonoids of Astragalus membranaceus protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in mice by inhibiting ferroptosis through SLC7A11/GPX-4 signaling pathway. Food Sci. Hum. Wellness 2024, 13, 414–420. [Google Scholar] [CrossRef]
- Coe, S.; Andreoli, D.; George, M.; Collett, J.; Reed, A.; Cossington, J.; Izadi, H.; Dixon, A.; Mansoubi, M.; Dawes, H. A feasibility study to determine whether the daily consumption of flavonoid-rich pure cocoa has the potential to reduce fatigue and fatigability in people with Parkinson’s (pwP). Clin. Nutr. ESPEN 2022, 48, 68–73. [Google Scholar] [CrossRef]
- Pang, H.; Xue, W.; Shi, A.; Li, M.; Li, Y.; Cao, G.; Yan, B.; Dong, F.; Xiao, W.; He, G.; et al. Multiple-Ascending-Dose Pharmacokinetics and Safety Evaluation of Baicalein Chewable Tablets in Healthy Chinese Volunteers. Clin. Drug Investig. 2016, 36, 713–724. [Google Scholar] [CrossRef]
- Adinolfi, S.; Patinen, T.; Jawahar Deen, A.; Pitkänen, S.; Härkönen, J.; Kansanen, E.; Küblbeck, J.; Levonen, A.L. The KEAP1-NRF2 pathway: Targets for therapy and role in cancer. Redox Biol. 2023, 63, 102726. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Cordaro, M.; Siracusa, R.; Fusco, R.; Peritore, A.F.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Interdonato, L.; Evangelista, M.; et al. Molecular targets for anti-oxidative protection of açaí berry against diabetes myocardial ischemia/reperfusion injury. Free Radic. Res. 2023, 57, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Thapa, K.; Khan, H.; Kanojia, N.; Singh, T.G.; Kaur, A.; Kaur, G. Therapeutic Insights on Ferroptosis in Parkinson’s disease. Eur. J. Pharmacol. 2022, 930, 175133. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Wakabayashi, N.; Misra, V.; Nilles, T.; Biswal, S.; Trush, M.A.; Kensler, T.W. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch. Biochem. Biophys. 2006, 454, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, T.A.; Zhang, W.Y.; Huang, S.R.; Hu, Y.; Sun, J. Rhein attenuates cerebral ischemia-reperfusion injury via inhibition of ferroptosis through NRF2/SLC7A11/GPX4 pathway. Exp. Neurol. 2023, 369, 114541. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Masaki, Y.; Izumi, Y.; Matsumura, A.; Akaike, A.; Kume, T. Protective effect of Nrf2-ARE activator isolated from green perilla leaves on dopaminergic neuronal loss in a Parkinson’s disease model. Eur. J. Pharmacol. 2017, 798, 26–34. [Google Scholar] [CrossRef]
- Liu, L.; Yang, S.; Wang, H. α-Lipoic acid alleviates ferroptosis in the MPP+-induced PC12 cells via activating the PI3K/Akt/Nrf2 pathway. Cell Biol. Int. 2021, 45, 422–431. [Google Scholar] [CrossRef]
- Mao, J.; Gao, H.; Bai, W.; Zeng, H.; Ren, Y.; Liu, Y.; Yang, X. Lipoic acid alleviates LPS-evoked PC12 cell damage by targeting p53 and inactivating the NF-κB pathway. Acta Neurobiol. Exp. 2021, 81, 375–385. [Google Scholar] [CrossRef]
- Li, J.; Yang, D.; Li, Z.; Zhao, M.; Wang, D.; Sun, Z.; Wen, P.; Dai, Y.; Gou, F.; Ji, Y.; et al. PINK1/Parkin-mediated mitophagy in neurodegenerative diseases. Ageing Res. Rev. 2023, 84, 101817. [Google Scholar] [CrossRef]
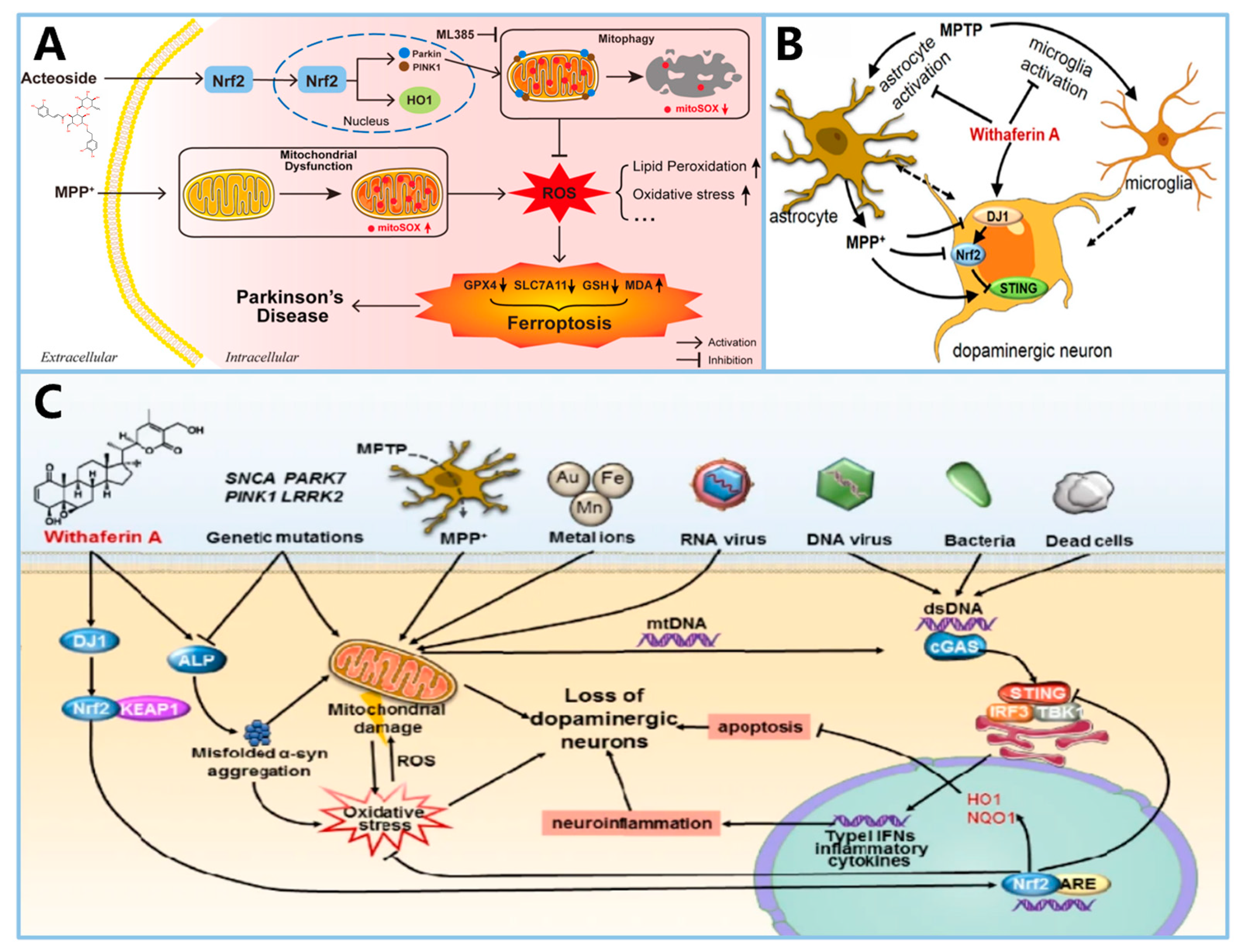
- Han, Z.; Wang, B.; Wen, Y.Q.; Li, Y.N.; Feng, C.X.; Ding, X.S.; Shen, Y.; Yang, Q.; Gao, L. Acteoside alleviates lipid peroxidation by enhancing Nrf2-mediated mitophagy to inhibit ferroptosis for neuroprotection in Parkinson’s disease. Free Radic. Biol. Med. 2024, 223, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, B.; Zhang, C.; Su, Z.; Guo, B.; Zhao, Y.; Zheng, R. The DJ1-Nrf2-STING axis mediates the neuroprotective effects of Withaferin A in Parkinson’s disease. Cell Death Differ. 2021, 28, 2517–2535. [Google Scholar] [CrossRef]
- Quek, H.; Luff, J.; Cheung, K.; Kozlov, S.; Gatei, M.; Lee, C.S.; Bellingham, M.C.; Noakes, P.G.; Lim, Y.C.; Barnett, N.L.; et al. A rat model of ataxia-telangiectasia: Evidence for a neurodegenerative phenotype. Hum. Mol. Genet. 2017, 26, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Nazmi, A.; Field, R.H.; Griffin, E.W.; Haugh, O.; Hennessy, E.; Cox, D.; Reis, R.; Tortorelli, L.; Murray, C.L.; Lopez-Rodriguez, A.B.; et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 2019, 67, 1254–1276. [Google Scholar] [CrossRef]
- Takahashi, M.; Yamada, T. Viral etiology for Parkinson’s disease--a possible role of influenza A virus infection. Jpn. J. Infect. Dis. 1999, 52, 89–98. [Google Scholar] [CrossRef]
- Holm, C.K.; Rahbek, S.H.; Gad, H.H.; Bak, R.O.; Jakobsen, M.R.; Jiang, Z.; Hansen, A.L.; Jensen, S.K.; Sun, C.; Thomsen, M.K.; et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 2016, 7, 10680. [Google Scholar] [CrossRef]
- Cleren, C.; Yang, L.; Lorenzo, B.; Calingasan, N.Y.; Schomer, A.; Sireci, A.; Wille, E.J.; Beal, M.F. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem. 2008, 104, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Storch, A.; Jost, W.H.; Vieregge, P.; Spiegel, J.; Greulich, W.; Durner, J.; Müller, T.; Kupsch, A.; Henningsen, H.; Oertel, W.H.; et al. Randomized, double-blind, placebo-controlled trial on symptomatic effects of coenzyme Q(10) in Parkinson disease. Arch. Neurol. 2007, 64, 938–944. [Google Scholar] [CrossRef]
- Müller, T.; Büttner, T.; Gholipour, A.F.; Kuhn, W. Coenzyme Q10 supplementation provides mild symptomatic benefit in patients with Parkinson’s disease. Neurosci. Lett. 2003, 341, 201–204. [Google Scholar] [CrossRef]
- Erb, M.; Hoffmann-Enger, B.; Deppe, H.; Soeberdt, M.; Haefeli, R.H.; Rummey, C.; Feurer, A.; Gueven, N. Features of idebenone and related short-chain quinones that rescue ATP levels under conditions of impaired mitochondrial complex I. PLoS ONE 2012, 7, e36153. [Google Scholar] [CrossRef]
- Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials 2018, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Sun, W.; Shen, M.; Zhang, Y.; Jiang, M.; Liu, A.; Ma, H.; Lai, X.; Wu, J. Idebenone improves motor dysfunction, learning and memory by regulating mitophagy in MPTP-treated mice. Cell Death Discov. 2022, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tan, Q.; Zhou, H.; Gu, Q.; Xu, J. Discovery of novel diphenylbutene derivative ferroptosis inhibitors as neuroprotective agents. Eur. J. Med. Chem. 2022, 231, 114151. [Google Scholar] [CrossRef]
- Oveisgharan, S.; Yu, L.; Barnes, L.L.; Agrawal, S.; Schneider, J.A.; Bennett, D.A.; Buchman, A.S. Association of Statins With Cerebral Atherosclerosis and Incident Parkinsonism in Older Adults. Neurology 2022, 98, e1976–e1984. [Google Scholar] [CrossRef]
- Kimura, Y.; Mochizuki, H. Targeting Glucagon-Like Peptide 1 Signaling: A Potential Disease Modifying Therapy for Parkinson’s Disease. Mov. Disord. 2024, 39, 1087. [Google Scholar] [CrossRef]
- Marathe, C.S.; Rayner, C.K.; Jones, K.L.; Horowitz, M. Glucagon-like peptides 1 and 2 in health and disease: A review. Peptides 2013, 44, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef]
- Lubomski, M.; Tan, A.H.; Lim, S.Y.; Holmes, A.J.; Davis, R.L.; Sue, C.M. Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. 2020, 267, 2507–2523. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Liu, X.; Wei, M.; Huang, H.; Cao, J.; Liu, S.; Bian, X.; Zhang, Y.; Xiao, F.; Xie, Y.; et al. Microbiota-derived lysophosphatidylcholine alleviates Alzheimer’s disease pathology via suppressing ferroptosis. Cell Metab. 2024. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, H.; Ding, L.; Gong, J.; Zhang, M.; Guo, W.; Xu, P.; Li, S.; Zhang, Y. Trojan Horse Delivery of 4,4′-Dimethoxychalcone for Parkinsonian Neuroprotection. Adv. Sci. 2021, 8, 2004555. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; Xu, K.; Ge, J.; Luo, X.; Wu, M.; Wang, N.; Zeng, J. Targeting Ferroptosis in Parkinson’s Disease: Mechanisms and Emerging Therapeutic Strategies. Int. J. Mol. Sci. 2024, 25, 13042. https://doi.org/10.3390/ijms252313042
Zhou M, Xu K, Ge J, Luo X, Wu M, Wang N, Zeng J. Targeting Ferroptosis in Parkinson’s Disease: Mechanisms and Emerging Therapeutic Strategies. International Journal of Molecular Sciences. 2024; 25(23):13042. https://doi.org/10.3390/ijms252313042
Chicago/Turabian StyleZhou, Minghao, Keyang Xu, Jianxian Ge, Xingnian Luo, Mengyao Wu, Ning Wang, and Jianfeng Zeng. 2024. "Targeting Ferroptosis in Parkinson’s Disease: Mechanisms and Emerging Therapeutic Strategies" International Journal of Molecular Sciences 25, no. 23: 13042. https://doi.org/10.3390/ijms252313042
APA StyleZhou, M., Xu, K., Ge, J., Luo, X., Wu, M., Wang, N., & Zeng, J. (2024). Targeting Ferroptosis in Parkinson’s Disease: Mechanisms and Emerging Therapeutic Strategies. International Journal of Molecular Sciences, 25(23), 13042. https://doi.org/10.3390/ijms252313042